Abstract
Bamboo is one of the most important non-timber forest resources worldwide. It has considerable economic value and unique flowering characteristics. The long juvenile phase in bamboo and unpredictable flowering time limit breeding and genetic improvement and seriously affect the productivity and application of bamboo forests. Members of SQUA-like subfamily genes play an essential role in controlling flowering time and floral organ identity. A comprehensive study was conducted to explain the functions of five SQUA-like subfamily genes in Phyllostachys edulis. Expression analysis revealed that all PeSQUAs have higher transcript levels in the reproductive period than in the juvenile phase. However, PeSQUAs showed divergent expression patterns during inflorescence development. The protein–protein interaction (PPI) patterns among PeSQUAs and other MADS-box members were analyzed by yeast two-hybrid (Y2H) experiments. Consistent with amino acid sequence similarity and phylogenetic analysis, the PPI patterns clustered into two groups. PeMADS2, 13, and 41 interacted with multiple PeMADS proteins, whereas PeMADS3 and 28 hardly interacted with other proteins. Based on our results, PeSQUA might possess different functions by forming protein complexes with other MADS-box proteins at different flowering stages. Furthermore, we chose PeMADS2 for functional analysis. Ectopic expression of PeMADS2 in Arabidopsis and rice caused early flowering, and abnormal phenotype was observed in transgenic Arabidopsis lines. RNA-seq analysis indicated that PeMADS2 integrated multiple pathways regulating floral transition to trigger early flowering time in rice. This function might be due to the interaction between PeMADS2 and homologous in rice. Therefore, we concluded that the five SQUA-like genes showed functional conservation and divergence based on sequence differences and were involved in floral transitions by forming protein complexes in P. edulis. The MADS-box protein complex model obtained in the current study will provide crucial insights into the molecular mechanisms of bamboo’s unique flowering characteristics.
1. Introduction
Bamboos are important members of the subfamily Bambusoideae and the family Poaceae, which are important timber, fiber, and food products worldwide [1]. Unlike other members of Poaceae, bamboo has unique flowering features and an unpredictable juvenile phase [2]. For instance, wood bamboo retains vegetative growth for a very long time (13–120 years) and usually dies after seed production [1]. The flowering incidence may be restricted to a few plants of a population, i.e., so-called sporadic flowering, whereas synchronous flowering may happen across populations in a community in which all plants flower and die within the same year [3]. The long juvenile phase in bamboo and unpredictable flowering time possibly limit breeding and genetic improvement and cause economic losses. Consequently, studying the mechanism of bamboo flowering has great scientific importance and economic value. Although many research groups tried to uncover the cause of bamboo flowering by genomes and transcriptomes [4,5,6], the molecular aspects of bamboo flowering remain unclear.
Phyllostachys edulis (Moso bamboo) genome and the transcriptomes of different bamboo species have been sequenced to characterize genes related to bamboo flowering; most of the cloned genes belonged to the MADS-box gene family [2,5,7,8]. For instance, 16 BeMADS genes were obtained from the flowering transcripts of Bambusa edulis, among which BeMADS1 could undergo co-transformation with other cytosol BeMADS proteins in the nucleus to function as transcription factors (TFs) [8]. PvSOC1 and PvMADS56 from P. violascens can significantly promote flowering in overexpression transgenic Arabidopsis thaliana or Oryza [9,10]. BoMADS50 could interact with SQUAMOSA (SQUA)-like proteins and positively promote flowering in B. oldhamii [6]. Based on previous results from our laboratory, 42 full-length P. edulis MADS-box members were identified, and the expression analysis revealed that several PeMADSs play roles in modulating flowering in Moso bamboo [11]. Ectopic expression of one of the candidate genes, namely, PeMADS5, could promote flowering in A. thaliana [11].
In plants, MADS-box gene family members reportedly play important roles in many aspects of development [12,13,14], particularly the regulation of reproductive development, including the flowering time, establishing floral organ identity, fruit ripening, seed pigmentation, and embryo development [15,16,17,18]. Members from the SQUA-like subfamily, also named as APETALA1/FRUITFULL (AP1/FUL), participate in floral meristem identity, floral organ specification, and flowering regulation [16]. There are four members of the Arabidopsis SQUA subfamily in which the roles of AP1, CAULIFLOWER (CAL), and FUL are crucial for the control of floral organ development and flowering time [19]. AP1 belongs to the A-class gene in the floral organ ABC model, and it is involved in the identification of sepal and petal development [20]. Meanwhile, AP1 and CAL share overlapping roles in floral meristem (FM) identity and determination [21]. FUL has an important role in Arabidopsis carpel and fruit development and is functionally redundant with AP1 and CAL in controlling inflorescence architecture [21,22]. The function of SQUA-like genes that control floral transition is somewhat conserved between eudicots and grass [23]. In rice, SQUA-like genes have redundant roles in controlling flowering time, and the ectopic expression of OsMADS14, OsMADS15, or OsMADS18 results in early flowering and dwarf habit [24,25]. In winter wheat and barley varieties, overexpression of the SQUA-like gene (VERNALIZATION 1) could promote flowering in response to vernalization [26,27]. Two bamboo SQUA-like subfamily genes, PpMADS1 and PpMADS2, were identified from P. praecox, and overexpression of these two genes significantly promotes early flowering in Arabidopsis [28].
Several studies support the important roles of SQUA-like genes in floral transition and inflorescence architecture. However, few studies investigated the regulatory roles of SQUA-like genes in P. edulis, which is the most commonly used species in China and is the only bamboo with whole-genome information released [18]. In this study, we identified five SQUA-like members in P. edulis. The expression patterns of PeMADSs at different flowering stages were comprehensively studied. Yeast two-hybrid (Y2H) assay showed that some SQUA-like members have extensive interactions with other MADS-box subfamilies. The Y2H assay showed that PeMADS2 has the highest number (11) of interaction proteins among the five PeSUQAs subgroup members, and PeAMDS2 was highly expressed during inflorescence initiation. Thus, we prioritize PeMADS2 for further functional study. The ectopic expression of PeMADS2 in Arabidopsis caused early flowering and abnormal phenotype in leaves and inflorescence. Furthermore, overexpression of PeMADS2 in rice also triggered early flowering time, and RNA-seq analysis indicated that PeMADS2 integrated multiple flower signaling pathways in transgenic plants. Our data provided the spatial expression analysis and protein–protein interaction (PPI) of SQUA-like genes with other bamboo MADS-box members. Our findings present new perspectives that contribute to the function of PeMADS2 in regulating the floral transition in transgenic Arabidopsis and rice.
2. Results
2.1. The Bioinformatics Analysis of Five SQUA-like Members in P. edulis
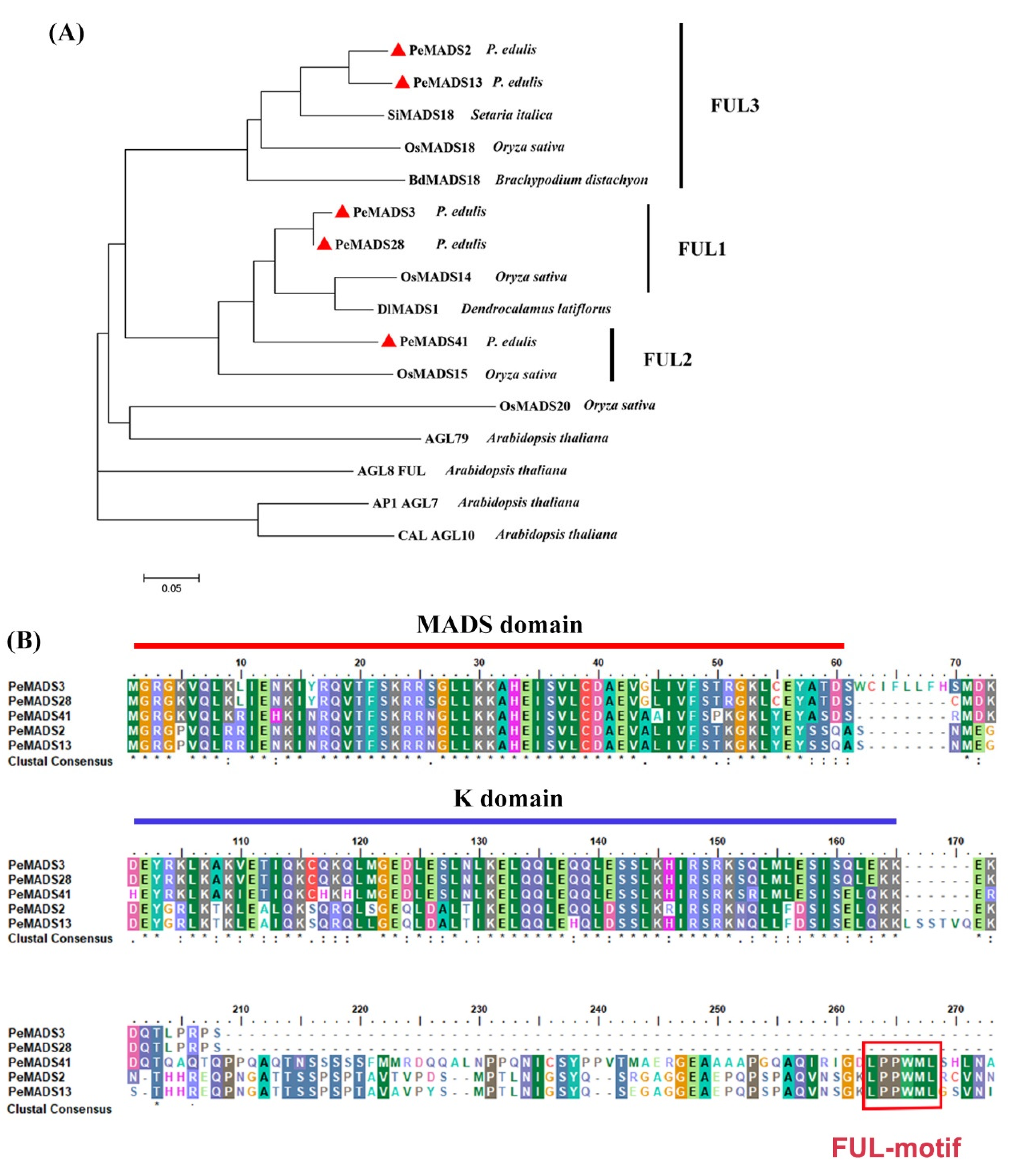
Five SQUA-like genes, namely, PeMADS2, PeMADS3, PeMADS13, PeMADS28, and PeMADS41, were obtained from our previous study [11]. The phylogenetic tree of SQUA-like proteins showed that these five P. edulis SQUA-like genes are clustered within monocot species (Oryza sativa, Brachypodium distachyon, Setaria italica, and D. latiflorus) and are separate from Arabidopsis (Figure 1A). PeMADS2 and PeMADS13 were clustered in one branch with rice OsMADS18, whereas PeMADS3 and PeMADS28 were gathered in the other branch with rice OsMADS14. However, PeMADS41 showed less homology with the other four genes. The alignment indicated that all five bamboo SQUA-like proteins contained a well-conserved MADS-domain in the N-terminus, followed by the less conserved K (keratin-like) domain, and a divergent C-terminal region (Figure 1B). Furthermore, a conserved FUL-like (LPPWML) motif was identified in the C-terminal region of PeMADS2, PeMADS13, and PeMADS41, which was highly conserved in most monocot SQUA-like genes [29]. However, this motif was absent in PeMADS3 and PeMADS28 due to the truncated C terminus (Figure 1B).
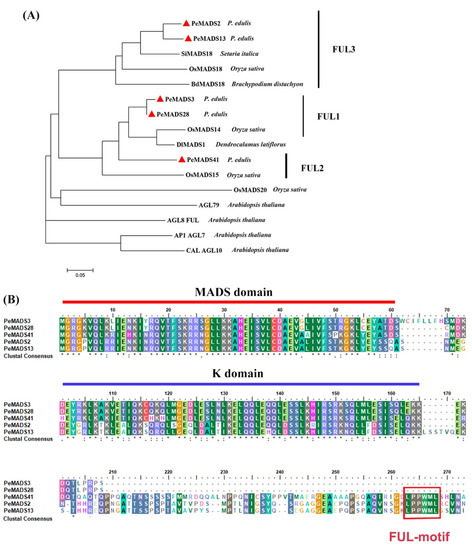
Figure 1.
Sequence analyses of PeSQUA-like. (A) A phylogenetic tree of Moso bamboo PeSQUA-like and SQUA-like proteins from other species. The phylogenetic tree was constructed with the neighbor-joining (NJ) method and evaluated by bootstrap analysis using MEGA soft (version 7.0). Sixteen SQUA-like proteins were used: four from A. thaliana (AGL7, AGL9, AGL10, and AGL79), four from rice (OsMADS14, OsMADS15, OsMADS18 and OsMADS20), one from Brachypodium distachyon (BdMADS18), one from Setaria italic (SiMADS18), and one from D. latiflorus (DlMADS1). P. edulis SQUA-like proteins are marked by red triangles. (B) Alignment of AA sequences of P. edulis SQUA-like proteins. The lined regions represent the conserved MADS domain and K domain. The red boxed region in the C-terminal represents the FUL-like motif. Residues that are identical in all sequences in the alignment are marked with “*” in the bottom row, conserved and semi-conserved substitutions with “:” and “.” respectively.
2.2. Expression Analysis of PeSQUA-like Genes during Different Flowering Stages
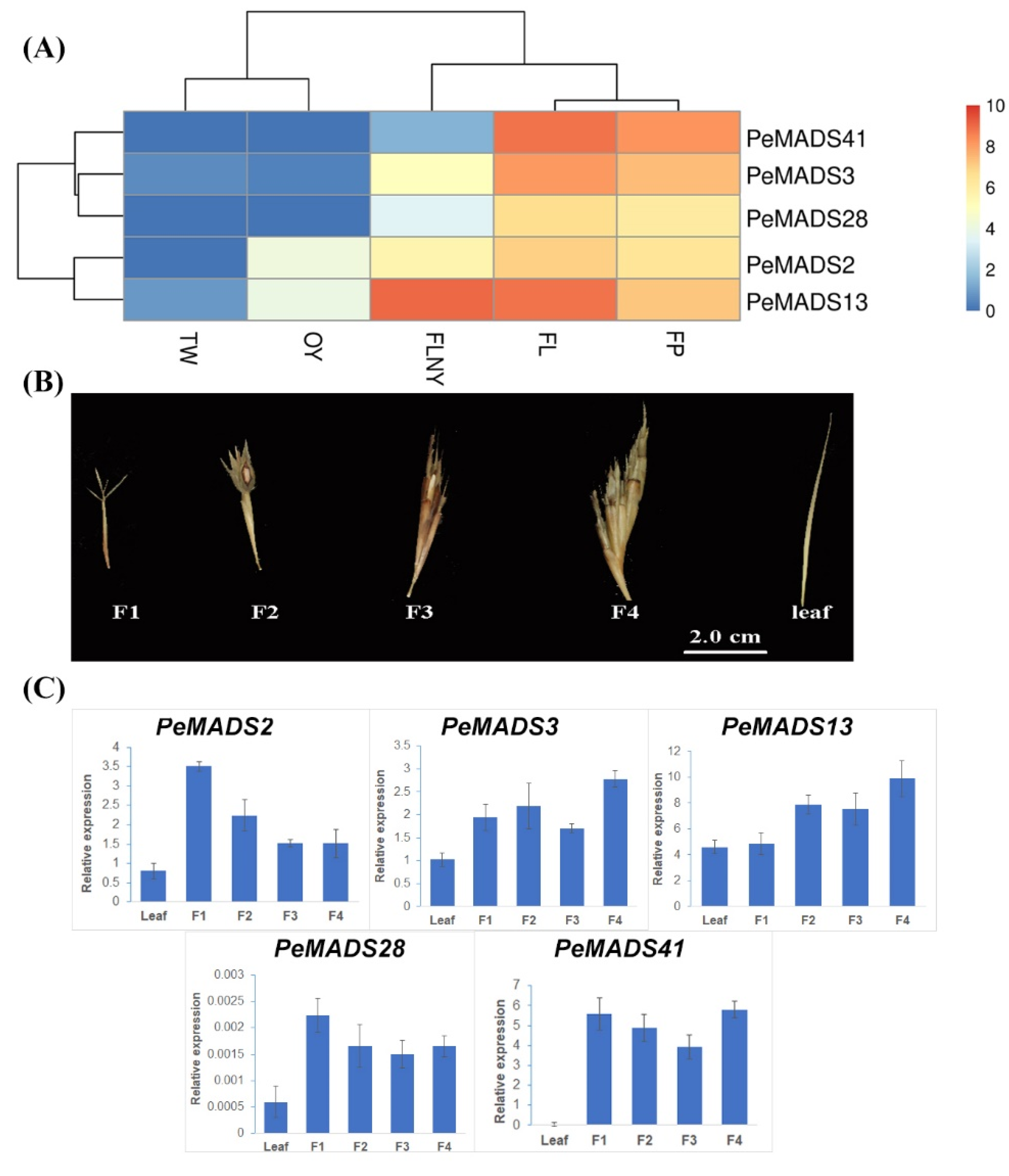
To better elucidate the roles of PeSQUA-like genes in the transition from vegetative growth to flowering, the expression levels were analyzed from online transcriptomes [30]. The transcriptomes include leaf samples from different developmental stages, as follows: 3-week-old seedlings (TW); 1-year-old plants (OY); plants that will flower in the next year (FLNY); and flowering plants (FL), and flower florets (FP) samples from the flowering stage. The PeSQUA-like genes were predominantly detected in leaves (FL) and florets (FP) from flowering plants and were moderately expressed in plants ready to flower (FLNY). However, they were poorly expressed in juvenile plants (TW and OY) (Figure 2A). Based on the expression patterns, the PeSQUAs were divided into two groups, which were similar to the homology tree (Figure 1A). Besides the flowering tissues, the transcripts of PeMADS2 and PeMADS13 were both high in the plants at the reproductive transition stage (FLNY).
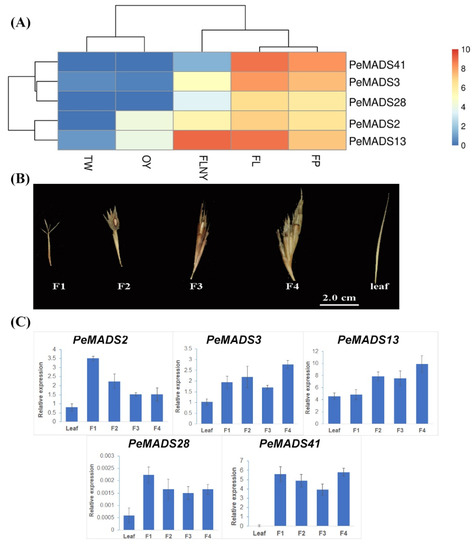
Figure 2.
The expression pattern of PeSQUA-like genes. (A) The expression levels of PeSQUAs during the transition from vegetative to reproductive growth. TW: leaves from 3-week-old seedlings, OY: leaves from 1-year-old plants, FLNY: leaves from plants that will flower in the next year, FL: leaves from flowering plants, and FP: flower florets. (B) The bamboo inflorescence development was divided into four stages: the first floral bud formation (F1), the initial stage of inflorescence development (F2), maturation of inflorescence (F3), and anthesis (F4). The leaf tissues (leaf) from the non-flowering plant. (C) qPCR analysis of PeSQUAs at different flowering stages. NTB or TIP41 was used as a reference gene; mean ± SD of three biological replicates is presented.
PeSQUA-like gene expression levels at different flowering developmental stages were also tested by qPCR. Bamboo inflorescence development (Figure 2B) had four stages, namely, the first floral bud formation (F1), the initial stage of inflorescence development (F2), maturation of inflorescence (F3), and anthesis (F4). The leaf tissues (leaf) from the non-flowering plant under the same growth environment as the flowering plants were used as the control. The transcripts of PeSQUAs were highly expressed in floral samples and less expressed in leaves. The expression of PeMADS2 exhibited a peak expression at the initial stage (F1) and decreased during inflorescence development, whereas PeAMDS3 and PeMADS13 showed an upregulated trend. The expression levels of PeMADS28 and PeMADS41 showed a mild change at different floral development stages.
2.3. PPI between PeSQUA-like Members and Other MADS-Box Proteins in P. edulis
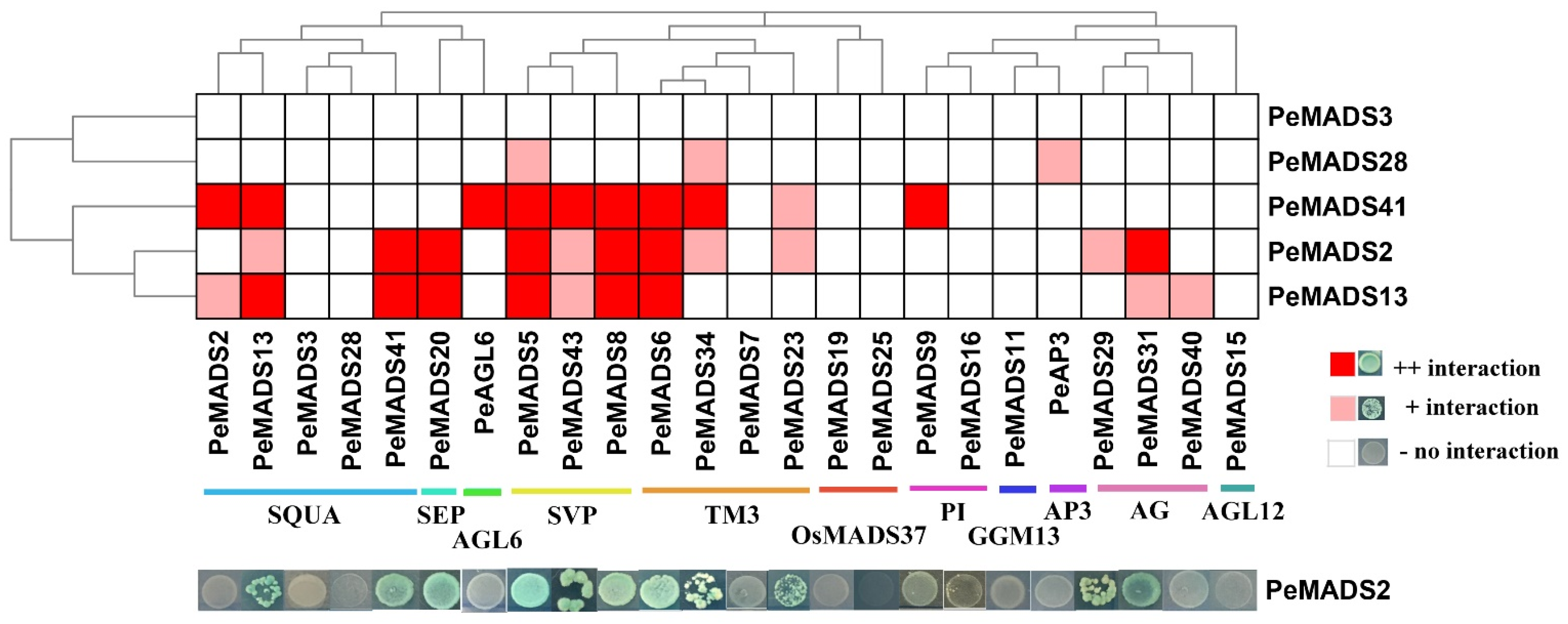
To investigate the interaction patterns of P. edulis SQUA-like members, a comprehensive Y2H assay was performed to clarify the protein interactions among bamboo MADS-box proteins (Figure 3). The five BD baits of PeSQUAs showed no auto-activation or toxicity (Figure 1). Among five genes, PeMADS2 had the highest number of interaction relationships with other MADS-box proteins, followed by PeMADS13 and PeMADS41. PeMADS13 could form a homologous dimer, whereas the other four PeSQUAs could not. Based on the PPI results, the PeSQUAs could be classified into two groups, thereby showing the variable PPI patterns. PeMADS2, 13, and 41 had an extensive interaction network among eight MADS-box subfamilies, whereas the PeMADS28 and PeMADS3 showed fewer or no interactions with other PeMADSs. Most of the interaction relationships were among SQUA-, SVP-, TM3-, and AG-like subgroups. The SVP-like proteins showed the strongest correlation with SQUA-like proteins, specifically with PeMADS2, 13, and 41.
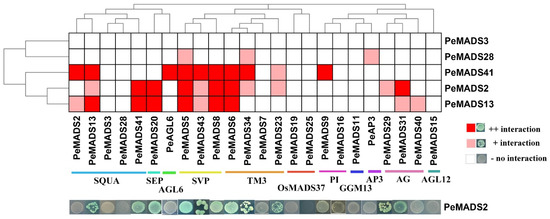
Figure 3.
Protein–protein interactions (PPI) among PeSQUAs and PeMADSs. A yeast two-hybrid assay was performed to verify the interaction among PeSQUAs and other MADS-box proteins. The result was displayed by the phylogenetic tree of the bamboo MADS-box gene family. The dark red represents a strong reaction, light red represents a mild reaction, and gray represents no reaction.
2.4. Ectopic Expression of PeMADS2 Accelerates Arabidopsis Flowering
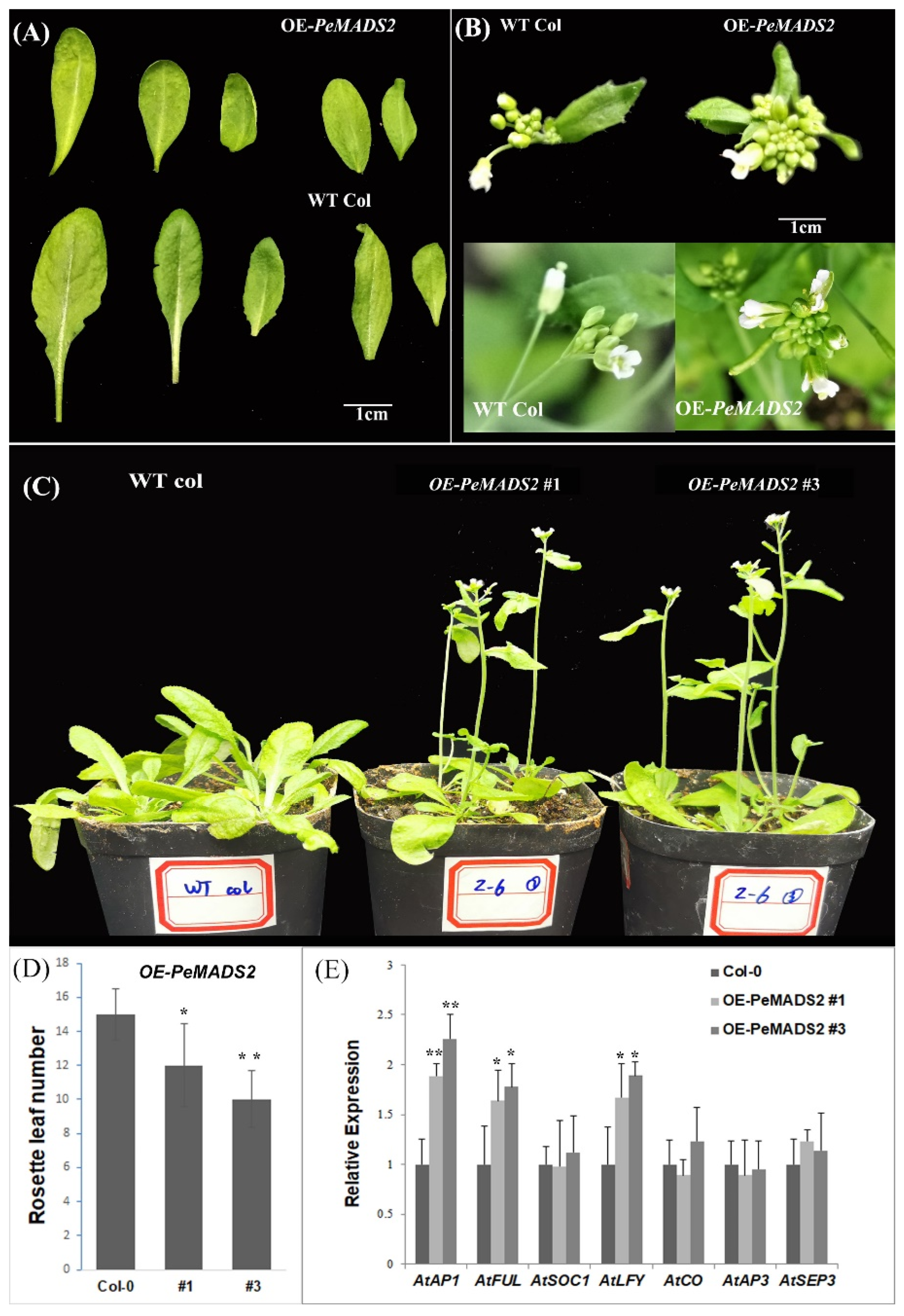
To explore the roles of bamboo SQUA-like genes in the regulation of flowering, we transformed PeAMDS2 into Arabidopsis. PeAMDS2 was highly expressed during inflorescence initiation and had the highest PPI potential. The ectopic expression of PeAMDS2 in Arabidopsis resulted in an early flowering phenotype under long-day (LD) conditions (Figure 4C), and the phenotypic variations were observed in leaves and floral organs (Figure 4A,B). The rosette and cauline leaves of OE-PeMADS2 plants were curled and smaller than those of the wild-type (WT) plants (Figure 4A). The inflorescences were densely clustered in the transgenic plants (Figure 4B). The flowering time was approximately 6–8 days earlier than WT plants, and the number of rosette leaves of OE-PeMADS2 plants was significantly fewer than that of WT Arabidopsis (Figure 4D). We further investigated the expression levels of important genes involved in flowering time and flower organ development. The expression levels of AtAP1, AtFUL, and LEAFY (AtLFY) were significantly upregulated, whereas the expression levels of the other four genes were insignificantly different between transgenic and WT plants (Figure 4E). PeMADS2 might regulate transgenic plant flowering by controlling the expression of its homology (AtAP1 and AtFUL) and AtLFY.
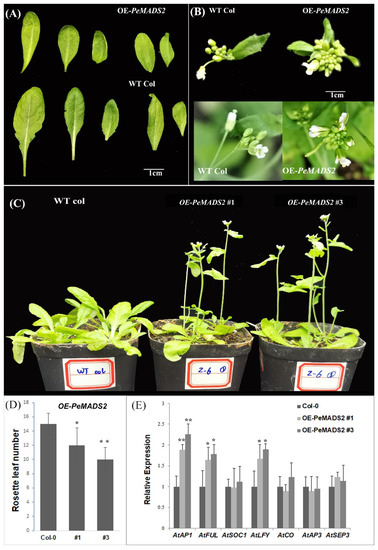
Figure 4.
The early flowering phenotype of OE-PeMADS2 in Arabidopsis. (A)The leaves of OE-PeMADS2 transgenic and WT Arabidopsis plants. (B) The floral organs of OE-PeMADS2 transgenic Arabidopsis and wild-type plants. (C) Phenotypes of overexpressing PeMADS2 transgenic lines (OE-PeMADS2#1, #3) and wild-type (WT) plants as control under LD conditions. (D) Flowering time was scored as the number of rosette leaves under LD conditions. (E) Transcription levels of AP1, FUL, LFY, CO, AP3, SOC1, and SEP3 in WT and transgenic plants. Arabidopsis Actin or TIP41 was used as the internal reference gene. Mean ± SD of three biological replicates is presented. Asterisks indicate a significant difference between transgenic and WT plants (* p ≤ 0.05, ** p ≤ 0.01, t’s test).
2.5. Ectopic Expression of PeMADS2 Promotes Rice Flowering
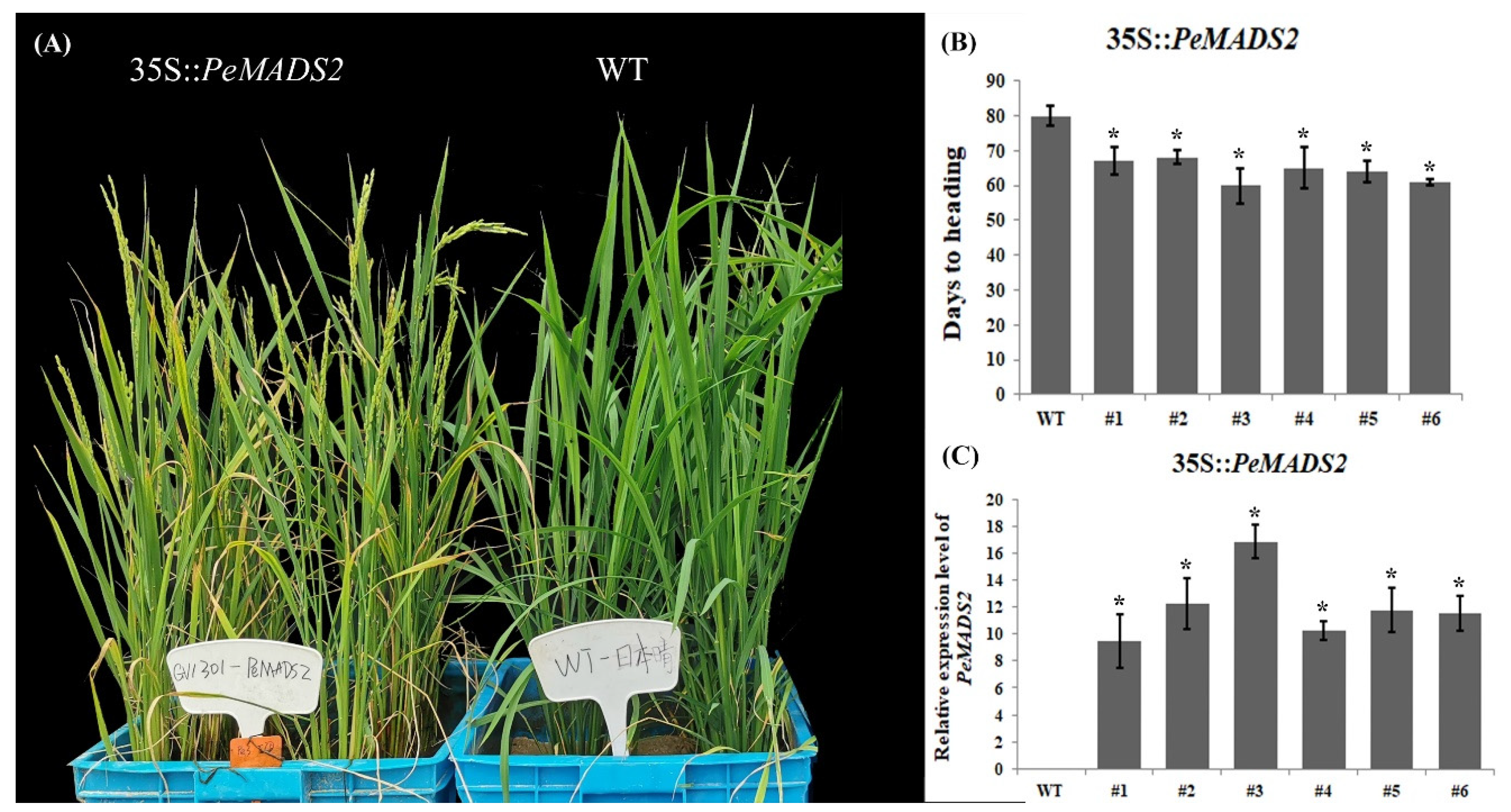
To further examine the function of hub genes, PeMADS2 was transformed into O. sativa (Dongjing), a member of the same grass family as bamboo. Twenty-four T1 OE-PeMADS2 transgenic rice plants were obtained. We analyzed the heading time and found that OE-PeMADS2 transgenic rice plants had a significantly early flowering phenotype (Figure 5A). In transgenic rice, the average relative expression level of PeMADS2 was ~12.05 ± 2.57 (Figure 5C). The heading time of transgenic plants (Figure 5B) was ~64.17 ± 3.18 d, which was 16 d earlier than that of the WT lines (80.62 ± 1.02 d). Furthermore, the highest level of PeMADS2 was detected in OE-PeMADS2 line #3, which showed the earliest heading, thereby indicating a negative association between the heading time and the expression level of PeMADS2.
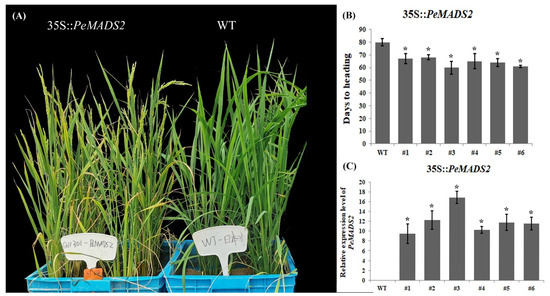
Figure 5.
Ectopic expression of PeMADS2 in Oryza. (A) Representative picture of WT and ectopic expression lines of PeMADS2 after heading. (B) Days to the heading of T3 homozygous transgenic plants. (C) Relative expression levels of PeMADS2 in transgenic rice plants and WT. Asterisks indicate a significant difference between transgenic and WT plants (* p ≤ 0.05, t’s test).
2.6. Analysis of Differential Gene Expression in OE-PeMADS2 Transgenic Rice
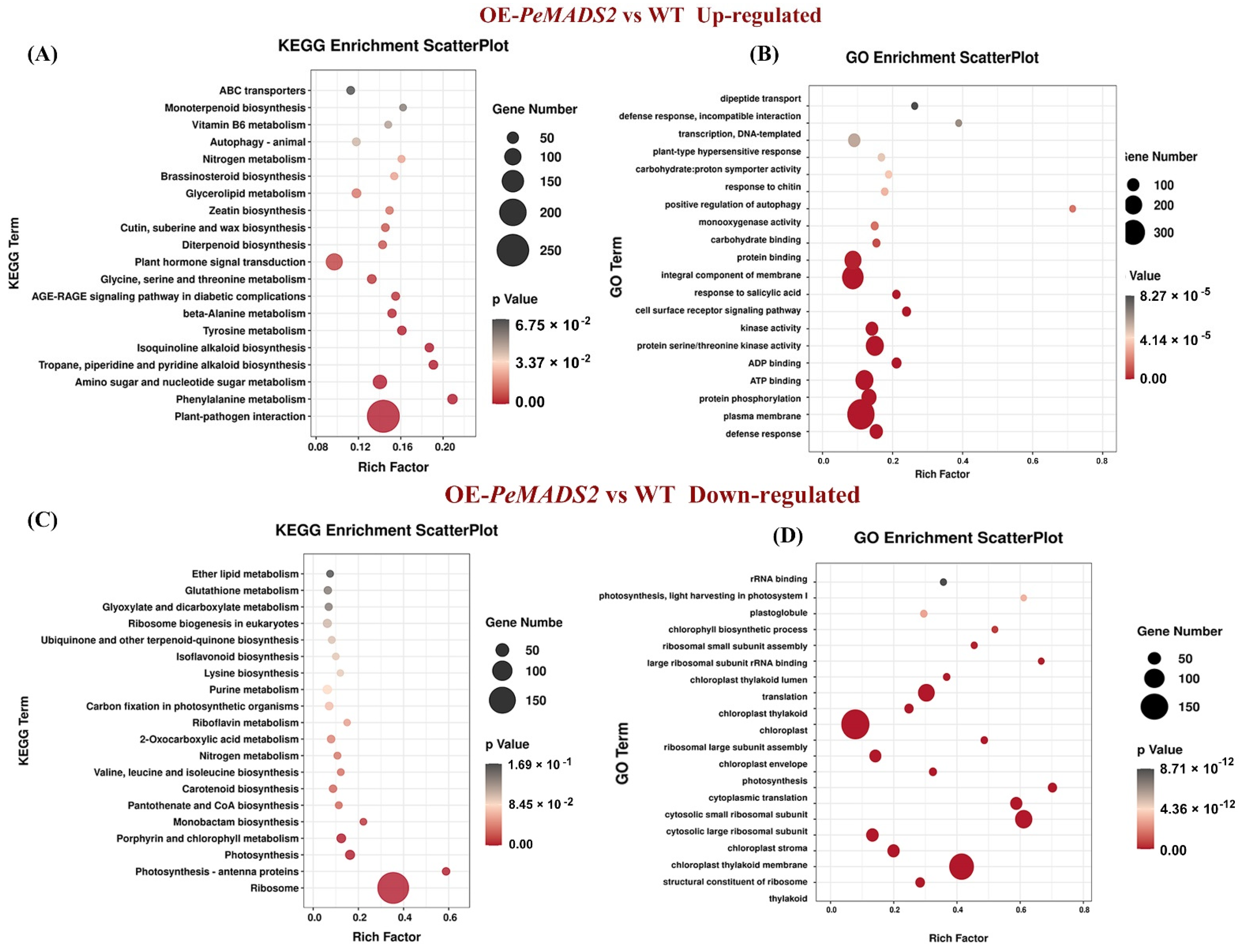
To explore the early heading mechanism in transgenic rice, the RNA-seq analyses of OE-PeMADS2 (line #3) and WT lines were performed. We pooled the short reads and aligned them to the Nipponbare reference genome to identify the transcripts. In total, we obtained 145,947,014 and 136,115,288 reads with three biological repeats from OE-PeMADS2 transgenic rice and wild-type rice libraries, respectively. The mapping rate for each library was above 95%. We obtained 39,333,958 (85.74%) and 38,097,230 (86.52%) uniquely mapped reads for further analysis (Supplemental Table S4). Among the 42,004 expressed unigenes, 27,213 have been GO annotated, and 14,606 have been KEGG annotated. Compared with WT lines, 3146 genes were differentially expressed, including 2021 that were upregulated and 1125 that were downregulated in OE-PeMADS2 lines. KEGG analysis indicated that the genes for plant–pathogen interaction and plant hormone signal transduction were significantly upregulated in transgenic lines (Figure 6A). In contrast, genes for ribosome and photosynthesis were significantly downregulated (Figure 6C). GO enrichment showed that genes upregulated in OE-PeMADS2 lines were enriched in defense response, plasma membrane, integral component of membrane, and ATP binding (Figure 6B). Differentially expressed genes (DEGs) in OE-PeMADS2 plants were enriched for functions related to floral development with overrepresented gene ontology (GO) terms such as “regulation of timing of the transition from vegetative to reproductive phase” (GO:0048510) and “flower development” (GO:0009908). Genes associated with plant hormone signal transduction, such as the salicylic acid- (GO:0009751, GO:0071446, and GO:2000031) and abscisic acid-activated signaling pathways (GO:0009738, GO:0010427), were overrepresented in OE-PeMADS2 (Supplemental Table S5). In contrast, downregulated GO terms were involved in the structural constituents of ribosome, chloroplast, and translation (Figure 6D, Supplemental Table S6).
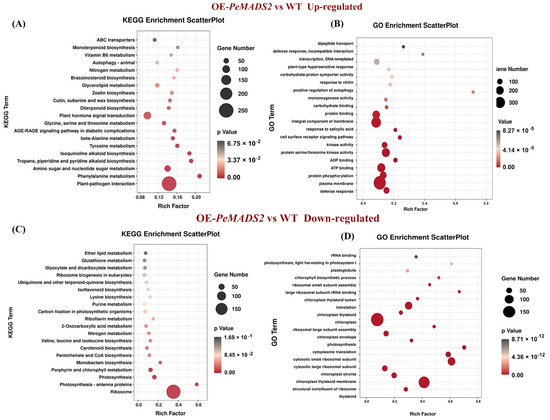
Figure 6.
Analysis of RNA-seq data of OE-PeMADS2 lines and wild-type rice plants. (A,B): GO and KEGG enrichment analysis of DEGs that were upregulated in OE-PeMADS2 plants. (C,D): GO and KEGG enrichment analysis of DEGs that were downregulated in OE-PeMADS2 plants. Low q-values are shown in the dark grey circle, and high q-values are shown in the red circle. The size of a circle represents DEG number.
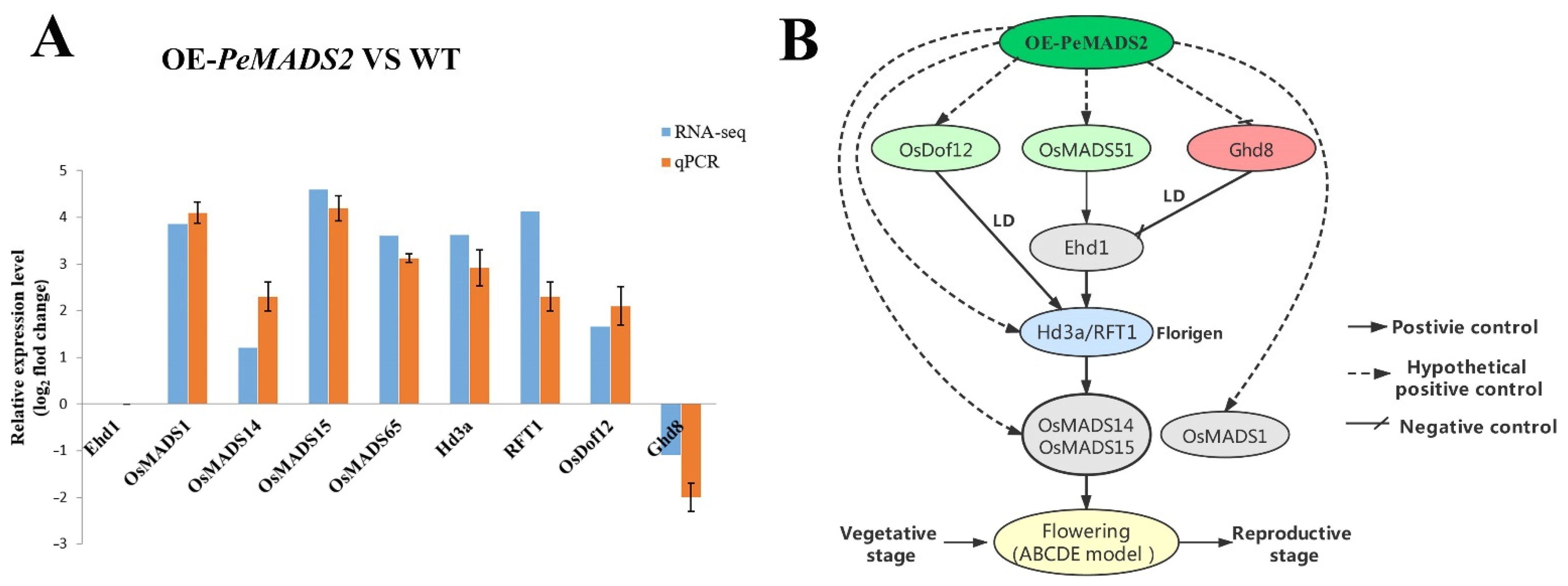
Thirty-seven genes involved in rice floral regulatory pathways were identified for further analysis based on transcription data (Supplemental Table S7). DEGs with a minimum fold change 1 (Log2 converted) and a q-value of <0.05 were identified and extracted (Table 1). OsMADS1, OsMADS15, OsMADS14, OsMADS51 (OsMADS65), DNA-BINDING WITH ONE FINGER (OsDof12), HEADING DATE 3a (Hd3a), and RICE FLOWERING LOCUS T 1 (RFT1) were all upregulated in the transgenic lines compared with WT. The expression levels of GRAIN NUMBER, PLANT HEIGHT, AND HEADING DATE 8 (Ghd8) were downregulated. The transcriptome results were further validated by qPCR analysis (Figure 7A), which showed similar expression trends with RNA-seq data.

Table 1.
DEGs related to rice floral pathway in transgenic plants.
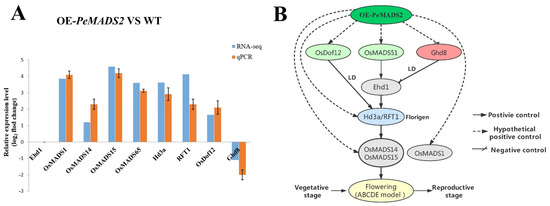
Figure 7.
Proposed working models of OE-PeMADS2 to induce the early flowering in transgenic rice. (A) Relative expression pattern analysis of rice floral-related genes by qRT–PCR analysis to validate RNA-seq data. Rice Ubq or EP were used as reference genes; mean ± SD of three biological replicates is presented. (B) The model of the early flowering signal pathway in OE-PeMADS2 transgenic rice. Arrows with solid lines indicate positive regulators of flowering, arrows with lines indicate negative regulators of flowering, and arrows with dotted lines indicate hypothetical positive control of flowering.
3. Discussion
3.1. Gene Expression Patterns and PPI Patterns Are Divergent among PeSQUA-like Genes
SQUA-like MADS-box genes are typical A-class floral organ identity genes that have essential roles in modulating floral transition and organ development [23]. Previous studies often focus on the extensive evolution and development studies of SQUA-like genes in rice and wheat [20,26], whereas relatively little information on their functions in bamboo is available. In the present study, five SQUA-like MADS-box genes were characterized from P. edulis, and these were grouped into two clusters, depending on the existence of the FUL-like motif in the C-terminal region (Figure 1). Sequence structures that have similar features are likely to share a relatively closer evolutionary relationship, especially if the features appear in a non-conserved region [31]. In our study, this FUL-like motif seems to be important for the function of bamboo SQUA-like proteins. The expression profiles revealed that PeSQUAs from the same lineage share a similar expression pattern, which is consistent with phylogenetic studies (Figure 2). Although the five PeSQUAs genes were highly expressed in leaves and florets of flowering plants, PeMADS2 and PeMADS13 have relatively higher transcript levels in the reproductive transformation period, thereby suggesting that PeMADS2 and PeMADS13 are involved in floral transition modulation. The conserved FUL-like motif consisting of hydrophobic amino acids might be crucial in PPIs [32]. Consistently, cluster I, which includes PeMADS2, 13, and 41 that harbor FUL-like motifs, could interact with multiple PeMADS proteins, whereas PeMADS3 and 28 have no FUL-like motif and hardly interact with other proteins (Figure 3). Although the importance of the FUL-like motif to protein function is unclear in bamboo, the differences of these sequences in the C-terminal region might explain the divergences in interacting and expression patterns of PeSQUAs [32]. Thus, PeMADS2, 13, and 41 might play more important roles than PeMADS3 and 28 in the floral development of P. edulis.
3.2. PPI Patterns of PeSQUAs Alternating from Vegetative to Reproductive Growth
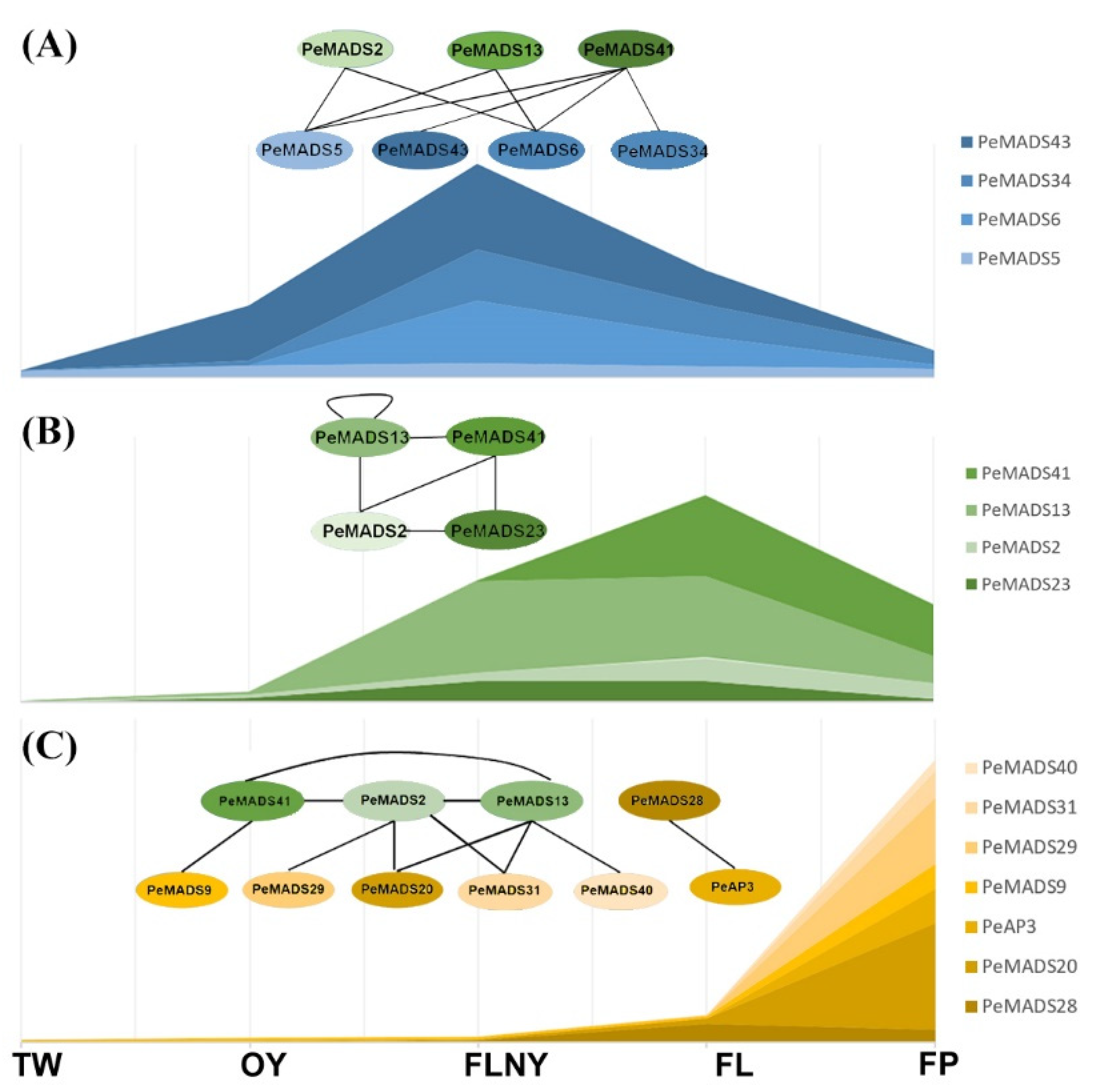
The change from vegetative to reproductive growth involves many genes. Understanding the PPI networks of the master floral developmental regulator is necessary to obtain the full picture of the phase transition in P. edulis. Thus, we proposed a hypothetical molecular model based on the PeSQUAs expression patterns and the PPI network during reproductive phase transitions (Figure 8). SQUA-like genes play key roles in several important developmental processes, especially in flower development [33]. In rice, the interaction of OsMADS18, OsMADS14, and OsMADS15 with other MADS proteins suggested that monocot SQUA-like genes accomplish a general role in floral transition and floral meristem identity by coordinating their functions [34]. Consistently, bamboo SQUA-like genes show high expression levels at the reproductive stage, and the potential interactors, i.e., SVP-like genes (PeMADS5 and 43) and TM3-like genes (PeMADS6 and 34), showed higher accumulation during the FLNY stage (Figure 8A). This finding suggested that the formation of dimers between SQUA-like and SVP-like genes or TM3-like genes triggers the initial stages of flower development. The expression levels of bamboo SQUA-like genes (PeMADS2, 13, 41, and 28) were higher in the leaves (FL/FLNY) than the floral organs (FP) at reproductive stages (Figure 8B). Rice SQUA-like genes (OsMADS14 and 15) were largely detected in the floral meristem and organs [35], whereas OsMADS18 was expressed in most tissues and increased during rice reproduction [25,34]. The PPI relationship among the SQUA-like subfamily was similar to rice in that the members could interact with one another [23,34]. Our results indicated that PeMADSs played important roles in the process of floral transition and morphological architecture due to the formation of PeSQUA homo- or heterodimer. In the floral organ (Figure 8C), PeSQUA proteins might interact with SEP-(PeMADS20), PI-(PeMADS9), AG-(PeMADS29, 31, and 40), and AP3-like (PeAP3) subfamily members; these genes are required for floral development in grasses corresponding to A-, B-, and C- classes of the ABC model [36]. These MADS protein complexes may play certain roles in floral meristem identity and inflorescence development. Based on our results, PeSQUAs might possess different functions by forming complicated protein complexes with other MADS-box proteins.
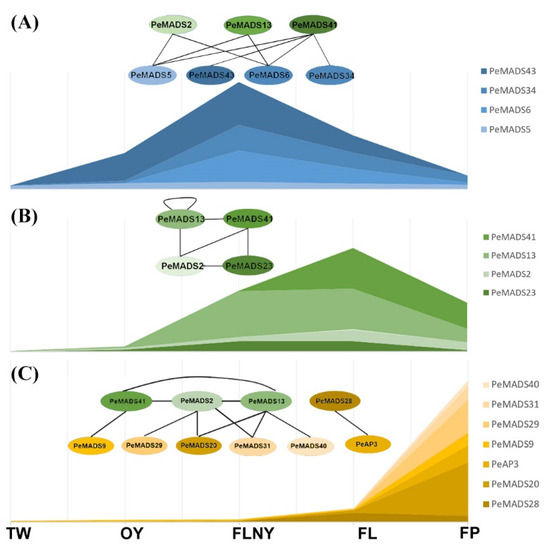
Figure 8.
A hypothesis of the molecular model of PeSQUAs in flower development. The stacked graphs represent bamboo MADS-box gene expression levels. The circle represents MADS-box proteins. The solid lines between different proteins indicate the PPI validated by Y2H. The model in the FLNY stage (A), the model in the FL (B), and the model in the FP (C). Each oval represents a different PeMADS protein.
3.3. PeMADS2 Acts as a Flowering Promoter in Transgenic Arabidopsis
Several studies have shown that the increased expression of SQUA-like genes in monocots is correlated with the floral transition and that the ectopic expression of all rice SQUA subfamily members results in early flowering and dwarfism [24,25]. This was in line with this study that ectopic expression of PeMADS2 had an early flowering phenotype in Arabidopsis. Ectopic expression of PeMADS2 led to the up-regulation of AtAP1, AtFUL, and AtLFY genes, which are associated with the flowering regulation (Figure 4E). These results suggest that the functions of PeMADS2 might be dependent on its homologs, AP1 and FUL, in Arabidopsis. However, the underlying mechanism mediated by PeMADS2 in Arabidopsis needs further investigation. The ectopic expression of TaVRN1, a SQUA-like member in wheat, caused the overexpression of Arabidopsis AP1, which resulted from the binding of TaVRN1 to CArG-box present on the AP1 promoter [37]. CArG-box was the binding site for the MADS domain transcription factors [38]. Therefore, in silico identification of the CArG-box motif was performed in the 3000 bp upstream of AtAP1, AtFUL, and AtLFY (Supplemental Table S8). At least two CArG-boxes were presented in the promoter regions of these three genes. According to the result, we speculated that PeMADS2 might promote transgenic Arabidopsis flowering by physically binding AtAP1, AtFUL, and AtLFY’s promoters to trigger the up-regulation of the genes’ expression, however, further “in vitro” and “in vivo” experimental verifications should be included in the future.
The ectopic expression of PeMADS2 in Arabidopsis and rice both showed early flower phenotype and caused up-regulation of flowering regulation genes (Figure 4E and Table 1). In both Arabidopsis and rice, the ectopic expression of PeMADS2 triggered the accumulation of its corresponding homologous genes. The upregulated Arabidopsis AtAP1 and AtFUL genes and rice OsMADS14 and OsMADS15 genes, belonged to the SQUA-like subgroup. Furthermore, the key flowering regulators, AtLFY in Arabidopsis and Hd3a/RFT1 in rice, were both significantly upregulated in the OE-PeMADS2 transgenic plants. The Arabidopsis LFY plays a key role in reproductive transitions via the regulation of floral homeotic MADS-box transcription factors [39]. The rice RFT1 and Hd3a are thought to encode the mobile flowering signal (florigens) proteins and promote floral transition [40]. However, the number of flowering-related DEGs is greater in transgenic rice than that in Arabidopsis, which might be due to the closer biological relationship between rice and Moso bamboo.
3.4. Ectopic Expression of PeMADS2 Triggered Multiple Flower Signaling Pathways to Induce Early Flowering in Rice
Genetic approaches in Moso bamboo are not feasible for now, and advanced experiments are performed in the most related model species and rice. The ectopic expression of PeMADS2 had an early flowering phenotype; the heading date was approximately 15 days earlier than in WT (Figure 5A). Based on transcriptomic data and qPCR, we proposed the working model of PeMADS2 to induce early flowering (Figure 7B). One of the regulation modules triggered is the OsMADS51/65-Hd3a/RFT1-OsMADS14/15 pathway. OsMADS51/OsMADS65, a type I MADS-box gene that can promote flowering through the Ehd1-dependent pathway via Hd3a and OsMADS14, was upregulated in OE-PeMADS2 transgenic rice [41]. Another flower promoter’s transcript level (OsDof12) also significantly increased in OE-PeMADS2 lines. OsDof12 might regulate flowering time by controlling the transcription levels of Hd3a and OsMADS14 independent of other flowering genes under LD [42]. PheDof12-1 is the homologous gene of OsDof12 in Moso bamboo; overexpressing PheDof12-1 in Arabidopsis causes early flowering under LD conditions [43]. In contrast, the flowering repressor DTH8/Ghd8 was repressed in OE-PeMADS2 lines compared with WT. DTH8 suppressed rice flowering by down-regulating the expressions of Ehd1 and Hd3a under LD conditions independent of Hd1 and Ghd7 [44]. In addition to upstream flowering regulators, rice florigens Hd3 and RFT1 were upregulated in OE-PeMADS2 transgenic rice plants [40]. These two florigens are fundamental for the flowering regulatory pathway because almost all flowering pathways finally target these two genes [45]. Furthermore, two SUQA clade MADS-box genes, namely, OsMADS14 and 15, were upregulated in OE-PeMADS2 plants; these are major genes located downstream of Hd3a and RFT1 that regulate the identity of floral meristem development [46]. PeMADS2 had high homology with OsMADS18 (SUQA-like clade), which can interact with OsMADS15 and OsMADS34 in the shoot apical meristem (SAM) in the reproductive transition [25,34]. Another rice MADS-box gene affected in OE-PeMADS2 transgenic rice was OsMADS1, which was expressed preferentially in flowers and played crucial roles in the determination and specification of floral organ identity in rice [47]. Ectopically expressed OsMADS1 in tobacco and rice caused early flowering [48]. OsMADS1 could interact with SUQA proteins (OsMADS14 and 15) [49].
These observations suggested that the ectopic expression of PeMADS2 in rice affected the expressions of upstream genes OsMADS51, OsDof12, and Ghd8, which were regulated by photoperiod. They either promote or restrain florigen gene expression, enhance the florigen genes Hd3a, RFT1 expression levels, and ultimately increase the expressions of the downstream genes OsMADS14, OsMADS15, and OsMADS1, which could control the transformation from SAM to inflorescence meristem (IM) [50]. Ehd1 is a key flowering time regulator in rice. The expression levels of the upstream and downstream genes involved in the Ehd1-dependent pathway showed significant changes (Figure 7A). However, the expression level of Ehd1 was not notably different between OE-PeMADS2 and WT plants. One of the assumptions is that PeMADS2 could directly interact with the upstream MADS proteins, OsMADS51/OsMADS65 or OsMADS1, in the regulation of flower development (independent from Ehd1) and increase the expression levels of downstream gene Hd3a/RFT1 and OsMADS14/15. Another assumption is that PeMADS2 could interact with the downstream MADS proteins, such as OsMADS14 and OsMADS15, thereby resulting in feedback regulations that upregulate the expression of upstream genes (Hd3a/RFT1 and OsMADS51/OsMADS65). A similar result was found in OsMADS15 ectopic expression plants, which was the ultimate downstream target of all the flowering regulators; however, the expression levels of the upstream regulators were upregulated in the transgenic lines [51]. The interactions among different flowering regulators are essential for their function in controlling flowering processes. In this study, PeMADS2 might be a crucial component of the interaction networks for floral transition in bamboo and could interact with other regulators to determine the flowering time in rice and Arabidopsis. The ectopic expression of PeMADS2 coordinately upregulated the other flowering regulators’ expression levels and might form protein complexes to promote the transition from the vegetative to the reproductive stage.
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
Flowering bamboo (P. edulis) materials were collected in Lingchuan Country, Guilin, China. Wild type and T-DNA-tagged Arabidopsis Colombia 0 (Col-0) were grown under long-day photoperiod conditions (16 h/8 h light/dark photoperiod). Rice (O. sativa) var. japonica “Dongjing” plants were cultivated in the field of Lin’an, Hangzhou, China.
4.2. Gene Cloning and Phylogenetic Analysis
Specific primers were designed to amplify the full-length ORF sequences of bamboo SQUA-like genes (Supplemental Table S1) using cDNA templates prepared from flower bud and leaf tissues. The amplified PCR products were cloned into the pMD18-T vector (Takara, Dalian, China). Five bamboo and related Oryza and Arabidopsis SQUA-like proteins were aligned by MAFFT version 7 (http://mafft.cbrc.jp/alignment/server/large.html, accessed on 3 October 2021) [52]. A phylogenetic tree was generated using the neighbor-joining method in the MEGA7 program [53,54]. The reliability of tree nodes was evaluated by bootstrap analysis for 1000 replicates.
4.3. DGE Data and Transcriptome Data Analysis
PeSQUAs expression profiles in bamboo at different inflorescence development stages (F1–F4) and non-flowering bamboo tissues (leaf) were analyzed by quantitative qPCR. For bamboo reproductive transition, the transcriptome data of leaf samples from the following developmental stages were determined: the juvenile stages of 3-week-old seedlings (TW) and 1-year-old plants (OY); the transition stage, i.e., plants that will flower in the next year (FLNY); and the flowering stage, including the flowering plants’ leaves (FL) and flower florets (FP) [30]. Accession number: SRR8053492-SRR8053506. We used HISAT2-2.0.4 (https://daehwankimlab.github.io/hisat2/ accessed on 14 December 2020) to map the reads to the Moso bamboo reference genome (http://www.bamboogdb.org/ accessed on 12 July 2021). StringTie and ballgown were used to estimate the expression levels of all transcripts and to determine the expression level of mRNAs by calculating FPKM [55].
4.4. Yeast Two-Hybrid Analysis
Yeast two-hybrid analyses were performed by the Matchmaker® Gold System (Clontech, Palo Alto, CA, USA). The protein-coding sequences of PeSQUAs were amplified by PCR using specific primers (Supplementary Table S2). PeSQUAs sequences were cloned into both pGBKT7 (bait) and pGADT7 (prey) vectors. Meanwhile, 19 bamboo MADS-box sequences from other subfamilies were also amplified and cloned into pGADT7 vectors separately. The resulting recombinant plasmids were introduced into yeast strains Y2HGold and Y187. Two-hybrid interactions were assayed on selective SD/-Trp/-Leu double-dropout (DDO) and SD/-Trp/-Leu/-His/-Ade/X-α-gal (40 mg/mL) media supplemented with Aureobasidin A (AbA).
4.5. Binary Plasmid Construction and Analysis of Transgenic Plants
The ORFs of full-length PeMADS2 was inserted into the binary vector pCAMBIA1301 using the Cauliflower mosaic virus (CaMV) 35S promoter to drive constitutive expression. Recombinant vectors were transferred to Agrobacterium tumefaciens strain GV3101 and used to transform Arabidopsis by the floral dip method [56]. Arabidopsis seeds were harvested after transformation and were selected with hygromycin. The flowering times were measured by counting rosette leaf numbers on main inflorescences. The recombinant vector pC1301-PeMADS2 was also transformed into O. sativa “Dongjing” plants. Calli induced from seeds were co-cultured with A. tumefaciens, and the putative transgenic rice was regenerated from calli [57]. The expression levels of PeMADS2 and flowering regulator genes in transgenic plants were analyzed by qPCR, the gene-specific primers for qPCR are listed in Supplemental Table S3.
4.6. RNA Extraction, and Quantitative RT–PCR (qPCR) Analysis
Total cellular RNA was extracted from frozen plant samples using TRIzol reagent kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocols. The purity and concentration of total RNA was measured by NanDrop 2000 spectrophotometer (Thermo Fisher, Wilmington, NC, USA) at a wavelength of 230, 260 and 280 nm. The first-strand cDNA was synthesized using the Prime-Script® RT reagent kit (Takara, Dalian, China) according to the manufacturer’s guidelines. The expression analysis of bamboo PeSQUAs at different flowering stages was performed by qPCR using SYBR® Premix Ex Taq II (Takara, Dalian, China) on a CFX-96-well Real-Time System (BioRad, Hercules, CA, USA). The relative expression levels of target genes were calculated using the 2−ΔΔCt method by normalizing NTB or TIP41 as the reference gene. The gene-specific primers are listed in Supplementary Table S3. Statistical analysis was conducted by two-tailed Student’s t-tests in Microsoft Excel 2011.
4.7. RNA-seq, KEGG, and GO Analysis
Leaves from WT and transgenic rice plants were collected when the first panicle appeared. After total RNA extraction according to the 4.6 protocols, sequencing libraries were generated using NEB-Next® Ultra™ RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA, USA). The libraries were sequenced on Illumina Novaseq™ 6000 (LC-Bio Technology CO., Ltd., Hangzhou, China) by following the recommended protocol. We used HISAT2-2.0.4 to map the reads to the Nipponbare reference annotation. The mapped reads of each sample were assembled using StringTie and merged to reconstruct a comprehensive transcriptome using gffcompare software [55]. StringTie and ballgown were used to estimate the expression levels of all transcripts and to determine the expression level of mRNAs by calculating FPKM [58]. The DEGs were selected with fold change >2 or fold change <0.5 and p-value < 0.05 by R package DESeq2. The expression levels of these selected DEGs’ expression were confirmed by qPCR. Gene ontology (GO) enrichment analysis of the DEGs was implemented by the GOseq R package [59] and further annotated against the Kyoto Encyclopedia of Genes and Genomes (KEGG) metabolic pathways database [60]. All the RNA-Seq raw data are available in the NCBI Sequence Read Archive (SRA) under the accessions number SRR16020759-SRR16020764.
5. Conclusions
Five PeSQUA-like genes were cloned from P. edulis and were further grouped into Cluster I (PeMADS2, 13, and 41) and Cluster II (PeMADS3 and 28). Five PeSQUAs had higher transcript levels in the reproductive period than the juvenile phase and had divergent expression patterns at different flowering stages.Y2H showed that three members of cluster I could interact with multiple PeMADS proteins, whereas PeMADS3 and 28 hardly interacted with other proteins. Based on our results, PeSQUA possessed different functions by forming the complicated protein complexes with other MADS-box proteins at different inflorescence development stages. Furthermore, we have chosen PeMADS2 for functional analysis. The ectopic expression of PeMADS2 in Arabidopsis and rice triggered early flowering, and the alteration of floral organ development was observed in Arabidopsis. RNA-seq and qRT–PCR analyses of plants overexpressing PeMADS2 indicated that PeMADS2 might integrate multiple flower signaling pathways to trigger early flowering time in rice. Our results provided valuable information on the interaction patterns of PeSQUA and other MADS-box members during P. edulis floral development. Moreover, to fully understand the PeSQUA subfamily member’s function related to flowering, additional transgenic investigation of other PeSQUAs members in Arabidopsis and rice should be conducted.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/ijms221910868/s1, Table S1: Primers for PeSQUA gene cloning; Table S2: List of primers used for yeast two-hybrid; Table S3: qPCR primers used for transgenic plants; Table S4: Summary of RNA-seq mapping; Table S5: Summary of significant enriched GO terms in OE-PeMADS2; Table S6: Summary of significant enriched GO terms in WT; Table S7: The flowering time-related genes in transgenic and wild type rice; Table S8: In silico identification of putative CArG-box motifs in Arabidopsis genes promoters.
Author Contributions
Conceptualization, Z.T. and Y.Z.; methodology, Y.Z. and M.S.; data curation, M.D.; writing—original draft preparation, Y.Z.; writing—review and editing, J.Z.; project administration, Z.T. and M.D.; funding acquisition, X.L. and M.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grants from the Natural Science Foundation of Zhejiang Province (LZ20C160002), the National Natural Science Foundation of China (31971735), and the State Key Laboratory of Subtropical Silviculture (ZY20180203). Scientific Research Development Fund of Zhejiang A&F University (W20190243) to Mingquan Ding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ramakrishnan, M.; Yrjälä, K.; Vinod, K.K.; Sharma, A.; Cho, J.; Satheesh, V.; Zhou, M. Genetics and Genomics of Moso Bamboo (Phyllostachys edulis): Current Status, Future Challenges, and Biotechnological Opportunities toward a Sustainable Bamboo Industry. Food Energy Secur. 2020, 9, e229. [Google Scholar] [CrossRef]
- Dutta, S.; Biswas, P.; Chakraborty, S.; Mitra, D.; Pal, A.; Das, M. Identification, Characterization and Gene Expression Analyses of Important Flowering Genes Related to Photoperiodic Pathway in Bamboo. BMC Genom. 2018, 19, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isagi, Y.; Shimada, K.; Kushima, H.; Tanaka, N.; Nagao, A.; Ishikawa, T.; Onodera, H.; Watanabe, S. Clonal Structure and Flowering Traits of a Bamboo [Phyllostachys pubescens (Mazel) Ohwi] Stand Grown from a Simultaneous Flowering as Revealed by AFLP Analysis. Mol. Ecol. 2004, 13, 2017–2021. [Google Scholar] [CrossRef]
- Wysocki, W.P.; Eduardo, R.S.; Yin, Y.; Duvall, M.R. The Floral Transcriptomes of Four Bamboo Species (Bambusoideae; Po-aceae): Support for Common Ancestry among Woody Bamboos. BMC Genom. 2016, 17, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, P.; Chakraborty, S.; Dutta, S.; Pal, A.; Das, M. Bamboo Flowering from the Perspective of Comparative Genomics and Transcriptomics. Front. Plant Sci. 2016, 7, 1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, D.; Li, L.; Ma, T.; Pei, J.; Zhao, Z.; Lu, M.; Wu, A.; Lin, X. The SOC1-like Gene BoMADS50 is Associated with the Flowering of Bambusa oldhamii. Hortic. Res. 2021, 8, 1–13. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, Y.; Zhang, C.; Qi, F.; Li, X.; Mu, S.; Peng, Z. Characterization of the Floral Transcriptome of Moso Bamboo (Phyllostachys edulis) at Different Flowering Developmental Stages by Transcriptome Sequencing and RNA-Seq Analysis. PLoS ONE 2014, 9, e98910. [Google Scholar] [CrossRef] [Green Version]
- Shih, M.C.; Chou, M.L.; Yue, J.J.; Hsu, C.T.; Chang, W.J.; Ko, S.S.; Liao, D.C.; Huang, Y.T.; Chen, J.J.; Yuan, J.L.; et al. BeMADS1 is a Key to Delivery MADSs into Nucleus in Reproductive Tissues-De Novo Characterization of Bambusa edulis Transcriptome and Study of MADS Genes in Bamboo Floral Development. BMC Plant Biol. 2014, 14, 179. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Qi, T.; Ma, J.; Ma, T.; Ma, L.; Lin, X. Ectopic Expression of a SOC1 Homolog from Phyllostachys Violascens Alters Flowering Time and Identity of Floral Organs in Arabidopsis thaliana. Trees 2016, 30, 2203–2215. [Google Scholar] [CrossRef]
- Liu, S.; Ma, T.; Ma, L.; Lin, X. Ectopic Expression of PvSOC1, a Homolog of SOC1 from Phyllostachys violascens, Promotes Flowering in Arabidopsis and Rice. Acta Physiol. Plant. 2016, 38, 1–9. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, D.; Lin, X.; Ding, M.; Tong, Z. Genome-Wide Identification of MADS-Box Family Genes in Moso Bamboo (Phyllostachys edulis) and a Functional Analysis of PeMADS5 in Flowering. BMC Plant Biol. 2018, 18, 176. [Google Scholar] [CrossRef]
- Zhang, H.; Forde, B.G. An Arabidopsis MADS Box Gene that Controls Nutrient-Induced Changes in Root Architecture. Science 1998, 279, 407–409. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Pelaz, S.; Liljegren, S.J.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; de Pouplana, L.R.; Martínez-Castilla, L.; Yanofsky, M.F. An Ancestral MADS-Box Gene Duplication Occurred before the Divergence of Plants and Animals. Proc. Natl. Acad. Sci. USA 2000, 97, 5328–5333. [Google Scholar] [CrossRef] [Green Version]
- Ng, M.; Yanofsky, M.F. Function and Evolution of the Plant MADS-box Gene Family. Nat. Rev. Genet. 2001, 2, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Gramzow, L.; Theissen, G. A Hitchhiker’s Guide to the MADS World of Plants. Genome Biol. 2010, 11, 214. [Google Scholar] [CrossRef] [Green Version]
- Gramzow, L.; Weilandt, L.; Theißen, G. MADS Goes Genomic in Conifers: Towards Determining the Ancestral Set of MADS-Box Genes in Seed Plants. Ann. Bot. 2014, 114, 1407–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Begcy, K.; Liu, K.; Folsom, J.J.; Wang, Z.; Zhang, C.; Walia, H. Heat Stress Yields a Unique MADS Box Transcription Factor in Determining Seed Size and Thermal Sensitivity. Plant Physiol. 2016, 171, 606–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.; Lu, Y.; Li, L.; Zhao, Q.; Feng, Q.; Gao, Z.; Lu, H.; Hu, T.; Yao, N.; Liu, K.; et al. The Draft Genome of the Fast-Growing Non-Timber Forest Species Moso Bamboo (Phyllostachys heterocycla). Nat. Genet. 2013, 45, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Litt, A.; Irish, V.F. Duplication and Diversification in the APETALA1/FRUITFULL Floral Homeotic Gene Lineage: Implications for the Evolution of Floral Development. J. Gen. 2003, 165, 821–833. [Google Scholar] [CrossRef]
- Wu, F.; Shi, X.; Lin, X.; Liu, Y.; Chong, K.; Theißen, G.; Meng, Z. The ABCs of Flower Development: Mutational Analysis of AP1/FUL-like Genes in Rice Provides Evidence for a Homeotic (A)-Function in Grasses. Plant J. 2016, 89, 310–324. [Google Scholar] [CrossRef]
- Ferrandiz, C.; Gu, Q.; Martienssen, R.; Yanofsky, M. Redundant Regulation of Meristem Identity and Plant Architecture by FRUITFULL, APETALA1 and CAULIFLOWER. Development 2000, 127, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Ferrandiz, C.; Yanofsky, M.; Martienssen, R. The FRUITFULL MADS-Box Gene Mediates Cell Differentiation During Arabidopsis Fruit Development. Development 1998, 125, 1509–1517. [Google Scholar] [CrossRef]
- Kobayashi, K.; Yasuno, N.; Sato, Y.; Yoda, M.; Yamazaki, R.; Kimizu, M.; Yoshida, H.; Nagamura, Y.; Kyozuka, J. Inflorescence Meristem Identity in Rice Is Specified by Overlapping Functions of Three AP1/FUL-like MADS Box Genes and PAP2, a SEPALLATA MADS Box Gene. Plant Cell 2012, 24, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.-S.; Lee, S.; Jung, K.H.; Yang, W.-S.; Yi, G.-H.; Oh, B.-G.; An, G. Production of Transgenic Rice Plants Showing Reduced Heading Date and Plant Height by Ectopic Expression of Rice MADS-box Genes. Mol. Breed. 2000, 6, 581–592. [Google Scholar] [CrossRef]
- Fornara, F.; Pařenicová, L.; Falasca, G.; Pelucchi, N.; Masiero, S.; Ciannamea, S.; Lopez-Dee, Z.; Altamura, M.M.; Colombo, L.; Kater, M.M. Functional Characterization of OsMADS18, a Member of the AP1/SQUA Subfamily of MADS Box Genes. Plant Physiol. 2004, 135, 2207–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Lin, H.; Chen, A.; Lau, M.; Jernstedt, J.; Dubcovsky, J. Wheat VRN1, FUL2 and FUL3 Play Critical and Redundant Roles in Spikelet Development and Spike Determinacy. Development 2019, 146, 175398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preston, J.C.; Kellogg, E.A. Discrete Developmental Roles for Temperate Cereal Grass VERNALIZATION1/FRUITFULL-like Genes in Flowering Competency and the Transition to Flowering. Plant Physiol. 2008, 146, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.P.; Peng, H.Z.; Jin, Q.Y.; Deng, M.J.; Li, T.; Xiao, X.C.; Hua, X.Q.; Wang, K.H.; Bian, H.W.; Han, N. Identification and characterization of two Bamboo (Phyllostachys praecox) AP1 / SQUA- like MADS-Box Genes during Floral Transition. Planta 2009, 231, 109–120. [Google Scholar] [CrossRef]
- Vandenbussche, M.; Theissen, G.; Van, P.; Gerats, T. Structural Diversification and Neo-Functionalization during Floral MADS-Box Gene Evolution by C-Terminal Frameshift Mutations. Nucleic Acids Res. 2003, 31, 4401–4409. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, H.; Wang, Y.; Xi, F.; Wang, H.; Kohnen, M.V.; Gao, P.; Wei, W.; Chen, K.; Liu, X.; et al. Whole-Genome Characterization of Chronological Age-Associated Changes in Methylome and Circular RNAs in Moso Bamboo (Phyllostachys edulis) from Vegetative to Floral Growth. Plant J. 2021, 106, 435–453. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tian, F.; Song, J.; Wei, S.; Xia, K.; Liao, J.; Zhang, M. Functional Conservation and Divergence of Four Ginger AP1/AGL9 MADS–Box Genes Revealed by Analysis of Their Expression and Protein–Protein Interaction, and Ectopic Expression of AhFUL Gene in Arabidopsis. PLoS ONE 2014, 9, e114134. [Google Scholar] [CrossRef] [Green Version]
- Mccarthy, E.W.; Abeer, M.; Amy, L. Functional Divergence of APETALA1 and FRUITFULL is Due to Changes in Both Regulation and Coding Sequence. Front. Plant Sci. 2015, 6, 1076. [Google Scholar] [CrossRef] [Green Version]
- Preston, J.C.; Kellogg, E.A. Conservation and Divergence of APETALA1/FRUITFULL-like Gene Function in Grasses: Evidence from Gene Expression Analyses. Plant J. 2007, 52, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Liu, X.; Xu, B.; Lu, P.; Dong, T. OsMADS18, a Membrane-Bound MADS-Box Transcription Factor, Modulates Plant Architecture and the ABA Response in Rice. J. Exp. Bot. 2019, 70, 3895–3909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callens, C.; Tucker, M.R.; Zhang, D.; Wilson, Z.A. Dissecting the Role of MADS-Box Genes in Monocot Floral Development and Diversity. J. Exp. Bot. 2018, 69, 2435–2459. [Google Scholar] [CrossRef] [PubMed]
- Immink, R.; Kaufmann, K.; Angenent, G. The ‘ABC’ of MADS Domain Protein Behaviour and Interactions. Semin. Cell Dev. Biol. 2010, 21, 87–93. [Google Scholar] [CrossRef]
- Adam, H.; Ouellet, F.; Kane, N.A.; Agharbaoui, Z.; Major, G.; Tominaga, Y.; Sarhan, F. Overexpression of TaVRN1 in Arabidopsis Promotes Early Flowering and Alters Development. Plant Cell Physiol. 2007, 48, 1192–1206. [Google Scholar] [CrossRef] [Green Version]
- Folter, S.D.; Angenent, G.C. Trans meets Cis in MADS Science. Trends Plant Sci. 2006, 11, 224–231. [Google Scholar] [CrossRef]
- Moyroud, E.; Kusters, E.; Monniaux, M.; Koes, R.; Parcy, F. LEAFY Blossoms. Trends Plant Sci. 2010, 15, 346–352. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, H.; Ren, D.; Tang, H.; Qiu, R.; Feng, J.; Long, Y.; Niu, B.; Chen, D.; Zhong, T.; et al. Genetic Interactions Between Diverged Alleles of Early Heading Date 1 (Ehd1) and Heading date 3a (Hd3a)/ RICE FLOWERING LOCUS T1 (RFT1) Control Differential Heading and Contribute to Regional Adaptation in Rice (Oryza sativa). New Phytol. 2015, 208, 936–948. [Google Scholar] [CrossRef]
- Kim, S.L.; Lee, S.; Kim, H.J.; Gil Nam, H.; An, G. OsMADS51 Is a Short-Day Flowering Promoter That Functions Upstream of Ehd1, OsMADS14, and Hd3a. Plant Physiol. 2007, 145, 1484–1494. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Yang, C.; Li, X.; Gan, Q.; Zhao, X.; Zhu, L. Functional Characterization of Rice OsDof12. Planta 2009, 229, 1159–1169. [Google Scholar] [CrossRef]
- Liu, J.; Cheng, Z.; Xie, L.; Li, X.; Gao, J. Multifaceted Role of PheDof12-1 in the Regulation of Flowering Time and Abiotic Stress Responses in Moso Bamboo (Phyllostachys edulis). Int. J. Mol. Sci. 2019, 20, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Xu, J.; Guo, H.; Jiang, L.; Chen, S.; Yu, C.; Zhou, Z.; Hu, P.; Zhai, H.; Wan, J. DTH8 Suppresses Flowering in Rice, Influencing Plant Height and Yield Potential Simultaneously. Plant Physiol. 2010, 153, 1747–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiroyuki, T.; Kenichiro, T.; Ko, S. Regulation of Flowering in Rice: Two Florigen Genes, a Complex Gene Network, and Natural Variation. Curr. Opin. Plant. Biol. 2011, 14, 45–52. [Google Scholar]
- Komiya, R.; Ikegami, A.; Tamaki, S.; Yokoi, S.; Shimamoto, K. Hd3a and RFT1 are Essential for Flowering in Rice. Development 2008, 135, 767–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Cai, Y.; Yan, H.; Jin, J.; You, X.; Wang, L.; Kong, F.; Zheng, M.; Wang, G.; Jiang, L.; et al. A Critical Role of OsMADS1 in the Development of the Body of the Palea in Rice. J. Plant Biol. 2018, 61, 11–24. [Google Scholar] [CrossRef]
- Jeon, J.S.; Jang, S.; Lee, S.; Nam, J.; Kim, C.; Lee, S.H.; Chung, Y.Y.; Kim, S.R.; Lee, Y.H.; Cho, Y.G.; et al. Leafy Hull Sterile1 Is a Homeotic Mutation in a Rice MADS Box Gene Affecting Rice Flower Development. Plant Cell 2000, 12, 871–884. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Moon, Y.H.; An, G.; Jang, S.K. Two Rice MADS Domain Proteins Interact with OsMADS1. Plant Mol. Biol. 2000, 44, 513–527. [Google Scholar] [CrossRef]
- Tsuji, H.; Taoka, K.I.; Shimamoto, K. Florigen in Rice: Complex Gene Network for Florigen Transcription, Florigen Activation Complex, and Multiple Functions. Curr. Opin. Plant Biol. 2013, 16, 228–235. [Google Scholar] [CrossRef]
- Lu, S.J.; Wei, H.; Wang, Y.; Wang, H.M.; Yang, R.F.; Zhang, X.B.; Tu, J.M. Overexpression of a Transcription Factor OsMADS15 Modifies Plant Architecture and Flowering Time in Rice (Oryza sativa L.). Plant Mol. Biol. Rep. 2012, 30, 1461–1469. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Wei, B.; Zhang, R.Z.; Guo, J.J.; Liu, D.M.; Li, A.L.; Fan, R.C.; Mao, L.; Zhang, X.-Q. Genome-Wide Analysis of the MADS-Box Gene Family in Brachypodium distachyon. PLoS ONE 2014, 9, e84781. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Doerks, T.; Bork, P. SMART: Recent Updates, New Developments and Status in 2015. Nucleic Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Clough, S.J.; Bent, A.F. Floral Dip: A Simplified Method for Agrobacterium-Mediated Transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-W.; Lu, C.-A.; Yu, T.-S.; Tseng, T.-H.; Wang, C.-S.; Yu, S.-M. Rice α-Amylase Transcriptional Enhancers Direct Multiple Mode Regulation of Promoters in Transgenic Rice. J. Biol. Chem. 2002, 277, 13641–13649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene Ontology Analysis for RNA-Seq: Accounting for Selection Bias. Genome Biol. 2010, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated Genome Annotation and Pathway Identification Using the KEGG Orthology (KO) as a Controlled Vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).