Disrupting Insulin and IGF Receptor Function in Cancer

Abstract
:1. Introduction
2. Rationale for Targeting the IGF System in Diseases
3. Methods of Targeting IGF1R and IR in Diseases
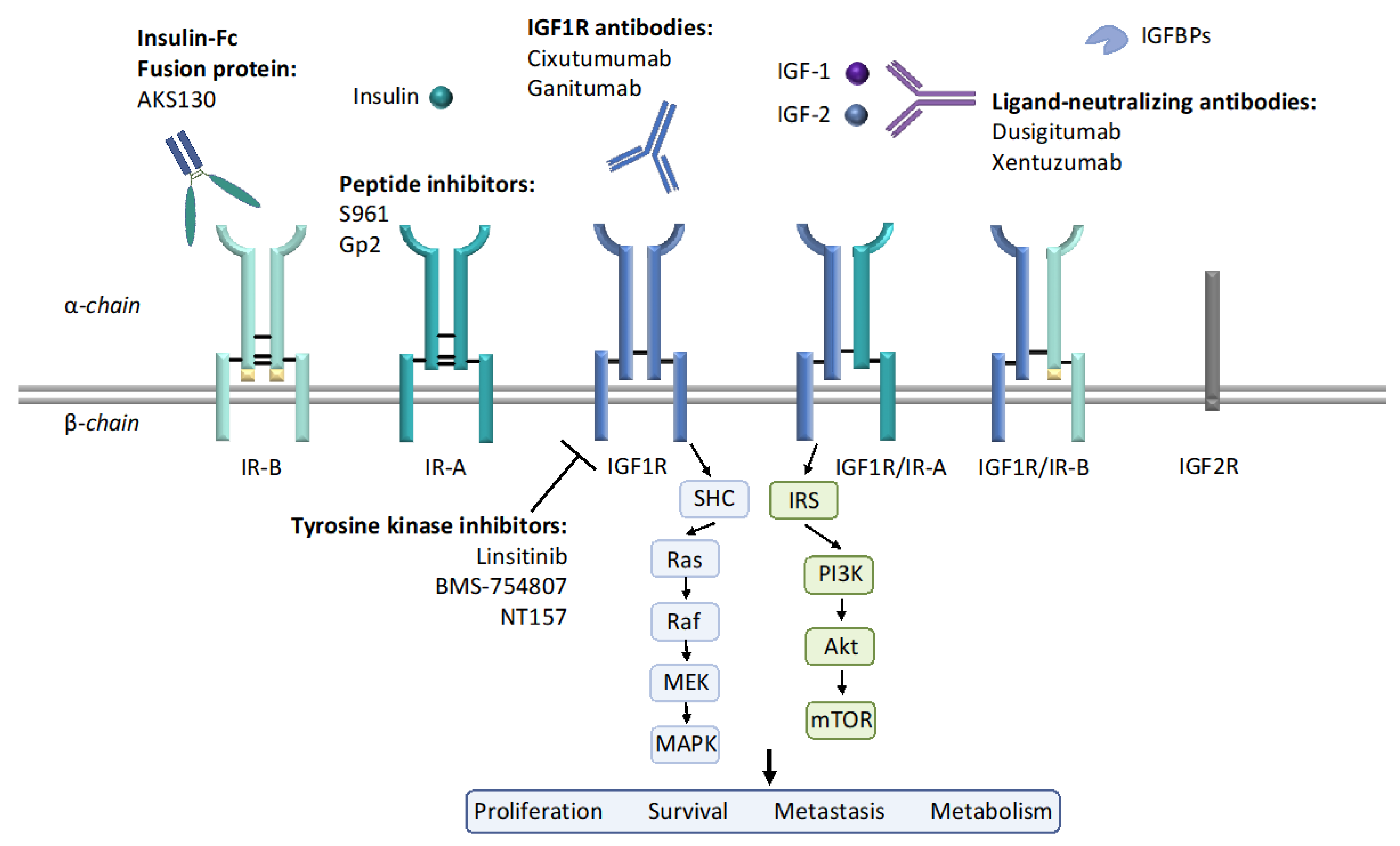
3.1. Monoclonal Antibodies
3.2. Tyrosine Kinase Inhibitors
3.3. Peptide Inhibitors
3.4. Ligand Neutralization
3.5. Receptor Downregulators
3.6. Antisense Oligonucleotides
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IGF | Insulin-like growth |
| IGFBP | IGF binding protein |
| IGF1R | Type I IGF receptor |
| IGF2R | Type II IGF receptor |
| IR | Insulin receptor |
| RTK | Receptor tyrosine kinase |
| TKD | Tyrosine kinase domain |
| IRS | Insulin receptor substrate |
| SHC | Src-homology collagen |
| PI3K | Phosphoinositide 3-kinase |
| MAPK | Mitogen-activated protein kinase |
| NAFLD | Nonalcoholic fatty liver disease |
| NSCLC | Non-small cell lung cancer |
| TED | Thyroid eye disease |
| TAO | Thyroid-associated ophthalmopathy |
| TSHR | Thyroid-stimulating hormone receptor |
| TSH | Thyroid-stimulating hormone |
| TNBC | Triple-negative breast cancer |
| mAb | Monoclonal antibody |
| HR | Hormone receptor |
| GH | Growth Hormone |
| TKI | Tyrosine kinase inhibitor |
| PPP | Picropodophyllin |
| HCC | Hepatocellular carcinoma |
| CDK | Cyclin dependent kinase |
| Mdm2 | Mouse double-minute 2 homolog |
| TamR | Tamoxifen-resistance |
| ASO | Antisense oligonucleotide |
References
- Hakuno, F.; Takahashi, S.I. IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeRoith, D.; Werner, H.; Beitner-Johnson, D.; Roberts, C.T., Jr. Molecular and Cellular Aspects of the Insulin-Like Growth Factor I Receptor. Endocr. Rev. 1995, 16, 143–163. [Google Scholar] [CrossRef]
- Yang, Y.; Yee, D. Targeting insulin and insulin-like growth factor signaling in breast cancer. J. Mammary Gland Biol. Neoplasia 2012, 17, 251–261. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- De Meyts, P. Insulin and its receptor: Structure, function and evolution. Bioessays 2004, 26, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- De Meyts, P.; Wallach, B.; Christoffersen, C.T.; Ursø, B.; Grønskov, K.; Latus, L.J.; Yakushiji, F.; Ilondo, M.M.; Shymko, R.M. The insulin-like growth factor-I receptor. Structure, ligand-binding mechanism and signal transduction. Horm. Res. 1994, 42, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Lou, M.; Garrett, T.P.J.; McKern, N.M.; Hoyne, P.A.; Epa, V.C.; Bentley, J.D.; Lovrecz, G.O.; Cosgrove, L.J.; Frenkel, M.J.; Ward, C.W. The first three domains of the insulin receptor differ structurally from the insulin-like growth factor 1 receptor in the regions governing ligand specificity. Proc. Natl. Acad. Sci. USA 2006, 103, 12429–12434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Choi, E.; Yu, H.; Bai, X.C. Structural basis of the activation of type 1 insulin-like growth factor receptor. Nat. Commun. 2019, 10, 4567. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.W.; Lawrence, M.C.; Streltsov, V.A.; Adams, T.E.; McKern, N.M. The insulin and EGF receptor structures: New insights into ligand-induced receptor activation. Trends Biochem. Sci. 2007, 32, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Liefers-Visser, J.A.L.; Meijering, R.A.M.; Reyners, A.K.L.; van der Zee, A.G.J.; de Jong, S. IGF system targeted therapy: Therapeutic opportunities for ovarian cancer. Cancer Treat. Rev. 2017, 60, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Slaaby, R.; Schaffer, L.; Lautrup-Larsen, I.; Andersen, A.S.; Shaw, A.C.; Mathiasen, I.S.; Brandt, J. Hybrid receptors formed by insulin receptor (IR) and insulin-like growth factor I receptor (IGF-IR) have low insulin and high IGF-1 affinity irrespective of the IR splice variant. J. Biol. Chem. 2006, 281, 25869–25874. [Google Scholar] [CrossRef] [Green Version]
- Kornfeld, S. Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu. Rev. Biochem. 1992, 61, 307–330. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, S.A. The Continuing Evolution of Insulin-like Growth Factor Signaling. F1000Research 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Jerome, L.; Shiry, L.; Leyland-Jones, B. Deregulation of the IGF axis in cancer: Epidemiological evidence and potential therapeutic interventions. Endocr. Relat. Cancer 2003, 10, 561–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef]
- Hellawell, G.O.; Turner, G.D.H.; Davies, D.R.; Poulsom, R.; Brewster, S.F.; Macaulay, V.M. Expression of the Type 1 Insulin-like Growth Factor Receptor Is Up-Regulated in Primary Prostate Cancer and Commonly Persists in Metastatic Disease. Cancer Res. 2002, 62, 2942–2950. [Google Scholar] [PubMed]
- Kim, J.S.; Kim, E.S.; Liu, D.; Lee, J.J.; Solis, L.; Behrens, C.; Lippman, S.M.; Hong, W.K.; Wistuba, I.I.; Lee, H.Y. Prognostic impact of insulin receptor expression on survival of patients with nonsmall cell lung cancer. Cancer 2012, 118, 2454–2465. [Google Scholar] [CrossRef] [Green Version]
- Law, J.H.; Habibi, G.; Hu, K.; Masoudi, H.; Wang, M.Y.; Stratford, A.L.; Park, E.; Gee, J.M.; Finlay, P.; Jones, H.E.; et al. Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res. 2008, 68, 10238–10246. [Google Scholar] [CrossRef] [Green Version]
- Adamek, A.; Kasprzak, A. Insulin-Like Growth Factor (IGF) System in Liver Diseases. Int. J. Mol. Sci. 2018, 19, 1308. [Google Scholar] [CrossRef] [Green Version]
- Yeo, C.D.; Park, K.H.; Park, C.K.; Lee, S.H.; Kim, S.J.; Yoon, H.K.; Lee, Y.S.; Lee, E.J.; Lee, K.Y.; Kim, T.J. Expression of insulin-like growth factor 1 receptor (IGF-1R) predicts poor responses to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in non-small cell lung cancer patients harboring activating EGFR mutations. Lung Cancer 2015, 87, 311–317. [Google Scholar] [CrossRef]
- Zhao, S.; Qiu, Z.; He, J.; Li, L.; Li, W. Insulin-like growth factor receptor 1 (IGF1R) expression and survival in non-small cell lung cancer patients: A meta-analysis. Int. J. Clin. Exp. Pathol. 2014, 7, 6694–6704. [Google Scholar]
- Bowers, L.W.; Rossi, E.L.; O’Flanagan, C.H.; deGraffenried, L.A.; Hursting, S.D. The Role of the Insulin/IGF System in Cancer: Lessons Learned from Clinical Trials and the Energy Balance-Cancer Link. Front. Endocrinol. (Lausanne) 2015, 6, 77. [Google Scholar] [CrossRef] [Green Version]
- The Endogenous Hormones and Breast Cancer Collaborative Group; Key, T.J.; Appleby, P.N.; Reeves, G.K.; Roddam, A.W. Insulin-like growth factor 1 (IGF1), IGF binding protein 3 (IGFBP3), and breast cancer risk: Pooled individual data analysis of 17 prospective studies. Lancet Oncol. 2010, 11, 530–542. [Google Scholar] [CrossRef] [Green Version]
- Murphy, N.; Knuppel, A.; Papadimitriou, N.; Martin, R.M.; Tsilidis, K.K.; Smith-Byrne, K.; Fensom, G.; Perez-Cornago, A.; Travis, R.C.; Key, T.J.; et al. Insulin-like growth factor-1, insulin-like growth factor-binding protein-3, and breast cancer risk: Observational and Mendelian randomization analyses with approximately 430 000 women. Ann. Oncol. 2020, 31, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Renehan, A.G.; Zwahlen, M.; Minder, C.; O’Dwyer, S.T.; Shalet, S.M.; Egger, M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: Systematic review and meta-regression analysis. Lancet 2004, 363, 1346–1353. [Google Scholar] [CrossRef]
- Pisani, P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch. Physiol. Biochem. 2008, 114, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Wolpin, B.M.; Meyerhardt, J.A.; Chan, A.T.; Ng, K.; Chan, J.A.; Wu, K.; Pollak, M.N.; Giovannucci, E.L.; Fuchs, C.S. Insulin, the insulin-like growth factor axis, and mortality in patients with nonmetastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J. New advances in understanding thyroid-associated ophthalmopathy and the potential role for insulin-like growth factor-I receptor. F1000Res. 2018, 7, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalek, K.; Morshed, S.A.; Latif, R.; Davies, T.F. TSH receptor autoantibodies. Autoimmun. Rev. 2009, 9, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Morshed, S.A.; Davies, T.F. Graves’ Disease Mechanisms: The Role of Stimulating, Blocking, and Cleavage Region TSH Receptor Antibodies. Horm. Metab. Res. 2015, 47, 727–734. [Google Scholar] [CrossRef] [Green Version]
- Douglas, R.S.; Gianoukakis, A.G.; Kamat, S.; Smith, T.J. Aberrant Expression of the Insulin-Like Growth Factor-1 Receptor by T Cells from Patients with Graves’ Disease May Carry Functional Consequences for Disease Pathogenesis. J. Immunol. 2007, 178, 3281–3287. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.; Han, R.; Horst, N.; Cruikshank, W.W.; Smith, T.J. Immunoglobulin activation of T cell chemoattractant expression in fibroblasts from patients with Graves’ disease is mediated through the insulin-like growth factor I receptor pathway. J. Immunol. 2003, 170, 6348–6354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weightman, D.R.; Perros, P.; Sherif, I.H.; Kendall-Taylor, P. Autoantibodies to IGF-1 binding sites in thyroid associated ophthalmopathy. Autoimmunity 1993, 16, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Tsui, S.; Naik, V.; Hoa, N.; Hwang, C.J.; Afifiyan, N.F.; Sinha Hikim, A.; Gianoukakis, A.G.; Douglas, R.S.; Smith, T.J. Evidence for an association between thyroid-stimulating hormone and insulin-like growth factor 1 receptors: A tale of two antigens implicated in Graves’ disease. J. Immunol. 2008, 181, 4397–4405. [Google Scholar] [CrossRef] [PubMed]
- Krieger, C.C.; Perry, J.D.; Morgan, S.J.; Kahaly, G.J.; Gershengorn, M.C. TSH/IGF-1 Receptor Cross-Talk Rapidly Activates Extracellular Signal-Regulated Kinases in Multiple Cell Types. Endocrinology 2017, 158, 3676–3683. [Google Scholar] [CrossRef] [Green Version]
- Brahmkhatri, V.P.; Prasanna, C.; Atreya, H.S. Insulin-like growth factor system in cancer: Novel targeted therapies. Biomed. Res. Int. 2015, 2015, 538019. [Google Scholar] [CrossRef] [Green Version]
- D’Ercole, A.J.; Stiles, A.D.; Underwood, L.E. Tissue concentrations of somatomedin C: Further evidence for multiple sites of synthesis and paracrine or autocrine mechanisms of action. Proc. Natl. Acad. Sci. USA 1984, 81, 935–939. [Google Scholar] [CrossRef] [Green Version]
- Livingstone, C.; Borai, A. Insulin-like growth factor-II: Its role in metabolic and endocrine disease. Clin. Endocrinol. (Oxf.) 2014, 80, 773–781. [Google Scholar] [CrossRef]
- Davison, Z.; de Blacquiere, G.E.; Westley, B.R.; May, F.E. Insulin-like growth factor-dependent proliferation and survival of triple-negative breast cancer cells: Implications for therapy. Neoplasia 2011, 13, 504–515. [Google Scholar] [CrossRef] [Green Version]
- Pacher, M.; Seewald, M.J.; Mikula, M.; Oehler, S.; Mogg, M.; Vinatzer, U.; Eger, A.; Schweifer, N.; Varecka, R.; Sommergruber, W.; et al. Impact of constitutive IGF1/IGF2 stimulation on the transcriptional program of human breast cancer cells. Carcinogenesis 2007, 28, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Cui, K.; Miyoshi, K.; Hennighausen, L.; Green, J.E.; Setser, J.; LeRoith, D.; Yakar, S. Reduced Circulating Insulin-like Growth Factor I Levels Delay the Onset of Chemically and Genetically Induced Mammary Tumors. Cancer Res. 2003, 63, 4384–4388. [Google Scholar] [PubMed]
- Samani, A.A.; Yakar, S.; LeRoith, D.; Brodt, P. The role of the IGF system in cancer growth and metastasis: Overview and recent insights. Endocr. Rev. 2007, 28, 20–47. [Google Scholar] [CrossRef] [PubMed]
- Manabe, T.; Yasuda, H.; Terai, H.; Kagiwada, H.; Hamamoto, J.; Ebisudani, T.; Kobayashi, K.; Masuzawa, K.; Ikemura, S.; Kawada, I.; et al. IGF2 Autocrine-Mediated IGF1R Activation Is a Clinically Relevant Mechanism of Osimertinib Resistance in Lung Cancer. Mol. Cancer. Res. 2020, 18, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Yamada, T.; Kita, K.; Taniguchi, H.; Arai, S.; Fukuda, K.; Terashima, M.; Ishimura, A.; Nishiyama, A.; Tanimoto, A.; et al. Transient IGF-1R inhibition combined with osimertinib eradicates AXL-low expressing EGFR mutated lung cancer. Nat. Commun. 2020, 11, 4607. [Google Scholar] [CrossRef]
- Fagan, D.H.; Uselman, R.R.; Sachdev, D.; Yee, D. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor: Implications for breast cancer treatment. Cancer Res. 2012, 72, 3372–3380. [Google Scholar] [CrossRef] [Green Version]
- Vaziri-Gohar, A.; Zheng, Y.; Houston, K.D. IGF-1 Receptor Modulates FoxO1-Mediated Tamoxifen Response in Breast Cancer Cells. Mol. Cancer. Res. 2017, 15, 489–497. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Sowers, J.Y.; Houston, K.D. IGFBP-1 Expression Promotes Tamoxifen Resistance in Breast Cancer Cells via Erk Pathway Activation. Front. Endocrinol. (Lausanne) 2020, 11, 233. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef]
- Fierz, Y.; Novosyadlyy, R.; Vijayakumar, A.; Yakar, S.; LeRoith, D. Insulin-sensitizing therapy attenuates type 2 diabetes-mediated mammary tumor progression. Diabetes 2010, 59, 686–693. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, E.J.; Alikhani, N.; Tobin-Hess, A.; Blank, J.; Buffin, N.J.; Zelenko, Z.; Tennagels, N.; Werner, U.; LeRoith, D. Insulin receptor phosphorylation by endogenous insulin or the insulin analog AspB10 promotes mammary tumor growth independent of the IGF-I receptor. Diabetes 2013, 62, 3553–3560. [Google Scholar] [CrossRef] [Green Version]
- Gradishar, W.J.; Yardley, D.A.; Layman, R.; Sparano, J.A.; Chuang, E.; Northfelt, D.W.; Schwartz, G.N.; Youssoufian, H.; Tang, S.; Novosiadly, R.; et al. Clinical and Translational Results of a Phase II, Randomized Trial of an Anti-IGF-1R (Cixutumumab) in Women with Breast Cancer That Progressed on Endocrine Therapy. Clin. Cancer. Res. 2016, 22, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.Y.; LaPara, K.; Yee, D. Disruption of insulin receptor function inhibits proliferation in endocrine-resistant breast cancer cells. Oncogene 2016, 35, 4235–4243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, T.; Claret, F.X. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baselga, J. Clinical trials of Herceptin(trastuzumab). Eur. J. Cancer 2001, 37, S18–S24. [Google Scholar] [CrossRef] [Green Version]
- Earl, H.M.; Hiller, L.; Vallier, A.; Loi, S.; McAdam, K.; Hughes-Davies, L.; Harnett, A.N.; Ah-See, M.; Simcock, R.; Rea, D.; et al. 6 versus 12 months of adjuvant trastuzumab for HER2-positive early breast cancer (PERSEPHONE): 4-year disease-free survival results of a randomised phase 3 non-inferiority trial. Lancet 2019, 393, 2599–2612. [Google Scholar] [CrossRef] [Green Version]
- Burtrum, D.; Zhu, Z.; Lu, D.; Anderson, D.M.; Prewett, M.; Pereira, D.S.; Bassi, R.; Abdullah, R.; Hooper, A.T.; Koo, H.; et al. A Fully Human Monoclonal Antibody to the Insulin-Like Growth Factor I Receptor Blocks Ligand-Dependent Signaling and Inhibits Human Tumor Growth in Vivo. Cancer Res. 2003, 63, 8912–8921. [Google Scholar] [PubMed]
- Ekyalongo, R.C.; Yee, D. Revisiting the IGF-1R as a breast cancer target. NPJ Precis. Oncol. 2017, 14. [Google Scholar] [CrossRef] [Green Version]
- Vijayakumar, A.; Novosyadlyy, R.; Wu, Y.; Yakar, S.; LeRoith, D. Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Horm. IGF Res. 2010, 20, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Yee, D. A tale of two receptors: Insulin and insulin-like growth factor signaling in cancer. Clin. Cancer Res. 2015, 21, 667–669. [Google Scholar] [CrossRef] [Green Version]
- Novello, S.; Scagliotti, G.; de Castro, G., Jr.; Kiyik, M.; Kowalyszyn, R.; Deppermann, K.M.; Arriola, E.; Bosquee, L.; Novosiadly, R.D.; Nguyen, T.S.; et al. An Open-Label, Multicenter, Randomized, Phase II Study of Cisplatin and Pemetrexed With or Without Cixutumumab (IMC-A12) as a First-Line Therapy in Patients with Advanced Nonsquamous Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 383–389. [Google Scholar] [CrossRef] [Green Version]
- Beltran, P.J.; Mitchell, P.; Chung, Y.A.; Cajulis, E.; Lu, J.; Belmontes, B.; Ho, J.; Tsai, M.M.; Zhu, M.; Vonderfecht, S.; et al. AMG 479, a fully human anti-insulin-like growth factor receptor type I monoclonal antibody, inhibits the growth and survival of pancreatic carcinoma cells. Mol. Cancer. Ther. 2009, 8, 1095–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kindler, H.L.; Richards, D.A.; Garbo, L.E.; Garon, E.B.; Stephenson, J.J., Jr.; Rocha-Lima, C.M.; Safran, H.; Chan, D.; Kocs, D.M.; Galimi, F.; et al. A randomized, placebo-controlled phase 2 study of ganitumab (AMG 479) or conatumumab (AMG 655) in combination with gemcitabine in patients with metastatic pancreatic cancer. Ann. Oncol. 2012, 23, 2834–2842. [Google Scholar] [CrossRef]
- Robertson, J.F.; Ferrero, J.; Bourgeois, H.; Kennecke, H.; de Boer, R.H.; Jacot, W.; McGreivy, J.; Suzuki, S.; Zhu, M.; McCaffery, I.; et al. Ganitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: A randomised, controlled, double-blind, phase 2 trial. Lancet Oncol. 2013, 14, 228–235. [Google Scholar] [CrossRef]
- Forest, A.; Amatulli, M.; Ludwig, D.L.; Damoci, C.B.; Wang, Y.; Burns, C.A.; Donoho, G.P.; Zanella, N.; Fiebig, H.H.; Prewett, M.C.; et al. Intrinsic Resistance to Cixutumumab Is Conferred by Distinct Isoforms of the Insulin Receptor. Mol. Cancer. Res. 2015, 13, 1615–1626. [Google Scholar] [CrossRef] [Green Version]
- Yee, D.; Paoloni, M.; van’t Veer, L.; Sanil, A.; Yau, C.; Forero, A.; Chien, A.J.; Wallace, A.M.; Moulder, S.; Albain, K.S.; et al. The evaluation of ganitumab/metformin plus standard neoadjuvant therapy in high-risk breast cancer: Results from the I-SPY 2 trial [abstract]. In Proceedings of the 2016 San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 6–10 December 2016; Volume 77. Abstract nr P6-11-04. [Google Scholar]
- Douglas, R.S. Teprotumumab, an insulin-like growth factor-1 receptor antagonist antibody, in the treatment of active thyroid eye disease: A focus on proptosis. Eye (Lond.) 2019, 33, 183–190. [Google Scholar] [CrossRef]
- Douglas, R.S.; Kahaly, G.J.; Patel, A.; Sile, S.; Thompson, E.H.Z.; Perdok, R.; Fleming, J.C.; Fowler, B.T.; Marcocci, C.; Marino, M.; et al. Teprotumumab for the Treatment of Active Thyroid Eye Disease. N. Engl. J. Med. 2020, 382, 341–352. [Google Scholar] [CrossRef]
- Smith, T.J.; Kahaly, G.J.; Ezra, D.G.; Fleming, J.C.; Dailey, R.A.; Tang, R.A.; Harris, G.J.; Antonelli, A.; Salvi, M.; Goldberg, R.A.; et al. Teprotumumab for Thyroid-Associated Ophthalmopathy. N. Engl. J. Med. 2017, 376, 1748–1761. [Google Scholar] [CrossRef]
- Mulvihill, M.J.; Cooke, A.; Rosenfeld-Franklin, M.; Buck, E.; Foreman, K.; Landfair, D.; O’Connor, M.; Pirritt, C.; Sun, Y.; Yao, Y.; et al. Discovery of OSI-906: A selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med. Chem. 2009, 1, 1153–1171. [Google Scholar] [CrossRef]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell. Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Awasthi, N.; Zhang, C.; Ruan, W.; Schwarz, M.A.; Schwarz, R.E. BMS-754807, a small-molecule inhibitor of insulin-like growth factor-1 receptor/insulin receptor, enhances gemcitabine response in pancreatic cancer. Mol. Cancer Ther. 2012, 11, 2644–2653. [Google Scholar] [CrossRef] [Green Version]
- Carboni, J.M.; Wittman, M.; Yang, Z.; Lee, F.; Greer, A.; Hurlburt, W.; Hillerman, S.; Cao, C.; Cantor, G.H.; Dell-John, J.; et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther. 2009, 8, 3341–3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girnita, A.; Girnita, L.; Prete, F.; Bartolazzi, A.; Larsson, O.; Axelson, M. Cyclolignans as inhibitors of the insulin-like growth factor-1 receptor and malignant cell growth. Cancer Res. 2004, 64, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stromberg, T.; Ekman, S.; Girnita, L.; Dimberg, L.Y.; Larsson, O.; Axelson, M.; Lennartsson, J.; Hellman, U.; Carlson, K.; Osterborg, A.; et al. IGF-1 receptor tyrosine kinase inhibition by the cyclolignan PPP induces G2/M-phase accumulation and apoptosis in multiple myeloma cells. Blood 2006, 107, 669–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarnowski, M.; Tkacz, M.; Zgutka, K.; Bujak, J.; Kopytko, P.; Pawlik, A. Picropodophyllin (PPP) is a potent rhabdomyosarcoma growth inhibitor both in vitro and in vivo. BMC Cancer 2017, 17, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Pan, J.; Lubet, R.A.; Wang, Y.; You, M. Targeting the insulin-like growth factor-1 receptor by picropodophyllin for lung cancer chemoprevention. Mol. Carcinog. 2015, 54 (Suppl. 1), E129–E137. [Google Scholar] [CrossRef]
- Vasilcanu, D.; Girnita, A.; Girnita, L.; Vasilcanu, R.; Axelson, M.; Larsson, O. The cyclolignan PPP induces activation loop-specific inhibition of tyrosine phosphorylation of the insulin-like growth factor-1 receptor. Link to the phosphatidyl inositol-3 kinase/Akt apoptotic pathway. Oncogene 2004, 23, 7854–7862. [Google Scholar] [CrossRef] [Green Version]
- Vasilcanu, R.; Vasilcanu, D.; Rosengren, L.; Natalishvili, N.; Sehat, B.; Yin, S.; Girnita, A.; Axelson, M.; Girnita, L.; Larsson, O. Picropodophyllin induces downregulation of the insulin-like growth factor 1 receptor: Potential mechanistic involvement of Mdm2 and beta-arrestin1. Oncogene 2008, 27, 1629–1638. [Google Scholar] [CrossRef]
- Aiken, R.; Axelson, M.; Harmenberg, J.; Klockare, M.; Larsson, O.; Wassberg, C. Phase I clinical trial of AXL1717 for treatment of relapsed malignant astrocytomas: Analysis of dose and response. Oncotarget 2017, 8, 81501–81510. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Clausen, M.H.; Nielsen, T.E. Allosteric small-molecule kinase inhibitors. Pharmacol. Ther. 2015, 156, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, T.; Gradler, U.; Bottcher, H.; Blaukat, A.; Shutes, A. Allosteric IGF-1R Inhibitors. ACS Med. Chem. Lett. 2010, 1, 199–203. [Google Scholar] [CrossRef] [Green Version]
- Reuveni, H.; Flashner-Abramson, E.; Steiner, L.; Makedonski, K.; Song, R.; Shir, A.; Herlyn, M.; Bar-Eli, M.; Levitzki, A. Therapeutic destruction of insulin receptor substrates for cancer treatment. Cancer Res. 2013, 73, 4383–4394. [Google Scholar] [CrossRef] [Green Version]
- Fenerich, B.A.; Fernandes, J.C.; Rodrigues Alves, A.P.N.; Coelho-Silva, J.L.; Scopim-Ribeiro, R.; Scheucher, P.S.; Eide, C.A.; Tognon, C.E.; Druker, B.J.; Rego, E.M.; et al. NT157 has antineoplastic effects and inhibits IRS1/2 and STAT3/5 in JAK2(V617F)-positive myeloproliferative neoplasm cells. Signal Transduct. Target. Ther. 2020, 5, 5. [Google Scholar] [CrossRef]
- Garofalo, C.; Capristo, M.; Mancarella, C.; Reunevi, H.; Picci, P.; Scotlandi, K. Preclinical Effectiveness of Selective Inhibitor of IRS-1/2 NT157 in Osteosarcoma Cell Lines. Front. Endocrinol. (Lausanne) 2015, 6, 74. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Chan, J.Y.; Temiz, N.A.; Yee, D. Insulin Receptor Substrate Suppression by the Tyrphostin NT157 Inhibits Responses to Insulin-Like Growth Factor-I and Insulin in Breast Cancer Cells. Horm. Cancer 2018, 9, 371–382. [Google Scholar] [CrossRef]
- Schaffer, L.; Brand, C.L.; Hansen, B.F.; Ribel, U.; Shaw, A.C.; Slaaby, R.; Sturis, J. A novel high-affinity peptide antagonist to the insulin receptor. Biochem. Biophys. Res. Commun. 2008, 376, 380–383. [Google Scholar] [CrossRef]
- Rostoker, R.; Bitton-Worms, K.; Caspi, A.; Shen-Orr, Z.; LeRoith, D. Investigating new therapeutic strategies targeting hyperinsulinemia’s mitogenic effects in a female mouse breast cancer model. Endocrinology 2013, 154, 1701–1710. [Google Scholar] [CrossRef] [Green Version]
- Vikram, A.; Jena, G. S961, an insulin receptor antagonist causes hyperinsulinemia, insulin-resistance and depletion of energy stores in rats. Biochem. Biophys. Res. Commun. 2010, 398, 260–265. [Google Scholar] [CrossRef]
- Knudsen, L.; Hansen, B.F.; Jensen, P.; Pedersen, T.A.; Vestergaard, K.; Schaffer, L.; Blagoev, B.; Oleksiewicz, M.B.; Kiselyov, V.V.; De Meyts, P. Agonism and antagonism at the insulin receptor. PLoS ONE 2012, 7, e51972. [Google Scholar] [CrossRef]
- Buck, E.; Gokhale, P.C.; Koujak, S.; Brown, E.; Eyzaguirre, A.; Tao, N.; Rosenfeld-Franklin, M.; Lerner, L.; Chiu, M.I.; Wild, R.; et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): Rationale for cotargeting IGF-1R and IR in cancer. Mol. Cancer Ther. 2010, 9, 2652–2664. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.Y.; Hackel, B.J.; Yee, D. Targeting Insulin Receptor in Breast Cancer Using Small Engineered Protein Scaffolds. Mol. Cancer Ther. 2017, 16, 1324–1334. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Chesebrough, J.W.; Cartlidge, S.A.; Ricketts, S.A.; Incognito, L.; Veldman-Jones, M.; Blakey, D.C.; Tabrizi, M.; Jallal, B.; Trail, P.A.; et al. Dual IGF-I/II-neutralizing antibody MEDI-573 potently inhibits IGF signaling and tumor growth. Cancer Res. 2011, 71, 1029–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haluska, P.; Menefee, M.; Plimack, E.R.; Rosenberg, J.; Northfelt, D.; LaVallee, T.; Shi, L.; Yu, X.Q.; Burke, P.; Huang, J.; et al. Phase I dose-escalation study of MEDI-573, a bispecific, antiligand monoclonal antibody against IGFI and IGFII, in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 4747–4757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedbichler, K.; Hofmann, M.H.; Kroez, M.; Ostermann, E.; Lamche, H.R.; Koessl, C.; Borges, E.; Pollak, M.N.; Adolf, G.; Adam, P.J. Pharmacodynamic and antineoplastic activity of BI 836845, a fully human IGF ligand-neutralizing antibody, and mechanistic rationale for combination with rapamycin. Mol. Cancer Ther. 2014, 13, 399–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, D.; Oliveira, M.; Iwata, H.; Gonçalves, A.; García-Corbacho, J.; Sablin, M.P.; Prat, A.; Hardebeck, M.C.; Puig, M.; Huang, D.C.; et al. A phase Ib multi-cohort study of xentuzumab and abemaciclib in patients (pts) with solid tumors and breast cancer (BC)—Initial report of four dose-finding cohorts [abstract]. In Proceedings of the 2019 San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 10–14 December 2019; Volume 80. Abstract nr P3-11-05. [Google Scholar] [CrossRef]
- Worrall, C.; Suleymanova, N.; Crudden, C.; Trocoli Drakensjo, I.; Candrea, E.; Nedelcu, D.; Takahashi, S.I.; Girnita, L.; Girnita, A. Unbalancing p53/Mdm2/IGF-1R axis by Mdm2 activation restrains the IGF-1-dependent invasive phenotype of skin melanoma. Oncogene 2017, 36, 3274–3286. [Google Scholar] [CrossRef] [Green Version]
- Fettig, L.; Zhang, X.; LaPara, K.; Murikipudi, S.; Delpero, A.R.; Lancaster, T.M.; Zion, T.C.; Yee, D. A novel long-acting insulin for cancer therapy reduces xenograft tumor growth [abstract]. In Proceedings of the 2019 San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 10–14 December 2019; Volume 80. Abstract nr P1-21-05. [Google Scholar] [CrossRef]
- Furukawa, J.; Wraight, C.J.; Freier, S.M.; Peralta, E.; Atley, L.M.; Monia, B.P.; Gleave, M.E.; Cox, M.E. Antisense oligonucleotide targeting of insulin-like growth factor-1 receptor (IGF-1R) in prostate cancer. Prostate 2010, 70, 206–218. [Google Scholar] [CrossRef]
- Salatino, M.; Schillaci, R.; Proietti, C.J.; Carnevale, R.; Frahm, I.; Molinolo, A.A.; Iribarren, A.; Charreau, E.H.; Elizalde, P.V. Inhibition of in vivo breast cancer growth by antisense oligodeoxynucleotides to type I insulin-like growth factor receptor mRNA involves inactivation of ErbBs, PI-3K/Akt and p42/p44 MAPK signaling pathways but not modulation of progesterone receptor activity. Oncogene 2004, 23, 5161–5174. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Drug Type | Compound | Preclinical Studies | Clinical Studies | Ongoing Clinical Trials |
|---|---|---|---|---|
| Monoclonal antibodies | Cixutumumab (IMC-A12) | [56] | [51,60] | |
| Ganitumab (AMG-479) | [61] | [62,63,65] | NCT03041701 NCT04129151 NCT01042379 | |
| Teprotumumab | [66,67,68] | |||
| Tyrosine kinase inhibitors | Linsitinib (OSI-906) | [69] | NCT01205685 | |
| BMS-754807 | [71,72] | NCT01225172 | ||
| Picropodophyllin | [73,74,75,76] | [79] | NCT01721577 | |
| NT157 | [82,83,84,85] | |||
| Peptide inhibitors | S961 | [86,87,88] | ||
| Gp2 | [91] | |||
| Ligand neutralization | Dusigitumab (MEDI-573) | [92] | NCT01446159 NCT01498952 | |
| Xentuzumab (BI 836845) | [94] | NCT02191891 NCT02123823 NCT02204072 | NCT03659136 NCT03099174 | |
| Receptor downregulators | Nutlin-3 | [96] | ||
| AKS-130 | [97] | |||
| Antisense oligonucleotides | ATL1101 | [98,99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, J.; Yee, D. Disrupting Insulin and IGF Receptor Function in Cancer. Int. J. Mol. Sci. 2021, 22, 555. https://doi.org/10.3390/ijms22020555
Cao J, Yee D. Disrupting Insulin and IGF Receptor Function in Cancer. International Journal of Molecular Sciences. 2021; 22(2):555. https://doi.org/10.3390/ijms22020555
Chicago/Turabian StyleCao, Jingran, and Douglas Yee. 2021. "Disrupting Insulin and IGF Receptor Function in Cancer" International Journal of Molecular Sciences 22, no. 2: 555. https://doi.org/10.3390/ijms22020555
APA StyleCao, J., & Yee, D. (2021). Disrupting Insulin and IGF Receptor Function in Cancer. International Journal of Molecular Sciences, 22(2), 555. https://doi.org/10.3390/ijms22020555