Structure-Activity Relationship Modeling and Experimental Validation of the Imidazolium and Pyridinium Based Ionic Liquids as Potential Antibacterials of MDR Acinetobacter baumannii and Staphylococcus aureus

 , ,
, ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Regression Models (Dataset I)
2.2. Regression Models (Dataset II)
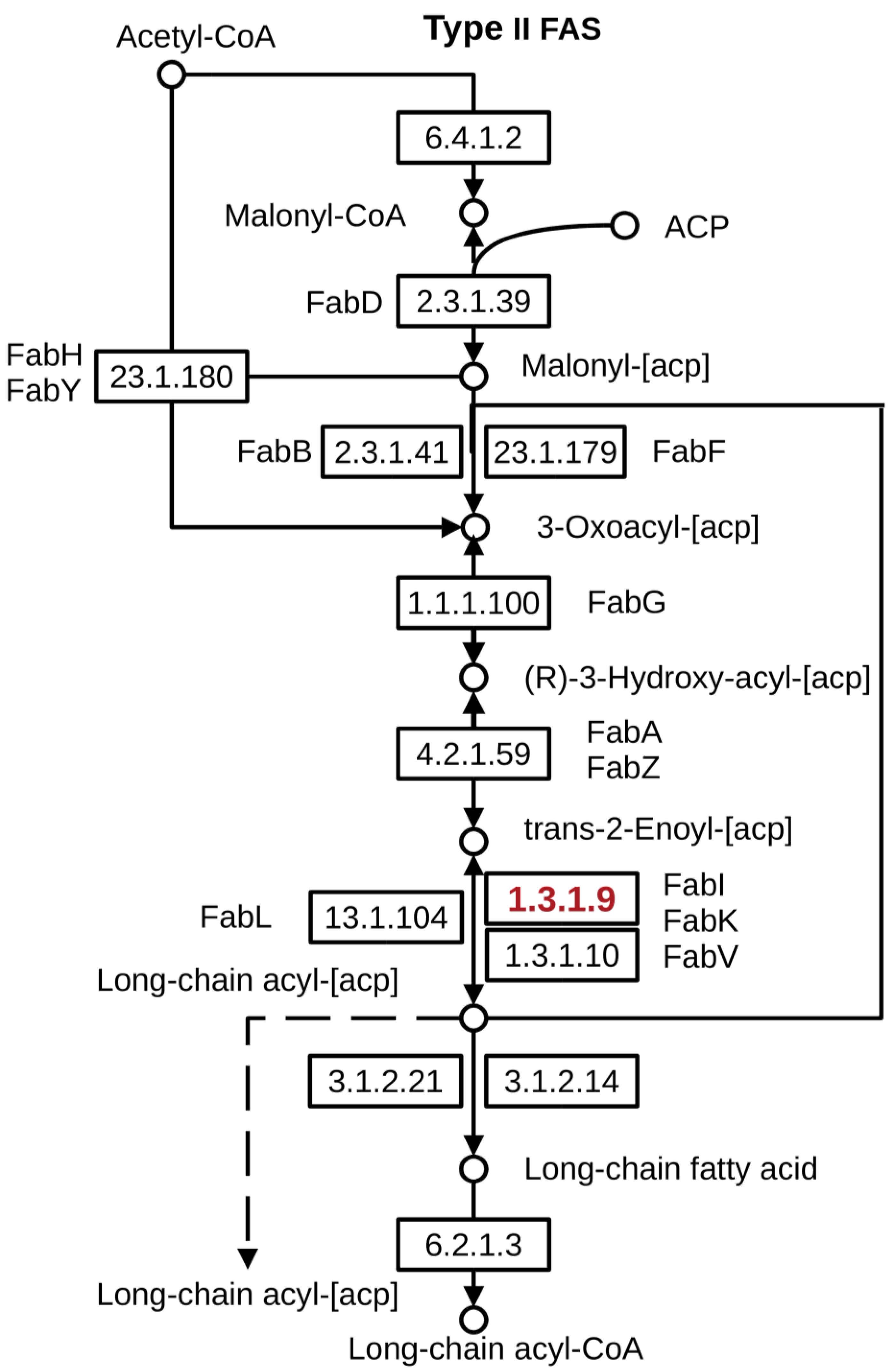
2.3. Evaluation of the Mode of Action (MoA) and Descriptor Importance
2.4. Evaluation Activity of New Compounds
2.5. Biology
Antibacterial Activity
3. Discussion
4. Materials and Methods
4.1. Data
4.2. On-Line Chemical Database and Modeling Environment
4.2.1. Methods
4.2.2. Associative Neural Network (ASNN)
4.2.3. Extreme Gradient Boosting (XGBoost)
4.2.4. Transformer Convolutional Neural Network (Trans-CNN)
4.2.5. Random Forest Regression (RFR)
4.2.6. Descriptors
4.2.7. Descriptor Preprocessing
4.2.8. Model Validation
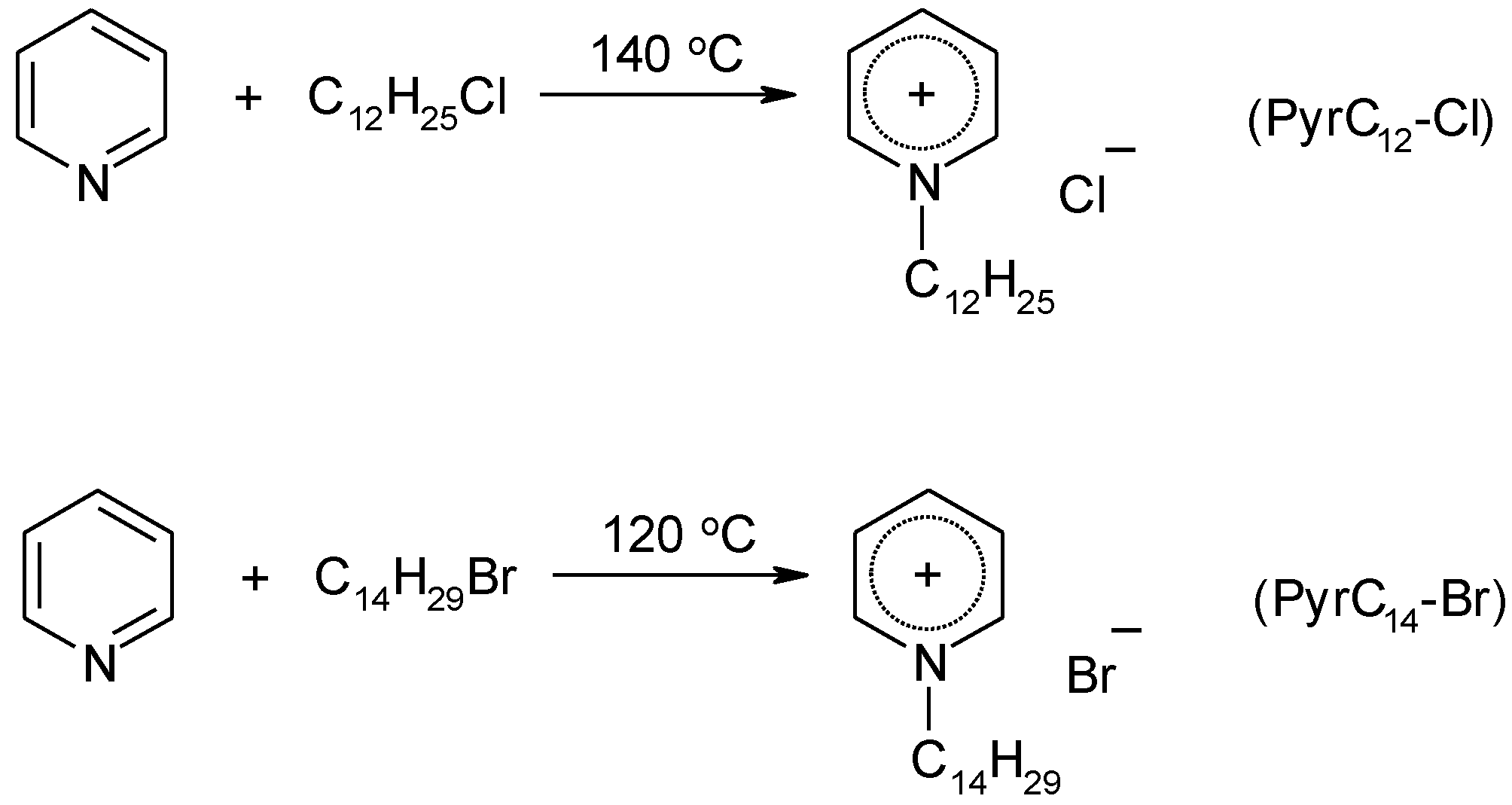
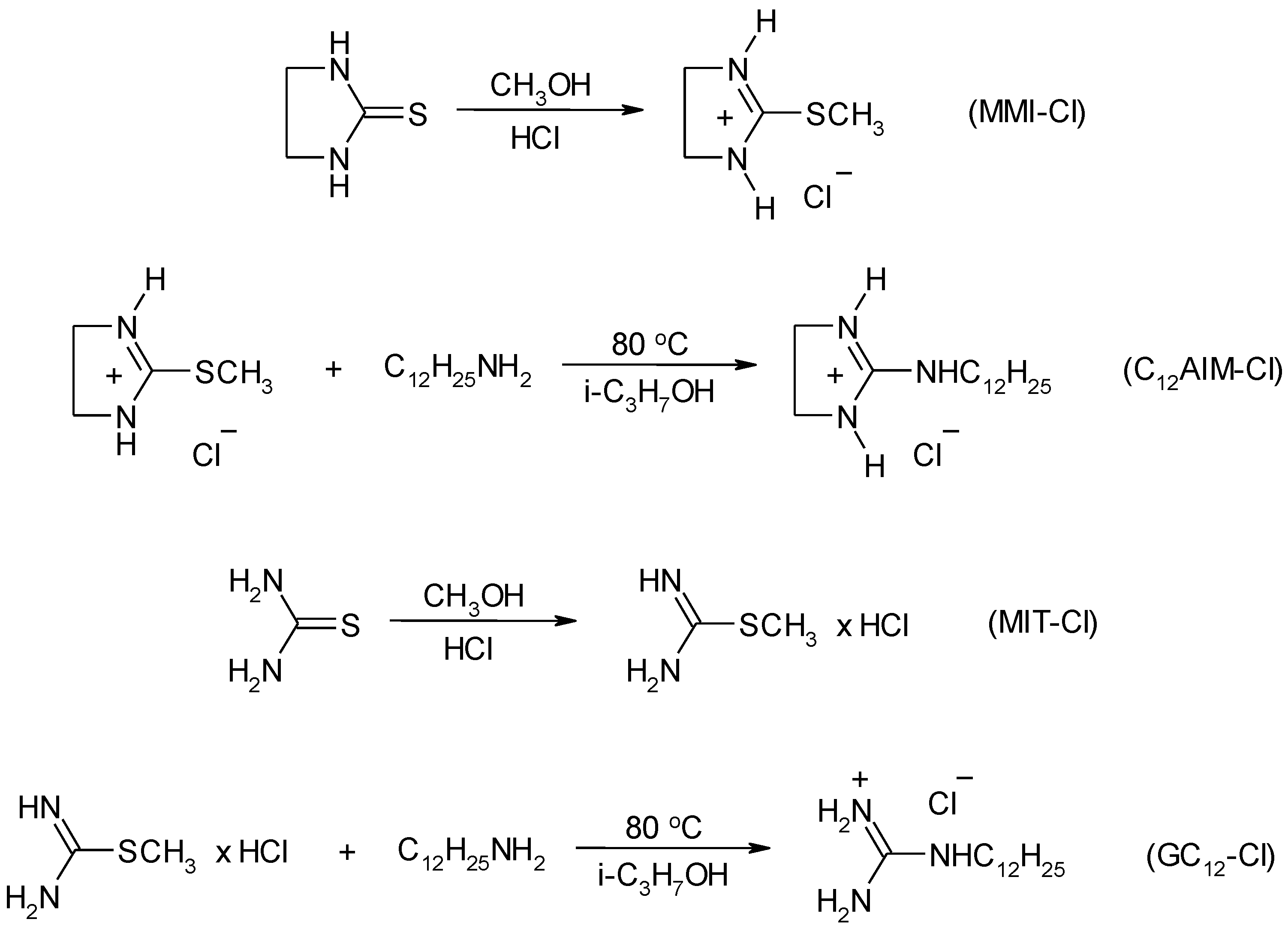
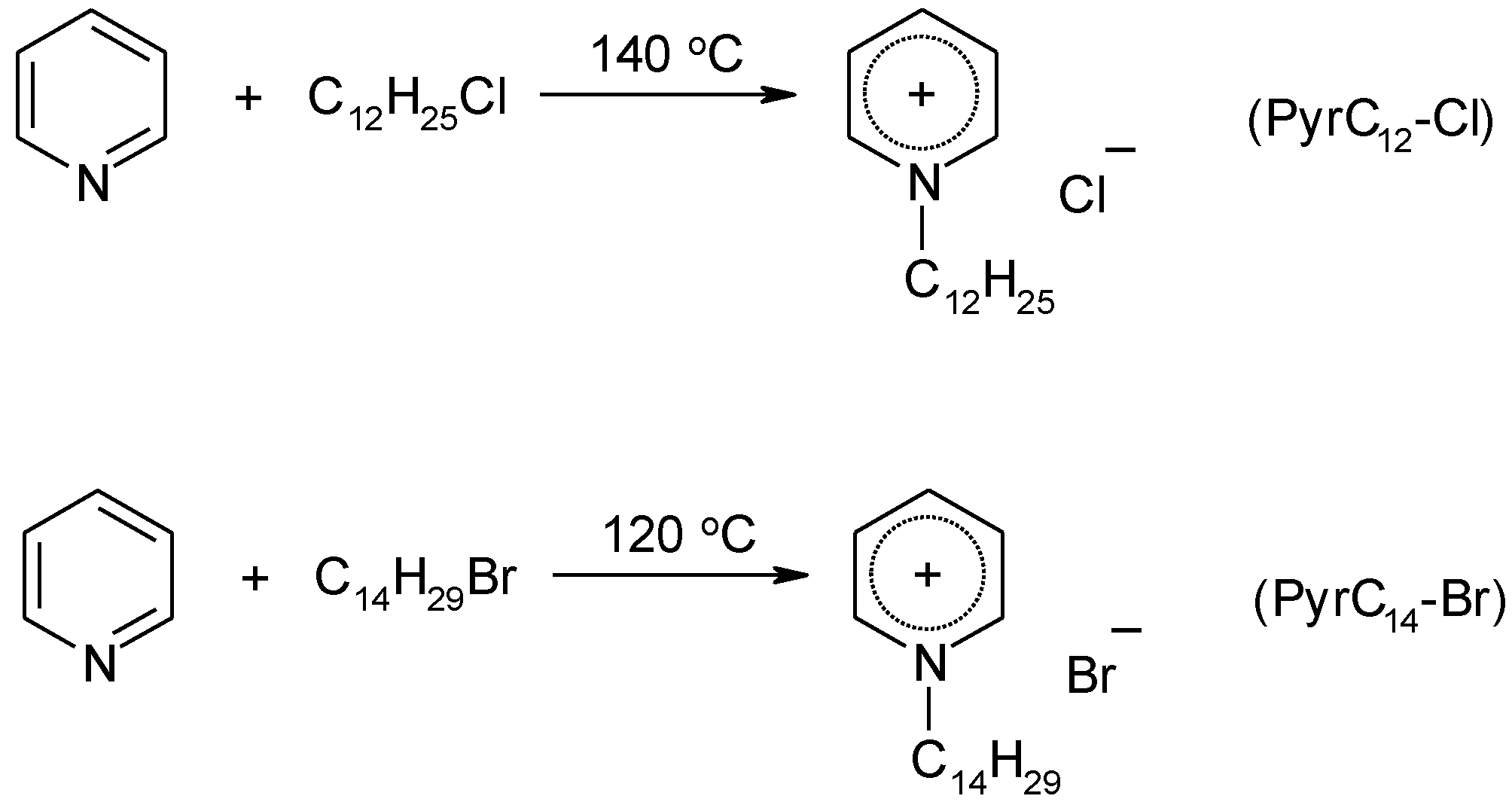
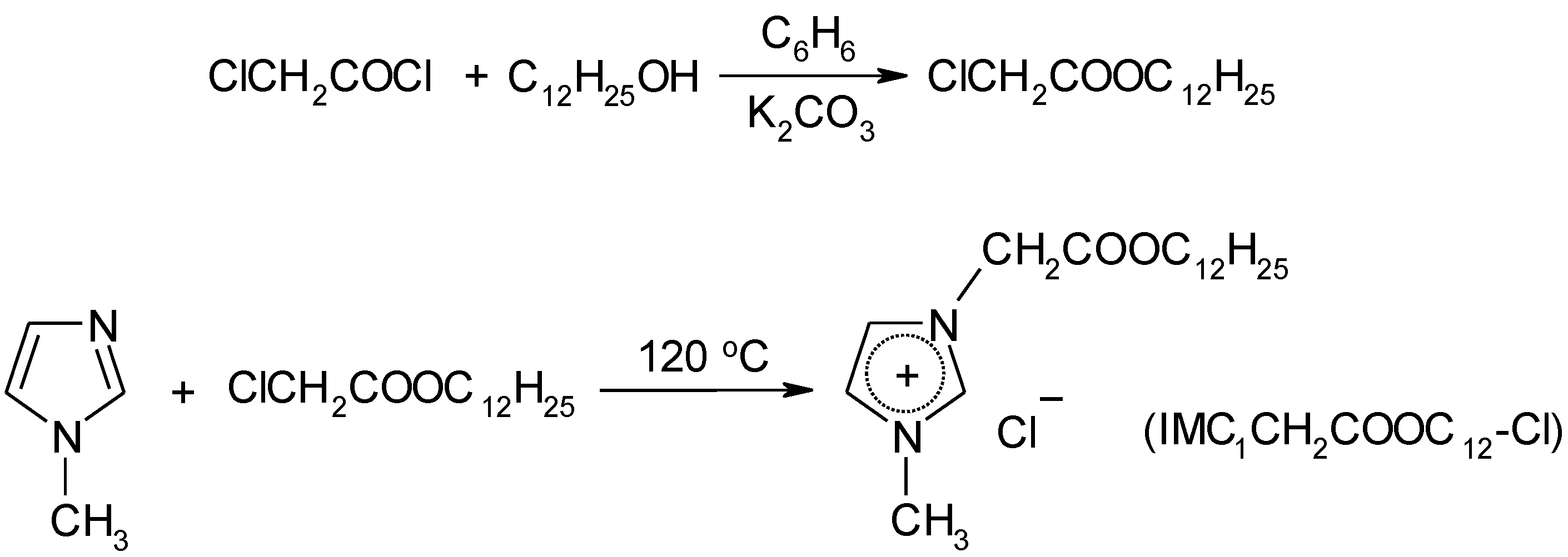
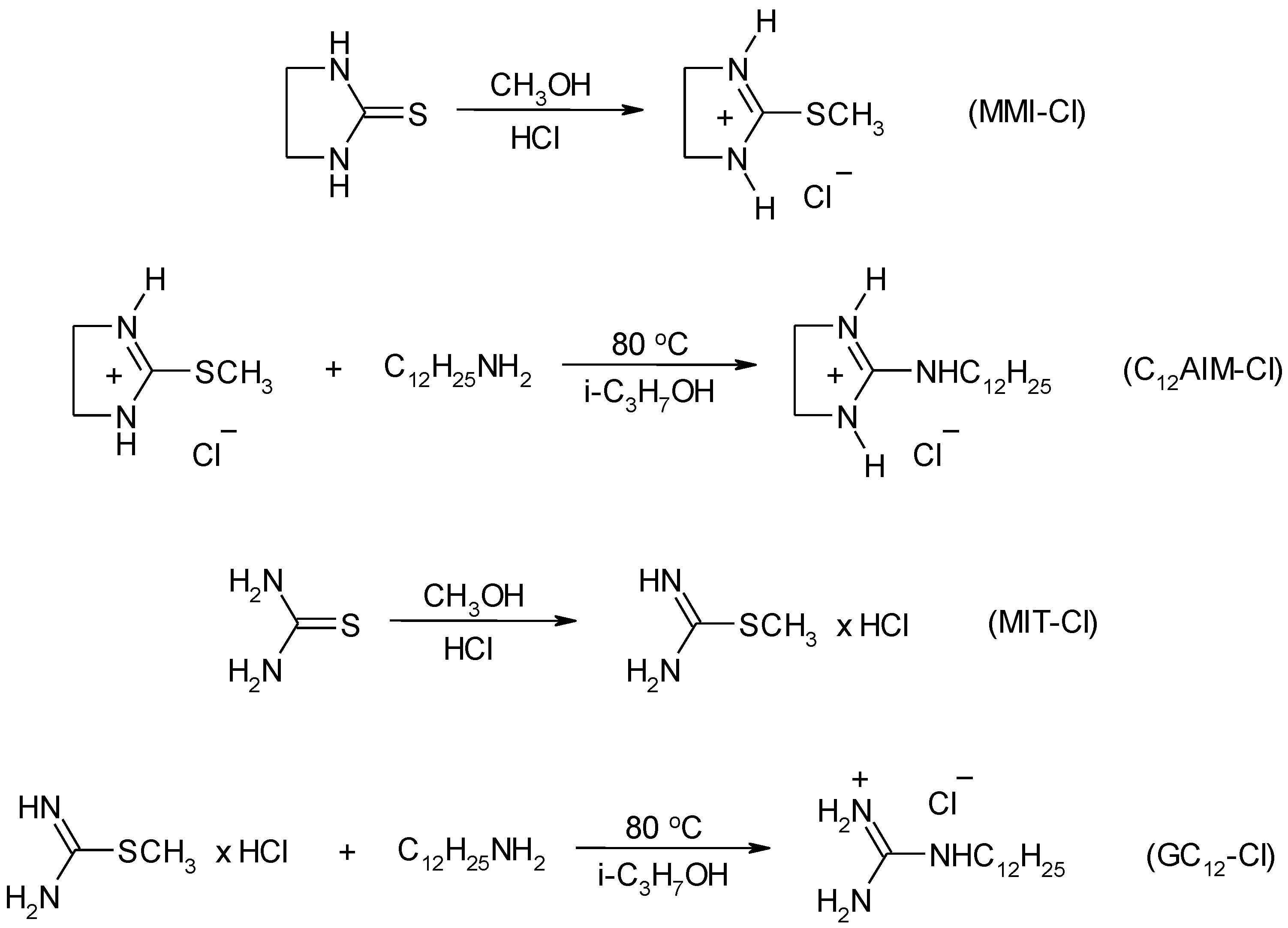
4.3. Synthesis
4.3.1. General
4.3.2. Synthesis of Ionic Liquids
4.4. Biology
4.5. Molecular Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| QSAR | Quantitative Structure–Activity Relationship |
| OCHEM | Online Chemical Modeling Environment |
| ASNN | Associative Neural Network |
| XGBOOST | Extreme Gradient Boosting |
| Trans-CNN | Transformer Convolutional Neural Network |
| RFR | Random Forest Regression |
| kNN | k-Nearest Neighbors |
| RMSE | Root Mean Squared Error |
| R2 | Square of correlation coefficient of determination |
| q2 | Coefficient of determination |
| MDR | Multi-drug resistant |
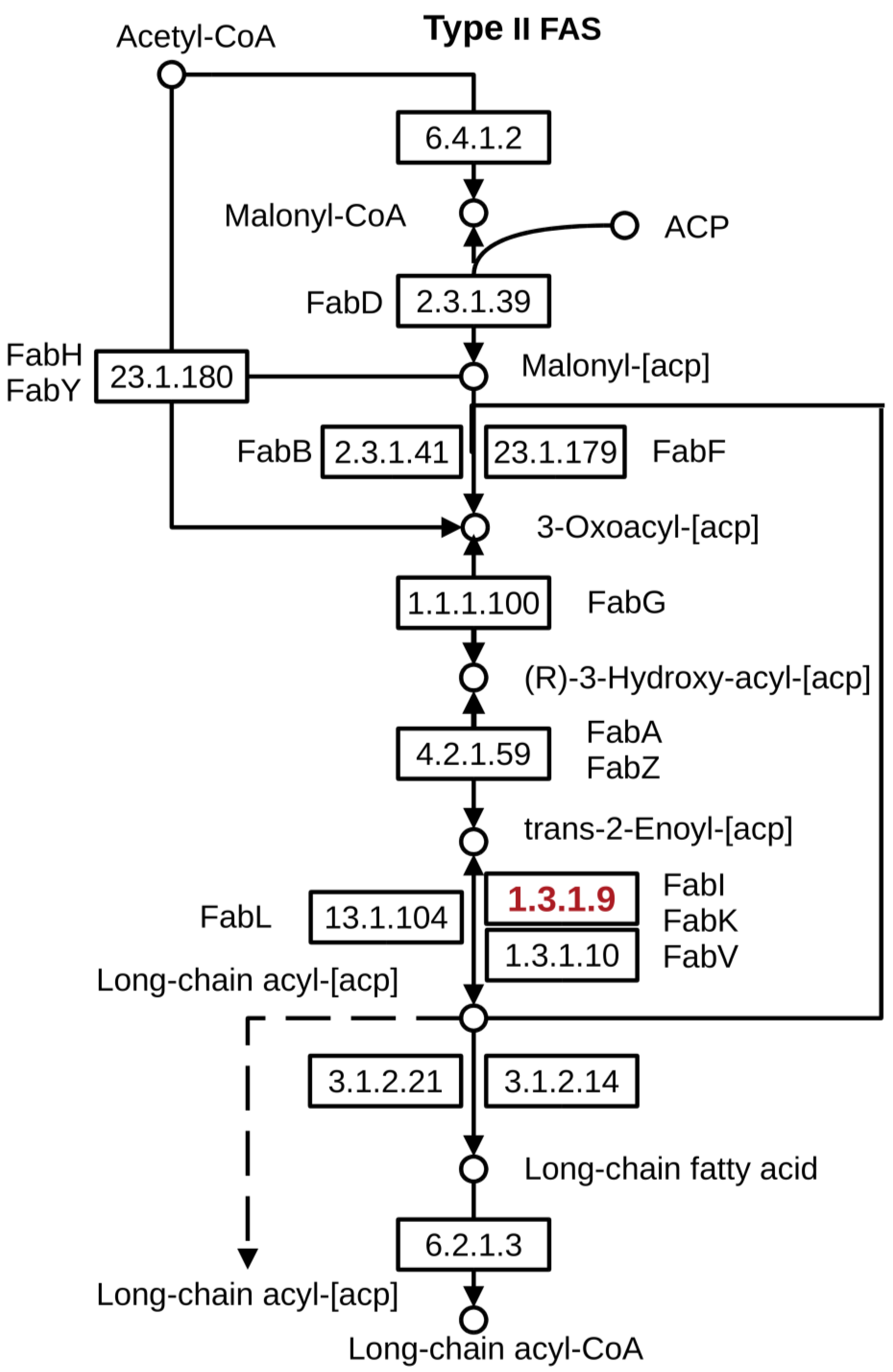
| FASII | Bacterial fatty acid biosynthesis type II |
| ACP | Acyl carrier protein |
| AChE | Acetylcholinesterase |
| AMP | Adenosine monophosphate |
| NADP | Nicotinamide adenine dinucleotide phosphate |
| ADT | AutoDock Tools |
| TCL | Triclosan |
| FabI | Enoyl-ACP reductase |
| AFabI | Enoyl-ACP reductase A.baumannii |
| SFabI | Enoyl-ACP reductase S. aureus |
| ATCC | American Type Culture Collection |
References
- List of Bacteria for Which New Antibiotics Are Urgently Needed. Available online: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 25 December 2020).
- Coates, R.; Moran, J.; Horsburgh, M.J. Staphylococci: Colonizers and pathogens of human skin. Future Microbiol. 2013, 9, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Martín-Aspas, A.; Guerrero-Sánchez, F.M.; García-Colchero, F.; Rodríguez-Roca, S.; Girón-González, J.A. Differential characteristics of Acinetobacter baumannii colonization and infection: Risk factors, clinical picture, and mortality. Infect. Drug Resist. 2018, 11, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trush, M.M.; Semenyuta, I.V.; Vdovenko, S.I.; Rogalsky, S.P.; Lobko, E.O.; Metelytsia, L.O. Synthesis, spectroscopic and molecular docking studies of imidazolium and pyridinium based ionic liquids with HSA as potential antimicrobial agents. J. Mol. Struct. 2017, 1137, 692–699. [Google Scholar] [CrossRef]
- Miskiewicz, A.; Ceranowicz, P.; Szymczak, M.; Bartuś, K.; Kowalczyk, P. The use of liquids ionic fluids as pharmaceutically active substances helpful in combating nosocomial infections induced by Klebsiella Pneumoniae new delhi strain, Acinetobacter Baumannii and Enterococcus species. Int. J. Mol. Sci. 2018, 19, 2779. [Google Scholar] [CrossRef] [Green Version]
- Ghanem, O.B.; Mutalib, M.; El-Harbawi, M.; Gonfa, G.; Kait, C.F.; Alitheen, N.B.M.; Lévêque, J.M. Effect of imidazolium-based ionic liquids on bacterial growth inhibition investigated via experimental and QSAR modelling studies. J. Hazard. Mater. 2015, 297, 198–206. [Google Scholar] [CrossRef]
- Wright, H.T.; Reynolds, K.A. Antibacterial targets in fatty acid biosynthesis. Curr. Opin. Microbiol. 2007, 10, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Enoyl-[acyl-carrier-protein] Reductase (NADH). Available online: https://enzyme.expasy.org/EC/1.3.1.9 (accessed on 25 December 2020).
- Stock, F.; Hoffmann, J.; Ranke, J.; Störmann, R.; Ondruschka, B.; Jastorff, B. Effects of ionic liquids on the acetylcholinesterase—A structure–activity relationship consideration. Green Chem. 2004, 6, 286–290. [Google Scholar] [CrossRef]
- Składanowski, A.C.; Stepnowski, P.; Kleszczyński, K.; Dmochowska, B. AMP deaminase in vitro inhibition by xenobiotics: A potential molecular method for risk assessment of synthetic nitro- and polycyclic musks, imidazolium ionic liquids and N-glucopyranosyl ammonium salts. Environ. Toxicol. Pharmacol. 2005, 19, 291–296. [Google Scholar] [CrossRef]
- Heitz, M.P.; Rupp, J.W. Determining mushroom tyrosinase inhibition by imidazolium ionic liquids: A spectroscopic and molecular docking study. Int. J. Biol. Macromol. 2018, 107, 1971–1981. [Google Scholar] [CrossRef]
- Dong, X.; Fan, Y.; Zhang, H.; Zhong, Y.; Yang, Y.; Miao, J.; Hua, S. Inhibitory effects of ionic liquids on the lactic dehydrogenase activity. Int. J. Biol. Macromol. 2016, 86, 155–161. [Google Scholar] [CrossRef]
- Heerding, D.A.; Chan, G.; DeWolf, W.E.; Fosberry, A.P.; Janson, C.A.; Jaworski, D.D.; McManus, E.; Miller, W.H.; Moore, T.D.; Payne, D.J.; et al. 1,4-Disubstituted imidazoles are potential antibacterial agents functioning as inhibitors of enoyl acyl carrier protein reductase (FabI). Bioorg. Med. Chem. Lett. 2001, 11, 2061–2065. [Google Scholar] [CrossRef]
- Desai, N.C.; Somani, H.; Trivedi, A.; Bhatt, K.; Nawale, L.; Khedkar, V.M.; Jha, P.C.; Sarkar, D. Synthesis, biological evaluation and molecular docking study of some novel indole and pyridine based 1,3,4-oxadiazole derivatives as potential antitubercular agents. Bioorg. Med. Chem. Lett. 2016, 26, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Karpov, P.; Godin, G.; Tetko, I.V. Transformer-CNN: Swiss knife for QSAR modeling and interpretation. J. Cheminform. 2020, 12, 17. [Google Scholar] [CrossRef] [Green Version]
- Tetko, I.V. Associative neural network. Methods Mol. Biol. 2008, 458, 185–202. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. arXiv 2016, arXiv:1603.02754. [Google Scholar] [CrossRef] [Green Version]
- Hall, L.H.; Kier, L.B. Electrotopological State Indexes for Atom Types—A Novel Combination of Electronic, Topological, and Valence State Information. J. Chem. Inf. Comput. Sci. 1995, 35, 1039–1045. [Google Scholar] [CrossRef]
- Tetko, I.V.; Tanchuk, V.Y. Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program. J. Chem. Inf. Comput. Sci. 2002, 42, 1136–1145. [Google Scholar] [CrossRef]
- Willighagen, E.L.; Mayfield, J.W.; Alvarsson, J.; Berg, A.; Carlsson, L.; Jeliazkova, N.; Kuhn, S.; Pluskal, T.; Rojas-Cherto, M.; Spjuth, O.; et al. The Chemistry Development Kit (CDK) v2.0: Atom typing, depiction, molecular formulas, and substructure searching. J. Cheminform. 2017, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; WILEY-VCH: Weinheim, Germany, 2000; p. 667. [Google Scholar]
- Tetko, I.V.; Sushko, I.; Pandey, A.K.; Zhu, H.; Tropsha, A.; Papa, E.; Oberg, T.; Todeschini, R.; Fourches, D.; Varnek, A. Critical assessment of QSAR models of environmental toxicity against Tetrahymena pyriformis: Focusing on applicability domain and overfitting by variable selection. J. Chem. Inf. Model. 2008, 48, 1733–1746. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Tetko, I.V.; Villa, A.E.; Livingstone, D.J. Neural network studies. 2. Variable selection. J. Chem. Inf. Comput. Sci. 1996, 36, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Kovalishyn, V.V.; Tetko, I.V.; Luik, A.I.; Kholodovych, V.V.; Villa, A.E.P.; Livingstone, D.J. Neural network studies. 3. Variable selection in the cascade-correlation learning architecture. J. Chem. Inf. Comput. Sci. 1998, 38, 651–659. [Google Scholar] [CrossRef]
- Sosnin, S.; Karlov, D.; Tetko, I.V.; Fedorov, M.V. Comparative Study of Multitask Toxicity Modeling on a Broad Chemical Space. J. Chem. Inf. Model. 2019, 59, 1062–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sushko, I.; Novotarskyi, S.; Körner, R.; Pandey, A.K.; Kovalishyn, V.V.; Prokopenko, V.V.; Tetko, I.V. Applicability domain for in silico models to achieve accuracy of experimental measurements. J. Chemom. 2010, 24, 202–208. [Google Scholar] [CrossRef]
- OECD. OECD Guidance Document on Acute Oral Toxicity Testing. In OECD Series on Testing and Assessment; Environment Directorate OECD: Paris, France, 2001. [Google Scholar] [CrossRef]
- OECD Guidelines for the Testing of Chemicals. Available online: https://www.oecd-ilibrary.org/environment/oecd-guidelines-for-the-testing-of-chemicals-section-4-health-effects_20745788 (accessed on 25 December 2020).
- Doria, O.F.; Castro, R.; Gutierrez, M.; Valenzuela, D.G.; Santos, L.; Ramirez, D.; Guzman, L. Novel alkylimidazolium ionic liquids as an antibacterial alternative to pathogens of the skin and soft tissue infections. Molecules 2018, 23, 2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Rock, C.O. Bacterial fatty acid metabolism in modern antibiotic discovery. Biochim. Biophys. Acta 2017, 1862, 1300–1309. [Google Scholar] [CrossRef]
- Massengo-Tiassé, R.P.; Cronan, J.E. Diversity in enoyl-acyl carrier protein reductases. Cell. Mol. Life Sci. 2009, 66, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Neves, Y.F.; Eloi, A.C.L.; de Freitas, H.M.M.; Soares, E.G.O.; Rivillo, D.; Demétrio da Silva, V.; Schrekker, H.S.; Badel, J.L. Imidazolium salts as alternative compounds to control diseases caused by plant pathogenic bacteria. J. Appl. Microbiol. 2020, 128, 1236–1247. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi, S.; Korner, R.; Pandey, A.K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V.V.; Tanchuk, V.Y.; et al. Online chemical modeling environment (OCHEM): Web platform for data storage, model development and publishing of chemical information. J. Comput. Aided. Mol. Des. 2011, 25, 533–554. [Google Scholar] [CrossRef] [Green Version]
- Villa, A.E.; Tetko, I.V.; Dutoit, P.; De Ribaupierre, Y.; De Ribaupierre, F. Corticofugal modulation of functional connectivity within the auditory thalamus of rat, guinea pig and cat revealed by cooling deactivation. J. Neurosci. Methods 1999, 86, 161–178. [Google Scholar] [CrossRef]
- Sadowski, J.; Gasteiger, J.; Klebe, G. Comparison of Automatic Three-Dimensional Model Builders Using 639 X-ray Structures. J. Chem. Inf. Comput. Sci. 1994, 34, 1000–1008. [Google Scholar] [CrossRef]
- Whitley, D.C.; Ford, M.G.; Livingstone, D.J. Unsupervised forward selection: A method for eliminating redundant variables. J. Chem. Inf. Comput. Sci. 2000, 40, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Sushko, I.; Novotarskyi, S.; Korner, R.; Pandey, A.K.; Cherkasov, A.; Li, J.; Gramatica, P.; Hansen, K.; Schroeter, T.; Muller, K.R.; et al. Applicability domains for classification problems: Benchmarking of distance to models for Ames mutagenicity set. J. Chem. Inf. Model. 2010, 50, 2094–2111. [Google Scholar] [CrossRef]
- Denk, M.K.; Ye, X. Alkylation of ethylenethiourea with alcohols: A convenient synthesis of S-alkyl-isothioureas without toxic alkylating agents. Tetrahedron Lett. 2005, 46, 7597–7599. [Google Scholar] [CrossRef]
- Bauer, A.W.; Kirby, W.M.; Sherris, J.C.; Turck, M. Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol. 1966, 45, 493–496. [Google Scholar] [CrossRef]
- Metelytsia, L.; Hodyna, D.; Dobrodub, I.; Semenyuta, I.; Zavhorodnii, M.; Blagodatny, V.; Kovalishyn, V.; Brazhko, O. Design of (quinolin-4-ylthio)carboxylic acids as new Escherichia coli DNA gyrase B inhibitors: Machine learning studies, molecular docking, synthesis and biological testing. Comput. Biol. Chem. 2020, 85, 107224. [Google Scholar] [CrossRef]
- Semenyuta, I.V.; Kobzar, O.L.; Hodyna, D.M.; Brovarets, V.S.; Metelytsia, L.O. In silico study of 4-phosphorylated derivatives of 1,3-oxazole as inhibitors of Candida albicans fructose-1,6-bisphosphate aldolase II. Heliyon 2019, 5, e01462. [Google Scholar] [CrossRef] [Green Version]
- Rose, P.W.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dimitropoulos, D.; Dutta, S.; Green, R.K.; Goodsell, D.S.; Prlić, A.; Quesada, M.; et al. The RCSB Protein Data Bank: New resources for research and education. Nucleic Acids Res. 2013, 41, D475–D482. [Google Scholar] [CrossRef]
- Discovery Studio Visualizer Software, Version 4.0. Available online: https://www.3ds.com/biovia/ (accessed on 25 December 2020).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comp. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- ChemAxon Marvin Sketch. Available online: http://www.chemaxon.com (accessed on 25 December 2020).
- Stewart, J.J. MOPAC2016; Stewart Computational Chemistry: Colorado Springs, CO, USA; Available online: http://openmopac.net (accessed on 25 December 2020).

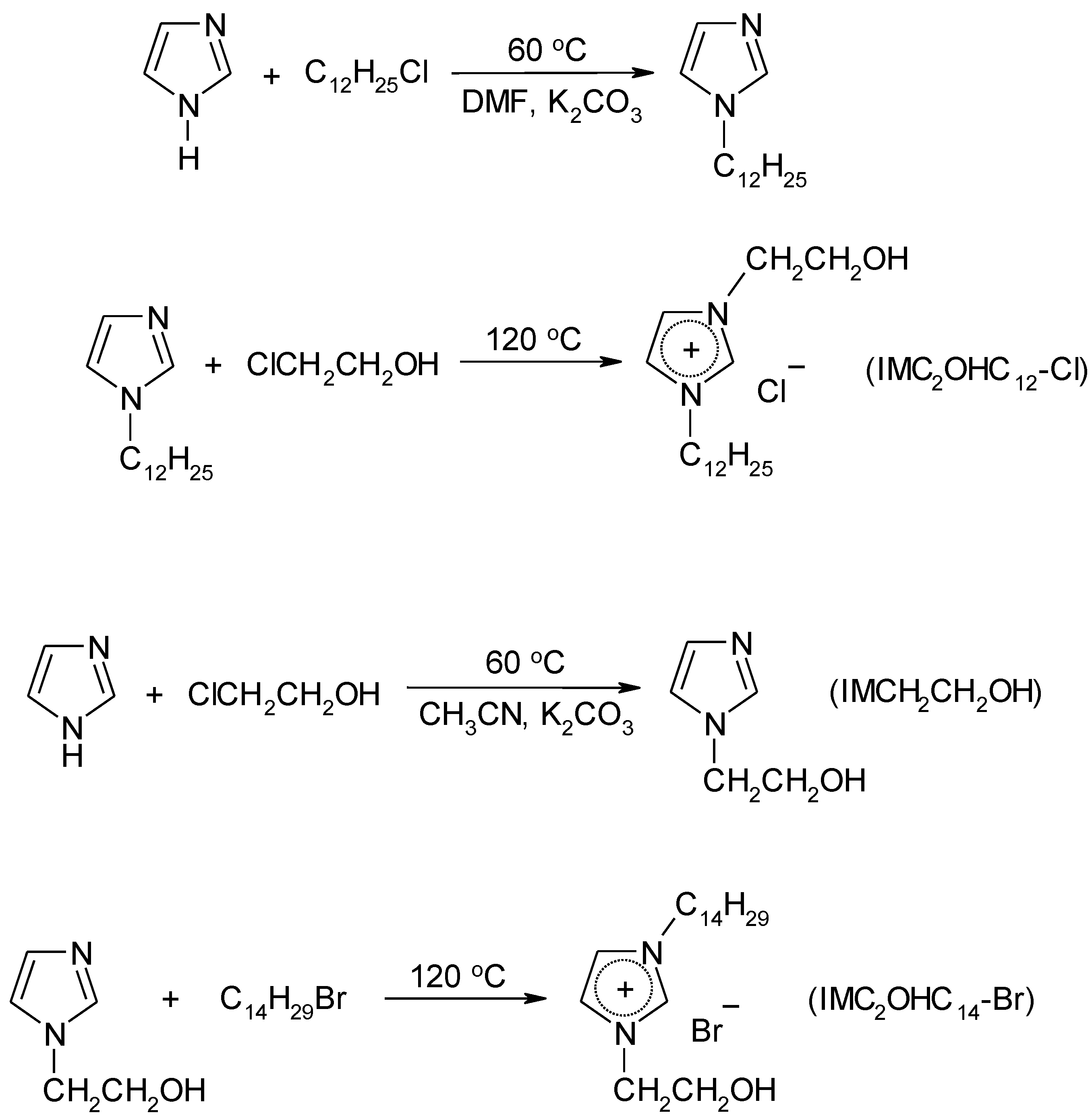

{kind=link}
{kind=link}
{kind=link}
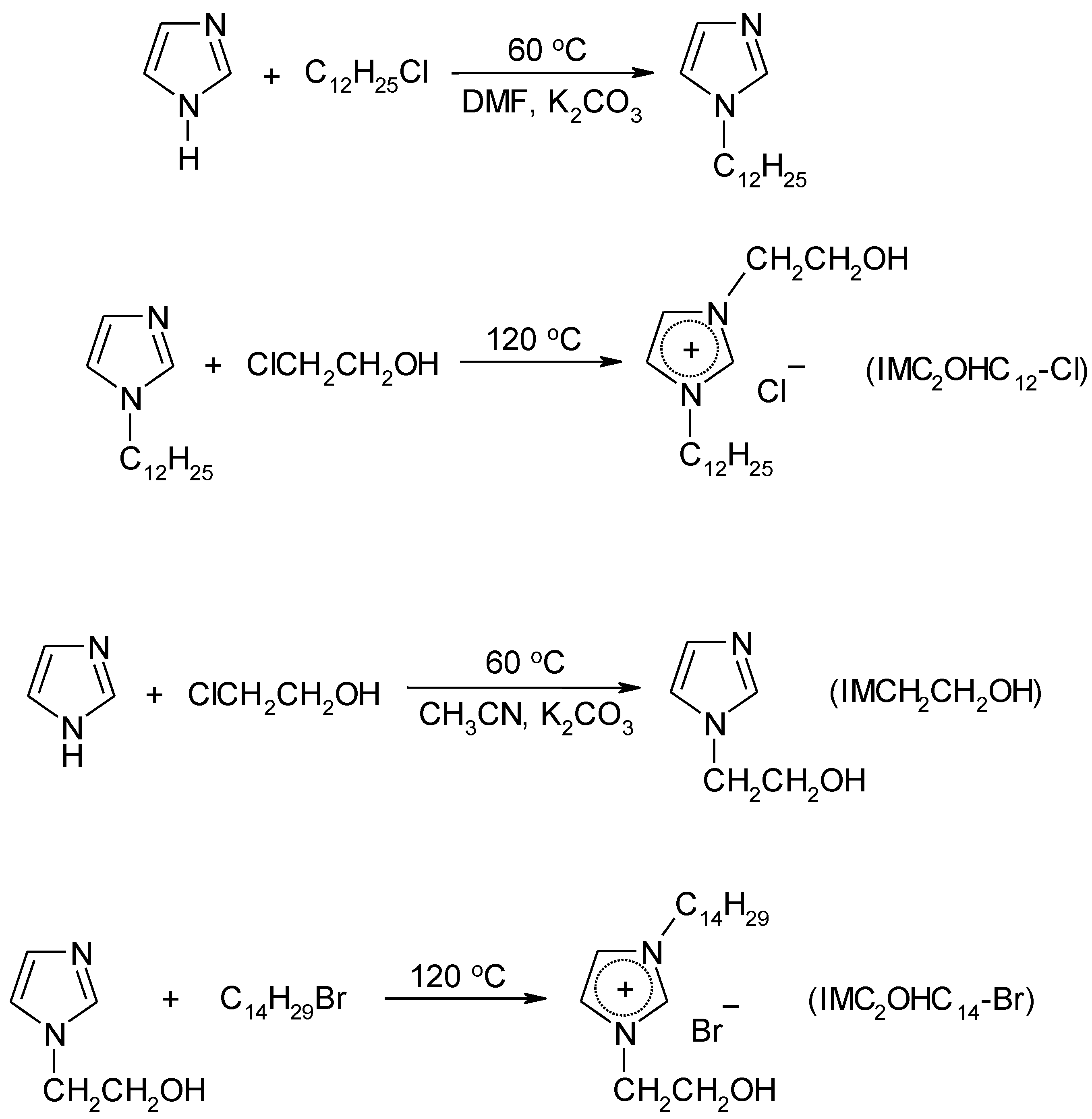{kind=link}
{kind=link}
| Method | Training Set a | Test Set a | ||||
|---|---|---|---|---|---|---|
| R2 | q2 | RMSE c | R2 | q2 | RMSE | |
| ASNN b | 0.74 ± 0.04 | 0.73 ± 0.05 | 0.55 ± 0.04 | 0.74 ± 0.06 | 0.70 ± 0.08 | 0.64 ± 0.09 |
| ASNN c | 0.70 ± 0.05 | 0.70 ± 0.05 | 0.59 ± 0.05 | 0.74 ± 0.06 | 0.69 ± 0.08 | 0.64 ± 0.09 |
| RFR b | 0.73 ± 0.04 | 0.73 ± 0.04 | 0.55 ± 0.04 | 0.73 ± 0.09 | 0.72 ± 0.09 | 0.6 ± 0.1 |
| Consensus d | 0.77 ± 0.04 | 0.77 ± 0.04 | 0.51 ± 0.04 | 0.77 ± 0.06 | 0.74 ± 0.07 | 0.6 ± 0.09 |
| Method | Training Set a | Test Set a | ||||
|---|---|---|---|---|---|---|
| R2 | q2 | RMSEc | R2 | q2 | RMSE | |
| ASNN b | 0.74 ± 0.04 | 0.73 ± 0.05 | 0.55 ± 0.04 | 0.74 ± 0.06 | 0.70± 0.08 | 0.64 ± 0.09 |
| ASNN c | 0.70 ± 0.05 | 0.70 ± 0.05 | 0.59 ± 0.05 | 0.74 ± 0.06 | 0.69± 0.08 | 0.64 ± 0.09 |
| RFR b | 0.73 ± 0.04 | 0.73 ± 0.04 | 0.55 ± 0.04 | 0.73 ± 0.09 | 0.72 ± 0.09 | 0.6 ± 0.1 |
| Consensus d | 0.77 ± 0.04 | 0.77 ± 0.04 | 0.51 ± 0.04 | 0.77 ± 0.06 | 0.74 ± 0.07 | 0.6 ± 0.09 |
| Compound | The Inhibition Zones Diameters (mm) | |
|---|---|---|
| A. baumannii | S. aureus | |
| 3 * | 20.3 ± 0.6 | 25.3 ± 0.6 |
| 4 * | 17.0 ± 0.9 | 19.7 ± 0.3 |
| 13 | 15.3 ± 0.6 | 14.0 ± 0.6 |
| 16 * | 16.3 ± 0.6 | 22.7 ± 0.9 |
| 17 | 17.0 ± 0.3 | 12.0 ± 0.3 |
| 20 | 15.7 ± 0.3 | 15.3 ± 0.3 |
| 22 | 11.0 ± 0.3 | 13.7 ± 0.6 |
| Ampicillin | n/a | n/a |
| Oxacillin | n/a | n/a |
| Ceftriaxone | 15.3 ± 0.3 | 15.7 ± 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semenyuta, I.V.; Trush, M.M.; Kovalishyn, V.V.; Rogalsky, S.P.; Hodyna, D.M.; Karpov, P.; Xia, Z.; Tetko, I.V.; Metelytsia, L.O. Structure-Activity Relationship Modeling and Experimental Validation of the Imidazolium and Pyridinium Based Ionic Liquids as Potential Antibacterials of MDR Acinetobacter baumannii and Staphylococcus aureus. Int. J. Mol. Sci. 2021, 22, 563. https://doi.org/10.3390/ijms22020563
Semenyuta IV, Trush MM, Kovalishyn VV, Rogalsky SP, Hodyna DM, Karpov P, Xia Z, Tetko IV, Metelytsia LO. Structure-Activity Relationship Modeling and Experimental Validation of the Imidazolium and Pyridinium Based Ionic Liquids as Potential Antibacterials of MDR Acinetobacter baumannii and Staphylococcus aureus. International Journal of Molecular Sciences. 2021; 22(2):563. https://doi.org/10.3390/ijms22020563
Chicago/Turabian StyleSemenyuta, Ivan V., Maria M. Trush, Vasyl V. Kovalishyn, Sergiy P. Rogalsky, Diana M. Hodyna, Pavel Karpov, Zhonghua Xia, Igor V. Tetko, and Larisa O. Metelytsia. 2021. "Structure-Activity Relationship Modeling and Experimental Validation of the Imidazolium and Pyridinium Based Ionic Liquids as Potential Antibacterials of MDR Acinetobacter baumannii and Staphylococcus aureus" International Journal of Molecular Sciences 22, no. 2: 563. https://doi.org/10.3390/ijms22020563
APA StyleSemenyuta, I. V., Trush, M. M., Kovalishyn, V. V., Rogalsky, S. P., Hodyna, D. M., Karpov, P., Xia, Z., Tetko, I. V., & Metelytsia, L. O. (2021). Structure-Activity Relationship Modeling and Experimental Validation of the Imidazolium and Pyridinium Based Ionic Liquids as Potential Antibacterials of MDR Acinetobacter baumannii and Staphylococcus aureus. International Journal of Molecular Sciences, 22(2), 563. https://doi.org/10.3390/ijms22020563