Structure–Activity Relationship (SAR) Study of Spautin-1 to Entail the Discovery of Novel NEK4 Inhibitors

 , ,
, , 
Abstract
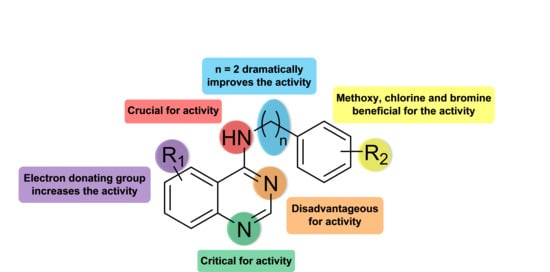
1. Introduction
2. Results
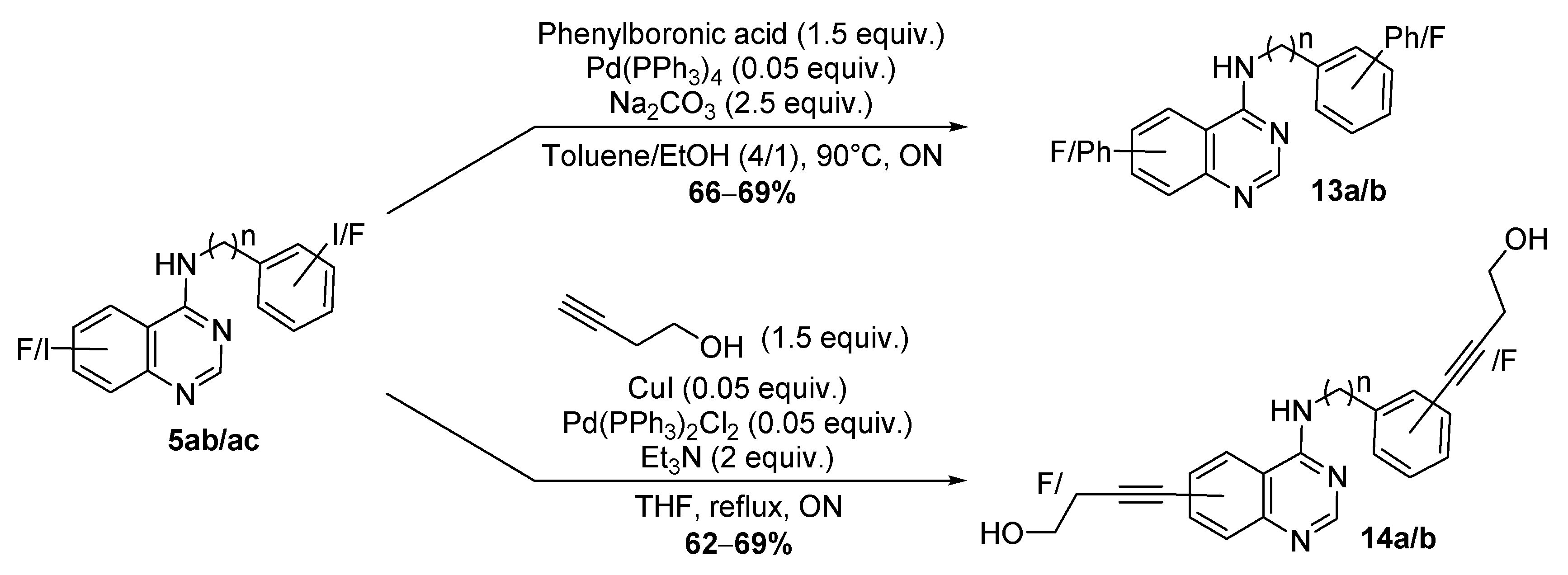
2.1. Chemistry
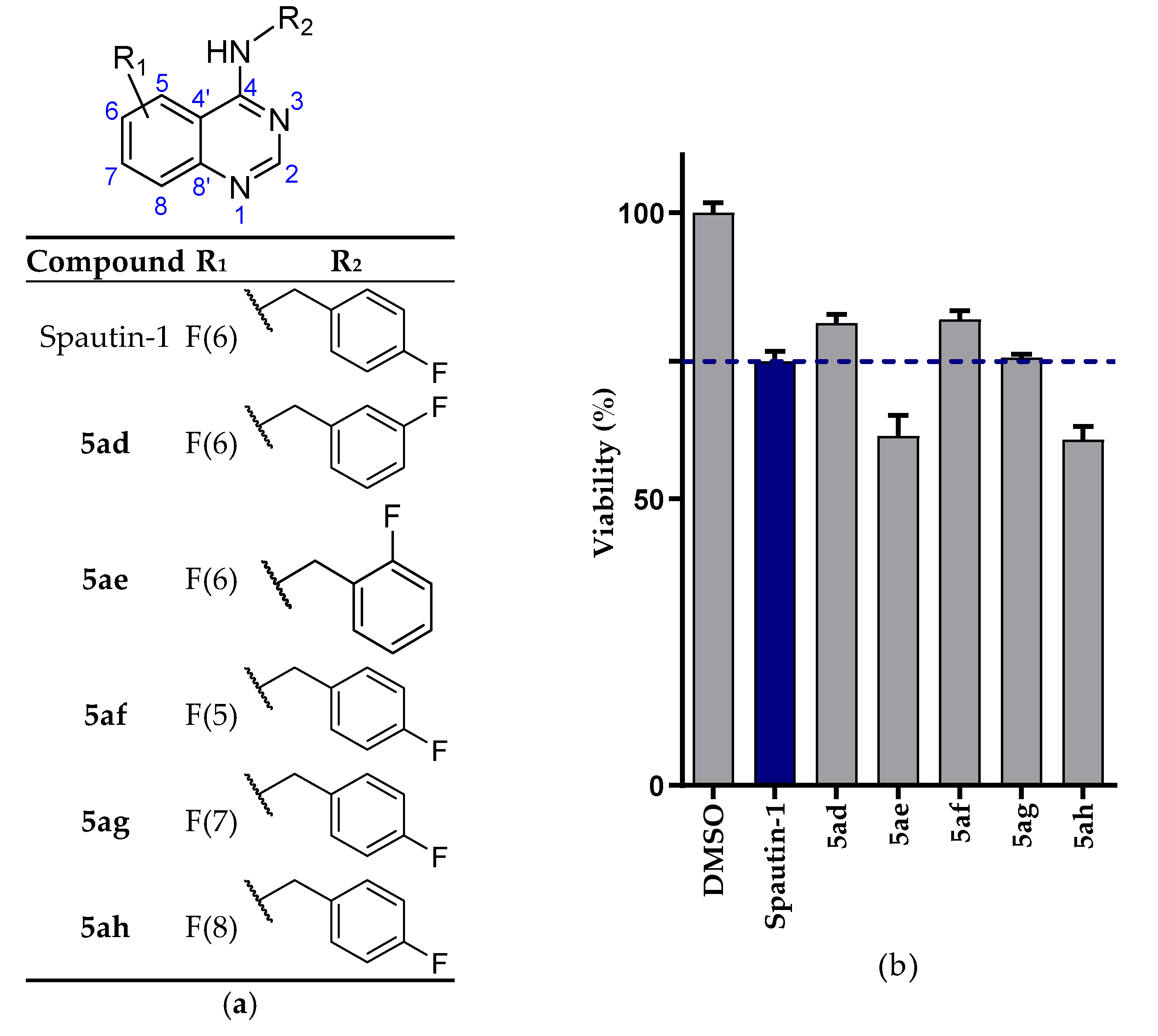
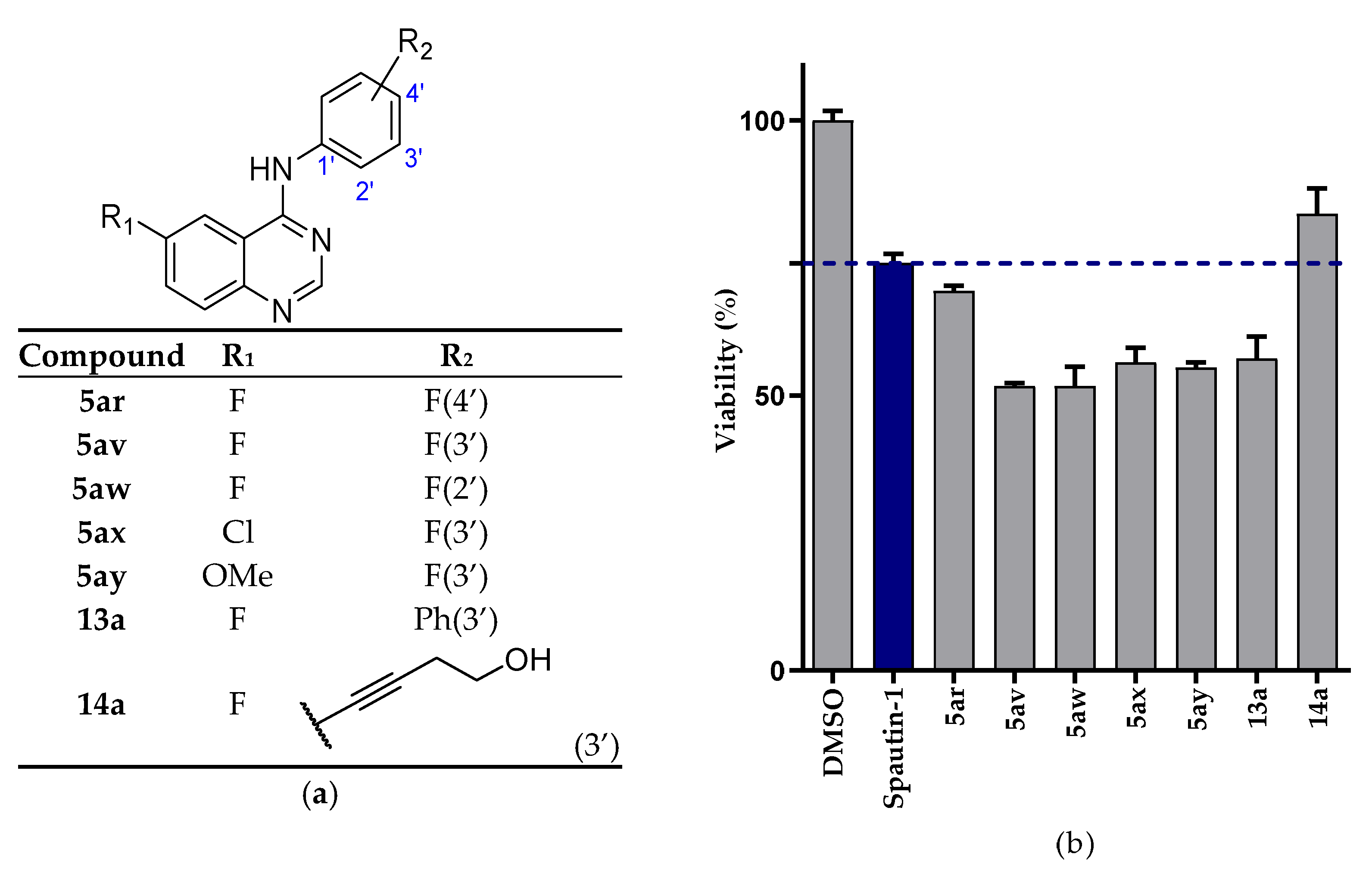
2.2. Biological Evaluation
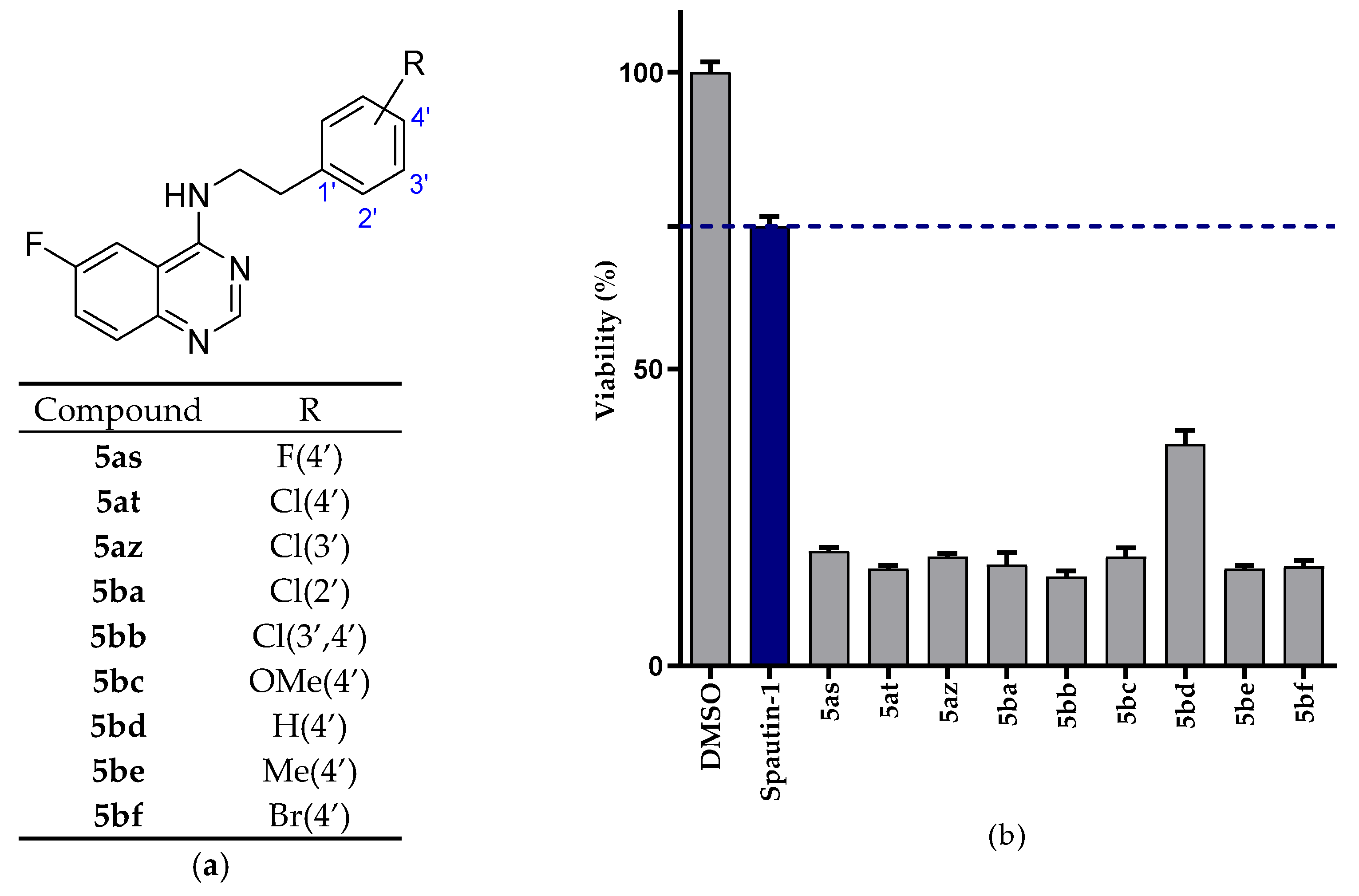
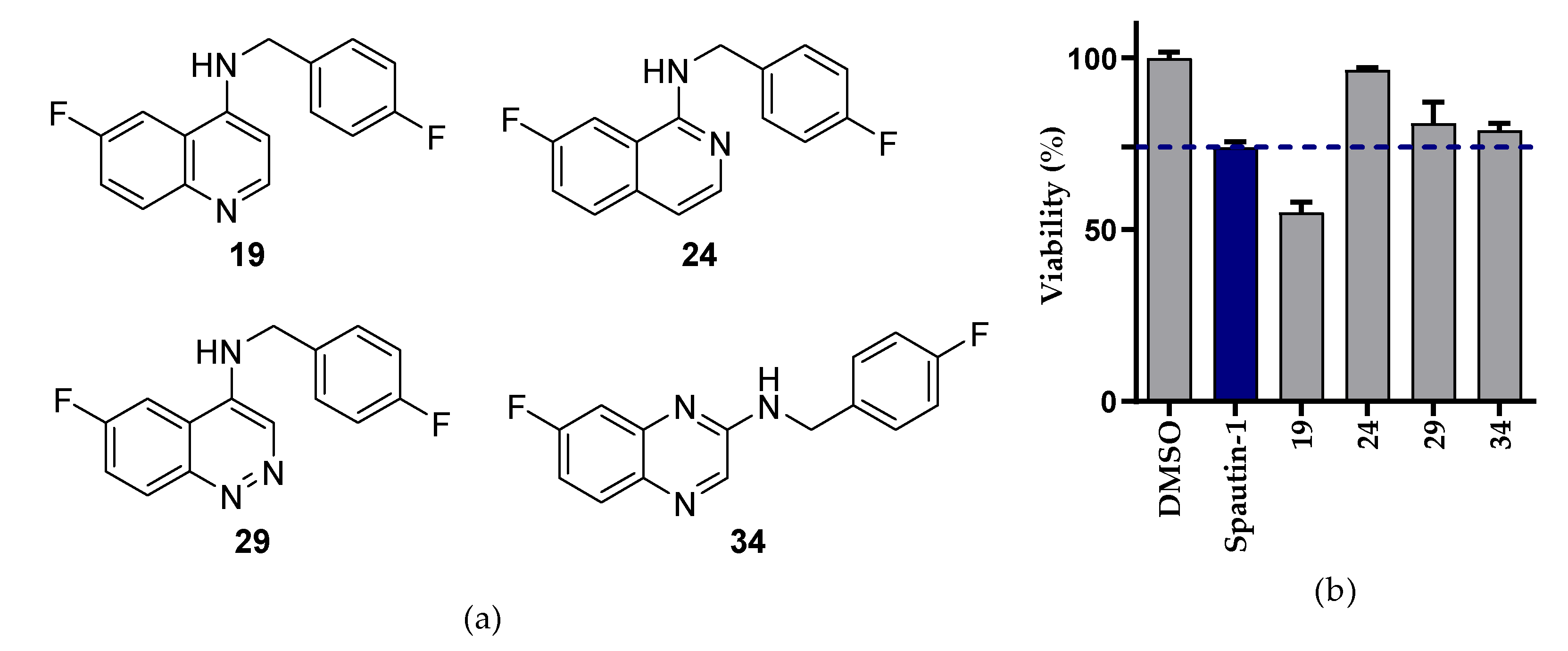
2.2.1. NSCLC Cell Viability Screening
2.2.2. ITC/TSA
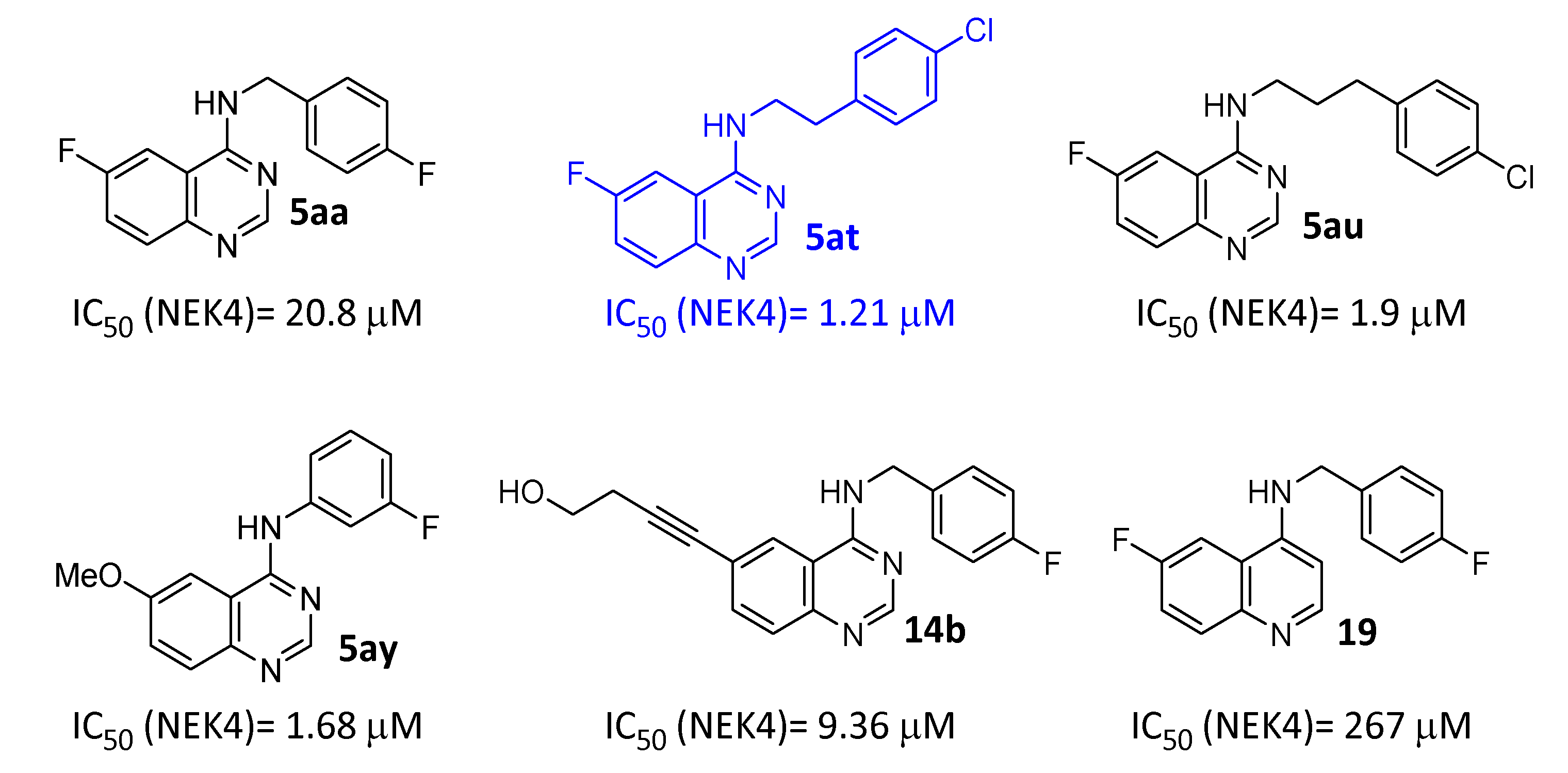
2.2.3. Kinase Screening
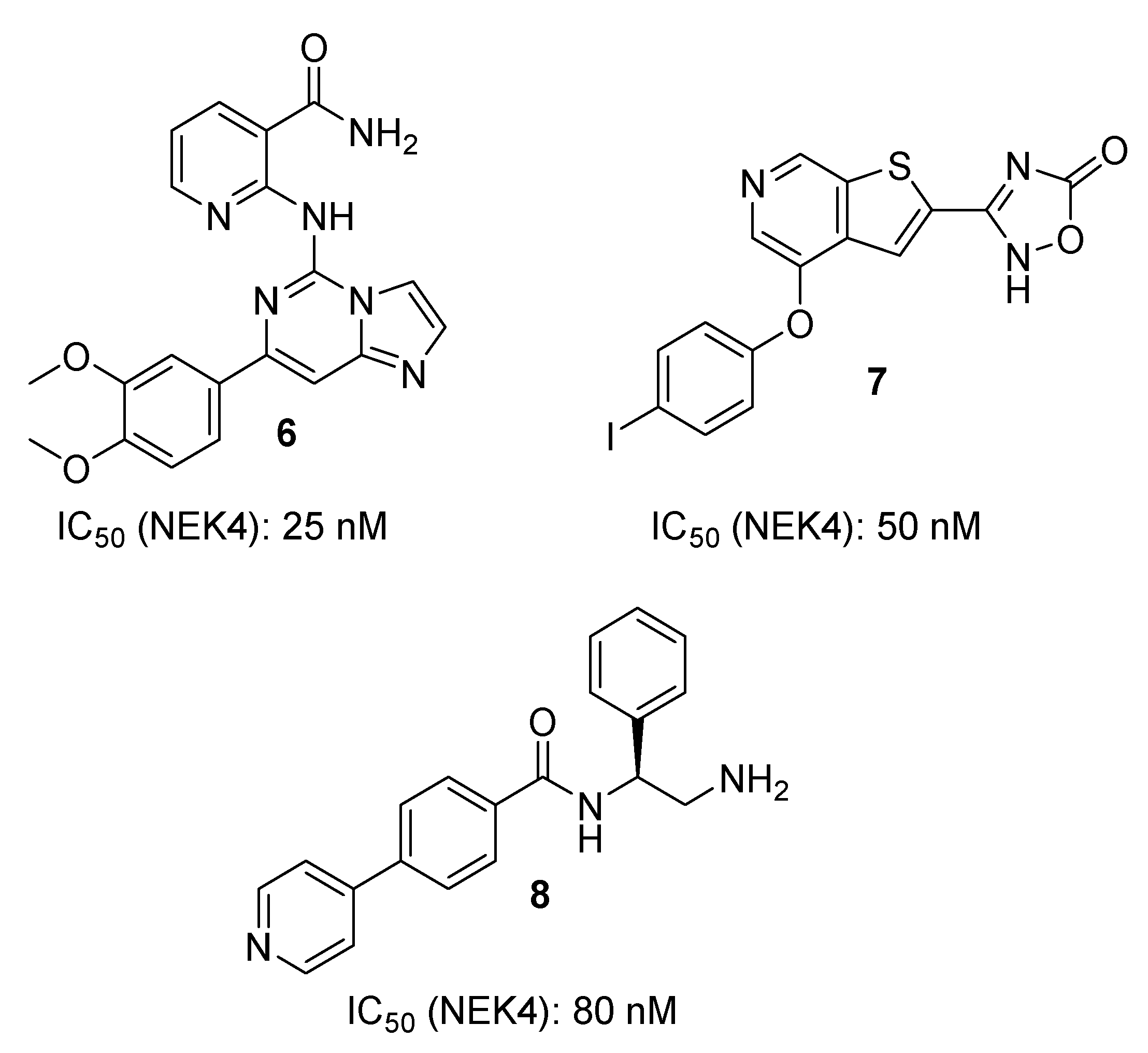
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Procedure for the Synthesis of Substituted-Quinazolin-4-One 11
Synthesis of 6-Fluoro-Quinazolin-4-One 11a
Synthesis of 5-Fluoro-Quinazolin-4-One 11b
Synthesis of 7-Fluoro-Quinazolin-4-One 11c
Synthesis of 8-Fluoro-Quinazolin-4-One 11d
Synthesis of 6-Chloro-Quinazolin-4-One 11e
Synthesis of 6-Methoxy-Quinazolin-4-One 11f
Synthesis of 6-Methyl-Quinazolin-4-One 11g
Synthesis of 6-Trifluoromethyl-Quinazolin-4-One 11h
Synthesis of 6-Nitro-Quinazolin-4-One 11i
Synthesis of 6-Iodo-Quinazolin-4-One 11j
4.1.2. General Procedure for the Synthesis of Substituted-4-Chloro-Quinazoline 12
Synthesis of 4-Chloro-6-Fluoroquinazoline 12a
Synthesis of 4-Chloro-5-Fluoroquinazoline 12b
Synthesis of 4-Chloro-7-Fluoroquinazoline 12c
Synthesis of 4-Chloro-8-Fluoroquinazoline 12d
Synthesis of 4,6-Dichloroquinazoline 12e
Synthesis of 4-Chloro-6-Methoxyquinazoline 12f
Synthesis of 4-Chloro-6-Methylquinazoline 12g
Synthesis of 4-Chloro-6-Trifluoromethyl-Quinazoline 12h
Synthesis of 4-Chloro-6-Nitroquinazoline 12i
Synthesis of 4-Chloro-6-Iodoquinazoline 12j
4.1.3. General Procedure for the Synthesis of Spautin-1 and Analogues 5
Synthesis of 6-Fluoro-N-(4-fluorobenzyl)-4-Quinazolinamine.TFA 5aa
Synthesis of 6-fluoro-N-(3-iodophenyl)-4-quinazolinamine.TFA 5ab
Synthesis of N-(4-Fluorobenzyl)-6-iodo-4-Quinazolinamine.TFA 5ac
Synthesis of 6-Fluoro-N-(3-Fluorobenzyl)-4-Quinazolinamine.TFA 5ad
Synthesis of 6-Fluoro-N-(2-Fluorobenzyl)-4-Quinazolinamine.TFA 5ae
Synthesis of 5-Fluoro-N-(4-Fluorobenzyl)-4-Quinazolinamine.TFA 5af
Synthesis of 7-Fluoro-N-(4-Fluorobenzyl)-4-Quinazolinamine.TFA 5ag
Synthesis of 8-Fluoro-N-(4-Fluorobenzyl)-4-Quinazolinamine.TFA 5ah
Synthesis of 6-Chloro-N-(4-Fluorobenzyl)-4-Quinazolinamine.TFA 5ai
Synthesis of N-(4-Fluorobenzyl)-6-Methoxy-4-Quinazolinamine.TFA 5aj
Synthesis of N-(4-Fluorobenzyl)-6-Methyl-4-Quinazolinamine.TFA 5ak
Synthesis of N-(4-Fluorobenzyl)-6-Trifluoromethyl-4-Quinazolinamine.TFA 5al
Synthesis of N-(4-Fluorobenzyl)-6-Nitro-4-Quinazolinamine.TFA 5am
Synthesis of N-(4-Chlorobenzyl)-6-Fluoro-4-Quinazolinamine.TFA 5an
Synthesis of 6-Fluoro-N-(4-Methoxybenzyl)-4-Quinazolinamine.TFA 5ao
Synthesis of 6-Fluoro-N-(4-(Trifluoromethyl)Benzyl)-4-Quinazolinamine.TFA 5ap
Synthesis of 6-Fluoro-N-(Phenylmethyl)-4-Quinazolinamine.TFA 5aq
Synthesis of 6-Fluoro-N-(4-Fluorophenyl)-4-Quinazolinamine.TFA 5ar
Synthesis of 6-Fluoro-N-[2-(4-Fluorophenyl)Ethyl]-4-Quinazolinamine.TFA 5as
Synthesis of N-[2-(4-Chlorophenyl)Ethyl]-6-Fluoro-4-Quinazolinamine.TFA 5at
Synthesis of 6-Fluoro-N-[3-(4-Chlorophenyl)Propyl]-4-Quinazolinamine.TFA 5au
Synthesis of 6-Fluoro-N-(3-Fluorophenyl)-4-Quinazolinamine.TFA 5av
Synthesis of 6-Fluoro-N-(2-Fluorophenyl)-4-Quinazolinamine.TFA 5aw
Synthesis of 6-Chloro-N-(3-Fluorophenyl)-4-Quinazolinamine.TFA 5ax
Synthesis of N-(3-Fluorophenyl)-6-Methoxy-4-Quinazolinamine.TFA 5ay
Synthesis of 6-Fluoro-N-(3-Chlorophenethyl)-4-Quinazolinamine.TFA 5az
Synthesis of 6-Fluoro-N-(2-Chlorophenethyl)-4-Quinazolinamine.TFA 5ba
Synthesis of 6-Fluoro-N-[2-(3,4-Dichlorophenyl)Ethyl]-4-Quinazolinamine.TFA 5bb
Synthesis of 6-Fluoro-N-[2-(4-Methoxyphenyl)Ethyl]-4-Quinazolinamine.TFA 5bc
Synthesis of 6-Fluoro-N-(2-Phenylethyl)-4-Quinazolinamine.TFA 5bd
Synthesis of 6-Fluoro-N-[2-(4-Methylphenyl)ethyl]-4-Quinazolinamine.TFA 5be
Synthesis of 6-Fluoro-N-[2-(4-Bromophenyl)ethyl]-4-Quinazolinamine.TFA 5bf
Synthesis of 6-Fluoro-N-(4-Fluorobenzyl)-N-Methyl-4-Quinazolinamine.TFA 5bg
Synthesis of 6-Fluoro-N-(4-Chlorophenethyl)-N-Methyl-4-Quinazolinamine.TFA 5bh
4.1.4. General Procedure for the Suzuki Reaction
Synthesis of N-(1, 1’-Biphenyl)-3-yl-6-Fluoro-4-Quinazolinamine.TFA 13a
Synthesis of N-(4-Fluorobenzyl)-6-Phenyl-4-Quinazolinamine.TFA 13b
4.1.5. General Procedure Sonogashira Reaction
Synthesis of 4-[3-[(6-Fluoro-4-Quinazolinyl)Amino]Phenyl]-3-Butyn-1-ol.TFA 14a
Synthesis of, 4-[4-[(4-Fluorobenzyl)Amino]-6-Quinazolinyl]-3-Butyn-1-ol.TFA 14b
4.1.6. Three-step Synthesis of 6-Fluoro-1-[(4-Fluorophenyl)Methyl]-1H-Benzimidazole 17
Synthesis of 4-Fluoro-N-(5-Fluoro-2-Nitrophenyl)-Benzenemethanamine 16
Synthesis of 4-Fluoro-N2-[(4-Fluorophenyl)Methyl]-1,2-Benzenediamine
Synthesis of 6-Fluoro-1-[(4-Fluorophenyl)Methyl]-1H-Benzimidazole 17
4.1.7. One-step Synthesis of 6-Fluoro-N-(4-Fluorobenzyl)-4-Quinolinamine 19
Synthesis of 6-Fluoro-N-(4-Fluorobenzyl)-4-Quinolinamine.TFA 19
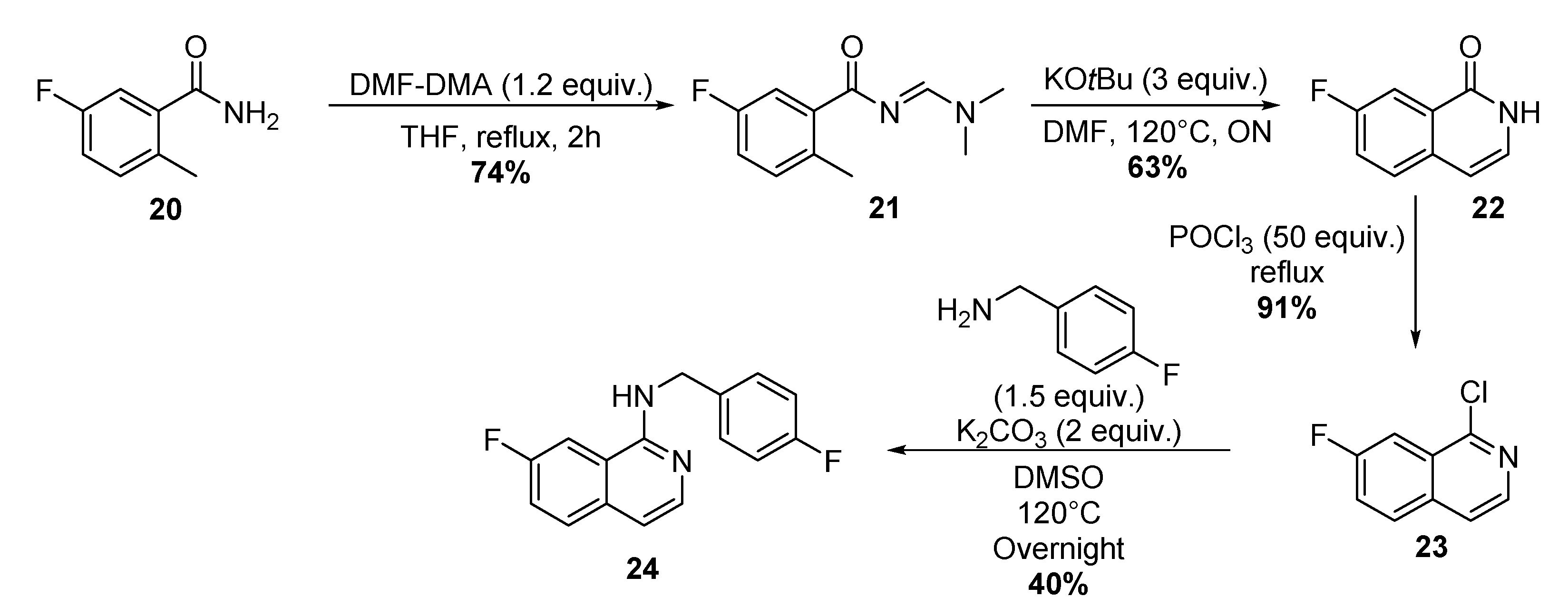
4.1.8. Four-step Synthesis of 7-Fluoro-N-(4-Fluorobenzyl)-1-Isoquinoline 24
Synthesis of (E)-N-((Dimethylamino)Methyl)-5-Fluoro-2-Methylbenzamide 21
Synthesis of 7-Fluoro-1-Isoquinolonone 22
Synthesis of 1-Chloro-7-Fluoroisoquinoline 23
Synthesis of 7-Fluoro-N-(4-Fluorobenzyl)-1-Isoquinoline.TFA 24
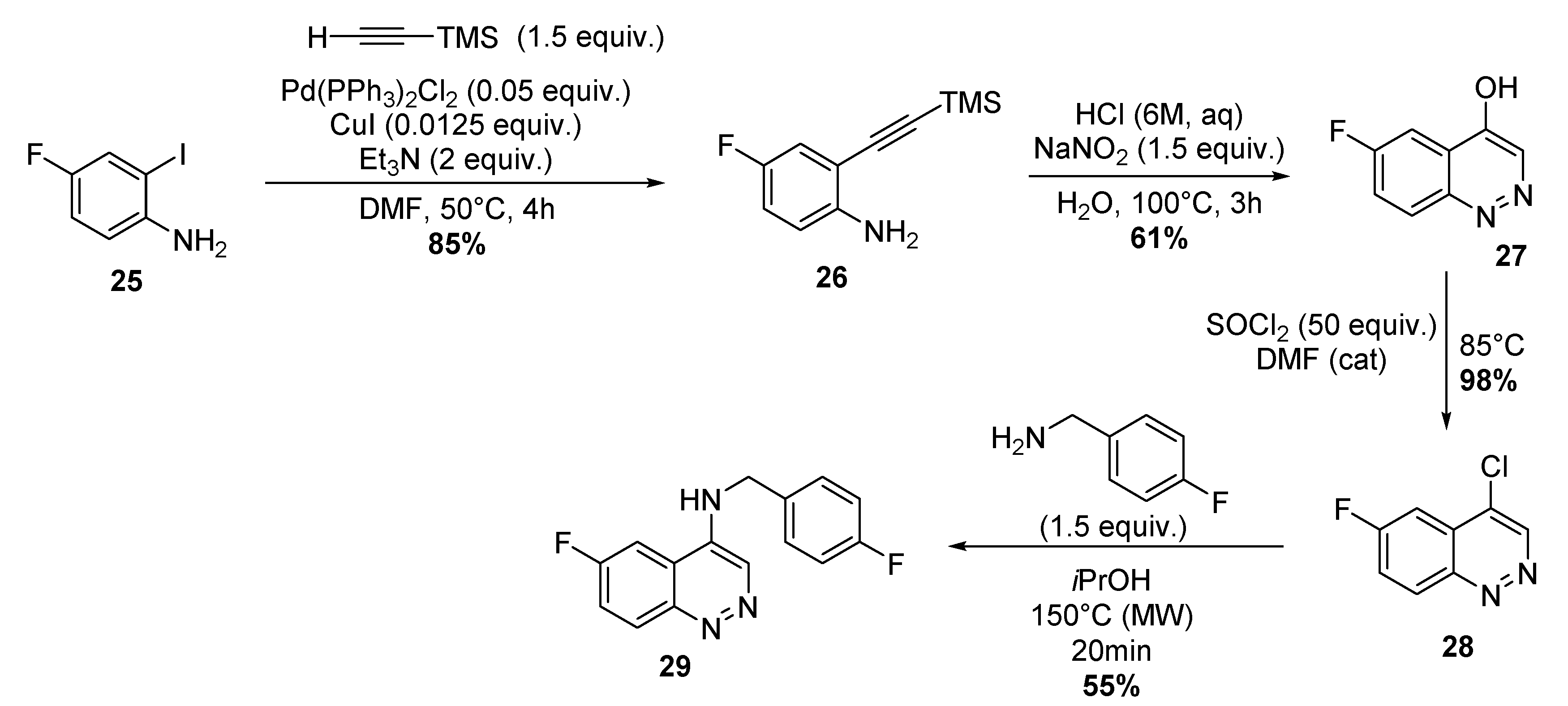
4.1.9. Four-step Synthesis of 6-Fluoro-N-(4-Fluorobenzyl)-4-Cinnolinamine 29
Synthesis of 4-Fluoro-2-(2-(Trimethylsilyl)Ethynyl)-Aniline 26
Synthesis of 6-FluoroCinnolin-4-ol 27
Synthesis of 4-Chloro-6-Fluoro-Cinnoline 28
Synthesis of 6-Fluoro-N-(4-Fluorobenzyl)-4-Cinnolinamine.TFA 29
4.1.10. Four-Step Synthesis of 7-Fluoro-N-(4-Fluorobenzyl)-2-Quinoxalinamine 34
Synthesis of Ethyl 2-(4-Fluoro-2-Nitrophenylamino)Acetate 31
Synthesis of 7-Fluoro-1H-Quinoxalin-2-One 32
Synthesis of 2-Chloro-7-Fluoro-Quinoxaline 33
Synthesis of 7-Fluoro-N-(4-Fluorobenzyl)-2-Quinoxalinamine.TFA 34
4.2. NSCLC Cells Viability Assay
4.3. Kinase Screening
4.4. ITC
4.5. TSA
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SAR | Structure–activity relationship |
| PACS | Pathophysiological cell signaling |
| USP13 | Ubiquitin Specific Protease 13 |
| EGFR | Epidermal growth factor receptor |
| NSCLC | Non-small cell lung cancer |
| ITC | Isothermal titration calorimetry |
| TSA | Thermal shift assay |
| DUBs | Deubiquitinating enzymes |
| NEK4 | Never in mitosis A related kinase 4 |
| TRAIL | Tumor necrosis factor-Related Apoptosis Inducing Ligand |
| EMT | Epithelial to mesenchymal transition |
| ON | Overnight |
| DMF | N,N-dimethylformamide |
| Tm | Melting temperature |
| ΔTm | Melting temperature difference |
| h | Human |
| TB | Terrific Broth |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| EDTA | Ethylenediaminetetraacetic acid |
| DTT | 1,4-dithiothreitol |
| IRP | Interdisciplinary Research Programmes |
| WFWG | Wetenschappelijk Fonds Willy Gepts |
| VUB | Vrije Universiteit Brussel |
References
- World Health Organization, International Agency for Research on Cancer. Globocan 2018: Lung Cancer. International Agency for Research on Cancer. Available online: http://gco.iarc.fr/today/data/factsheets/cancers/15-Lung-fact-sheet.pdf (accessed on 25 March 2020).
- D’Addario, G.; Fruh, M.; Reck, M.; Baumann, P.; Klepetko, W.; Felip, E.; On behalf of the ESMO Guidelines Working Group Metastatic Non-Small-Cell Lung Cancer. ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2010, 21, v116–v119. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.-J.; Sun, J.-M.; Lee, S.-H.; Ahn, J.S.; Park, K. EGFR TKI Combination with Immunotherapy in Non-Small Cell Lung Cancer. Expert Opin. Drug Saf. 2017, 16, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance Mechanisms to Osimertinib in EGFR-Mutated Non-Small Cell Lung Cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef]
- Martín-Bernabé, A.; Balcells, C.; Tarragó-Celada, J.; Foguet, C.; Bourgoin-Voillard, S.; Seve, M.; Cascante, M. The Importance of Post-Translational Modifications in Systems Biology Approaches to Identify Therapeutic Targets in Cancer Metabolism. Curr. Opin. Syst. Biol. 2017, 3, 161–169. [Google Scholar] [CrossRef]
- Hsu, J.-M.; Li, C.-W.; Lai, Y.-J.; Hung, M.-C. Posttranslational Modifications of PD-L1 and Their Applications in Cancer Therapy. Cancer Res. 2018, 78, 6349–6353. [Google Scholar] [CrossRef]
- Sharma, B.S.; Prabhakaran, V.; Desai, A.P.; Bajpai, J.; Verma, R.J.; Swain, P.K. Post-Translational Modifications (PTMs), from a Cancer Perspective: An Overview. Oncogen 2019, 2, 12. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, Functions, Mechanisms. Biochim. Biophys. Acta BBA Mol. Cell Res. 2004, 1695, 55–72. [Google Scholar] [CrossRef]
- Clague, M.J.; Heride, C.; Urbé, S. The Demographics of the Ubiquitin System. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef]
- Castagnoli, L.; Mandaliti, W.; Nepravishta, R.; Valentini, E.; Mattioni, A.; Procopio, R.; Iannuccelli, M.; Polo, S.; Paci, M.; Cesareni, G.; et al. Selectivity of the CUBAN Domain in the Recognition of Ubiquitin and NEDD8. FEBS J. 2019, 286, 653–677. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, P.; Wei, Y.; Piao, H.; Wang, W.; Maddika, S.; Wang, M.; Chen, D.; Sun, Y.; Hung, M.-C.; et al. Deubiquitylation and Stabilization of PTEN by USP13. Nat. Cell Biol. 2013, 15, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Man, X.; Piao, C.; Lin, X.; Kong, C.; Cui, X.; Jiang, Y. USP13 Functions as a Tumor Suppressor by Blocking the NF-KB-Mediated PTEN Downregulation in Human Bladder Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 259. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Zhang, R.; Su, M.; Liu, W. USP13 Serves as a Tumor Suppressor via the PTEN/AKT Pathway in Oral Squamous Cell Carcinoma. Cancer Manag. Res. 2019, 11, 9175–9183. [Google Scholar] [CrossRef]
- Fang, X.; Zhou, W.; Wu, Q.; Huang, Z.; Shi, Y.; Yang, K.; Chen, C.; Xie, Q.; Mack, S.C.; Wang, X.; et al. Deubiquitinase USP13 Maintains Glioblastoma Stem Cells by Antagonizing FBXL14-Mediated Myc Ubiquitination. J. Exp. Med. 2017, 214, 245–267. [Google Scholar] [CrossRef]
- Han, C.; Yang, L.; Choi, H.H.; Baddour, J.; Achreja, A.; Liu, Y.; Li, Y.; Li, J.; Wan, G.; Huang, C.; et al. Amplification of USP13 Drives Ovarian Cancer Metabolism. Nat. Commun. 2016, 7, 13525. [Google Scholar] [CrossRef]
- Han, C.; Lu, X.; Nagrath, D. Regulation of Protein Metabolism in Cancer. Mol. Cell. Oncol. 2018, 5, e1285384. [Google Scholar] [CrossRef]
- Li, Y.; Luo, K.; Yin, Y.; Wu, C.; Deng, M.; Li, L.; Chen, Y.; Nowsheen, S.; Lou, Z.; Yuan, J. USP13 Regulates the RAP80-BRCA1 Complex Dependent DNA Damage Response. Nat. Commun. 2017, 8, 15752. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, M.; Jing, Y.; Yin, X.; Ma, P.; Zhang, Z.; Wang, X.; Di, W.; Zhuang, G. Deubiquitinase USP13 Dictates MCL1 Stability and Sensitivity to BH3 Mimetic Inhibitors. Nat. Commun. 2018, 9, 215. [Google Scholar] [CrossRef]
- Zhao, X.; Fiske, B.; Kawakami, A.; Li, J.; Fisher, D.E. Regulation of MITF Stability by the USP13 Deubiquitinase. Nat. Commun. 2011, 2, 414. [Google Scholar] [CrossRef]
- Wang, Y.; Ou, Z.; Sun, Y.; Yeh, S.; Wang, X.; Long, J.; Chang, C. Androgen Receptor Promotes Melanoma Metastasis via Altering the MiRNA-539-3p/USP13/MITF/AXL Signals. Oncogene 2017, 36, 1644–1654. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Guo, Z.; Xia, X.; Liu, Y.; Huang, C.; Jiang, L.; Wang, X.; Liu, J.; Huang, H. Inhibition of EGFR Signaling with Spautin-1 Represents a Novel Therapeutics for Prostate Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 157. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, Y.; Liu, C.; Zhang, Y.; Wang, D.; Wang, S.; Wu, Y.; Liu, F.; Li, Q.; Liu, X.; et al. Amplification of USP13 Drives Non-Small Cell Lung Cancer Progression Mediated by AKT/MAPK Signaling. Biomed. Pharmacother. 2019, 114, 108831. [Google Scholar] [CrossRef] [PubMed]
- Giron, P.; Eggermont, C.; Noeparast, A.; Vandenplas, H.; Teugels, E.; Forsyth, R.; De Wever, O.; Aza-Blanc, P.; Gutierrez, G.J.; De Grève, J. Targeting USP13 Mediated Drug Tolerance Increases the Efficacy of EGFR Inhibition of Mutant EGFR in Non-Small Cell Lung Cancer. Int. J. Cancer 2020. [Google Scholar] [CrossRef]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 Controls the Levels of P53 by Regulating the Deubiquitination Activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef]
- Aly, R.M.; Serya, R.A.; Amira, M.; Al-Ansary, G.H.; Abou El Ella, D.A. Quinoline-Based Small Molecules as Effective Protein Kinases Inhibitors. J. Am. Sci. 2016, 12, 10–32. [Google Scholar]
- Cortes, J.E.; Kantarjian, H.M.; Brümmendorf, T.H.; Kim, D.-W.; Turkina, A.G.; Shen, Z.-X.; Pasquini, R.; Khoury, H.J.; Arkin, S.; Volkert, A.; et al. Safety and Efficacy of Bosutinib (SKI-606) in Chronic Phase Philadelphia Chromosome–Positive Chronic Myeloid Leukemia Patients with Resistance or Intolerance to Imatinib. Blood 2011, 118, 4567–4576. [Google Scholar] [CrossRef]
- Elisei, R.; Schlumberger, M.J.; Müller, S.P.; Schöffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in Progressive Medullary Thyroid Cancer. J. Clin. Oncol. 2013, 31, 3639–3646. [Google Scholar] [CrossRef]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor Activity of HKI-272, an Orally Active, Irreversible Inhibitor of the HER-2 Tyrosine Kinase. Cancer Res. 2004, 64, 3958–3965. [Google Scholar] [CrossRef]
- Collins, I.; Caldwell, J.; Fonseca, T.; Donald, A.; Bavetsias, V.; Hunter, L.-J.K.; Garrett, M.D.; Rowlands, M.G.; Aherne, G.W.; Davies, T.G.; et al. Structure-Based Design of Isoquinoline-5-Sulfonamide Inhibitors of Protein Kinase B. Bioorg. Med. Chem. 2006, 14, 1255–1273. [Google Scholar] [CrossRef]
- Larocque, E.; Naganna, N.; Ma, X.; Opoku-Temeng, C.; Carter-Cooper, B.; Chopra, G.; Lapidus, R.G.; Sintim, H.O. Aminoisoquinoline Benzamides, FLT3 and Src-Family Kinase Inhibitors, Potently Inhibit Proliferation of Acute Myeloid Leukemia Cell Lines. Future Med. Chem. 2017, 9, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, A.W.; Adler, M.; Aubele, D.L.; Bowers, S.; Franzini, M.; Goldbach, E.; Lorentzen, C.; Neitz, R.J.; Probst, G.D.; Quinn, K.P.; et al. Novel Cinnoline-Based Inhibitors of LRRK2 Kinase Activity. Bioorg. Med. Chem. Lett. 2013, 23, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Barlaam, B.; Cadogan, E.; Campbell, A.; Colclough, N.; Dishington, A.; Durant, S.; Goldberg, K.; Hassall, L.A.; Hughes, G.D.; MacFaul, P.A.; et al. Discovery of a Series of 3-Cinnoline Carboxamides as Orally Bioavailable, Highly Potent, and Selective ATM Inhibitors. ACS Med. Chem. Lett. 2018, 9, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Szumilak, M.; Stanczak, A. Cinnoline Scaffold—A Molecular Heart of Medicinal Chemistry? Molecules 2019, 24, 2271. [Google Scholar] [CrossRef]
- El Newahie, A.M.S.; Ismail, N.S.M.; Abou El Ella, D.A.; Abouzid, K.A.M. Quinoxaline-Based Scaffolds Targeting Tyrosine Kinases and Their Potential Anticancer Activity: Quinoxaline-Based Scaffolds Targeting Tyrosine Kinases. Arch. Pharm. (Weinheim) 2016, 349, 309–326. [Google Scholar] [CrossRef]
- Park, S.J.; Jo, D.S.; Jo, S.-Y.; Shin, D.W.; Shim, S.; Jo, Y.K.; Shin, J.H.; Ha, Y.J.; Jeong, S.-Y.; Hwang, J.J.; et al. Inhibition of Never in Mitosis A (NIMA)-Related Kinase-4 Reduces Survivin Expression and Sensitizes Cancer Cells to TRAIL-Induced Cell Death. Oncotarget 2016, 7, 65957–65967. [Google Scholar] [CrossRef]
- Ding, N.-H.; Zhang, L.; Xiao, Z.; Rong, Z.-X.; Li, Z.; He, J.; Chen, L.; Ou, D.-M.; Liao, W.-H.; Sun, L.-Q. NEK4 Kinase Regulates EMT to Promote Lung Cancer Metastasis. J. Cell. Mol. Med. 2018, 22, 5877–5887. [Google Scholar] [CrossRef]
- Wells, C.I.; Kapadia, N.R.; Couñago, R.M.; Drewry, D.H. In Depth Analysis of Kinase Cross Screening Data to Identify Chemical Starting Points for Inhibition of the Nek Family of Kinases. MedChemComm 2018, 9, 44–66. [Google Scholar] [CrossRef]
- Yamamoto, N.; Takeshita, K.; Shichijo, M.; Kokubo, T.; Sato, M.; Nakashima, K.; Ishimori, M.; Nagai, H.; Li, Y.-F.; Yura, T.; et al. The Orally Available Spleen Tyrosine Kinase Inhibitor 2-[7-(3,4-Dimethoxyphenyl)-Imidazo[1,2-c]Pyrimidin-5-Ylamino]Nicotinamide Dihydrochloride (BAY 61-3606) Blocks Antigen-Induced Airway Inflammation in Rodents. J. Pharmacol. Exp. Ther. 2003, 306, 1174–1181. [Google Scholar] [CrossRef]
- George, D.; Friedman, M.; Allen, H.; Argiriadi, M.; Barberis, C.; Bischoff, A.; Clabbers, A.; Cusack, K.; Dixon, R.; Fix-Stenzel, S.; et al. Discovery of Thieno[2,3-c]Pyridines as Potent COT Inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4952–4955. [Google Scholar] [CrossRef]
- Mack, H.; Teusch, N.; Mueller, B.K.; Hornberger, W.; Jarvis, M.F.; Sauer, D. Preparation of 4-(4-Pyridyl)-Benzamides as Rho Kinases Inhibitors. WO2009027392, 5 March 2009. [Google Scholar]
- Zayed, M.; Ihmaid, S.; Ahmed, H.; El-Adl, K.; Asiri, A.; Omar, A. Synthesis, Modelling, and Anticonvulsant Studies of New Quinazolines Showing Three Highly Active Compounds with Low Toxicity and High Affinity to the GABA-A Receptor. Molecules 2017, 22, 188. [Google Scholar] [CrossRef] [PubMed]
- Asadi, P.; Khodarahmi, G.; Jahanian-Najafabadi, A.; Saghaie, L.; Hassanzadeh, F. Biologically Active Heterocyclic Hybrids Based on Quinazolinone, Benzofuran and Imidazolium Moieties: Synthesis, Characterization, Cytotoxic and Antibacterial Evaluation. Chem. Biodivers. 2017, 14, e1600411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, Y.; Zhang, B.; Lu, B.; Li, J. Design, Synthesis and Biological Evaluation of Novel Histone Deacetylase Inhibitors Incorporating 4-Aminoquinazolinyl Systems as Capping Groups. Bioorg. Med. Chem. Lett. 2017, 27, 4885–4888. [Google Scholar] [CrossRef]
- Felts, A.S.; Saleh, S.A.; Le, U.; Rodriguez, A.L.; Weaver, C.D.; Conn, P.J.; Lindsley, C.W.; Emmitte, K.A. Discovery and SAR of 6-Substituted-4-Anilinoquinazolines as Non-Competitive Antagonists of MGlu5. Bioorg. Med. Chem. Lett. 2009, 19, 6623–6626. [Google Scholar] [CrossRef]
- Liu, Q.; Douglas, D.G.; Delucca, G.V.; George, V.; Shi, Q.; Tebben, A.J. Preparation of Carbazole Carboxamide Compounds Useful as Kinase Inhibitors. U.S. Patent Application Publication No. 20100160303A1, 24 June 2010. [Google Scholar]
- Liu, L.T.; Yuan, T.-T.; Liu, H.-H.; Chen, S.-F.; Wu, Y.-T. Synthesis and Biological Evaluation of Substituted 6-Alkynyl-4-Anilinoquinazoline Derivatives as Potent EGFR Inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 6373–6377. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhuang, C.; Zhu, L.; Zhang, Y.; Yao, J.; Dong, G.; Wang, S.; Liu, Y.; Chen, H.; Sheng, C.; et al. Structure–Activity Relationship and Antitumor Activity of Thio-Benzodiazepines as P53–MDM2 Protein–Protein Interaction Inhibitors. Eur. J. Med. Chem. 2012, 56, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.W.; Chesworth, R.; Boriack-Sjodin, P.A.; Munchhof, M.J.; Jin, L.; Penebre, E.; Barbash, O.L. Prmt5 Inhibitors and Uses Thereof. WO2016022605A1, 11 February 2016. [Google Scholar]
- Berger, M.; Albrecht, B.; Berces, A.; Ettmayer, P.; Neruda, W.; Woisetschläger, M. S(+)-4-(1-Phenylethylamino)Quinazolines as Inhibitors of Human Immunoglobuline E Synthesis: Potency Is Dictated by Stereochemistry and Atomic Point Charges at N-1. J. Med. Chem. 2001, 44, 3031–3038. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Xu, W.; Xu, W. Method for Synthesizing 4-Bromo-7-Fluoroisoquinoline. CN108314648, 24 July 2018. [Google Scholar]
- Fisher, B.; Johnson, M.G.; Brian, L.; Shin, Y.; Kaizerman, J. Preparation of 5-Cyano-4,6-Diaminopyrimidine or 6-Aminopurine Derivatives as PI3K-Delta Inhibitors. WO 2012061696, 10 May 2012. [Google Scholar]
- Ismail, R.S.M.; Ismail, N.S.M.; Abuserii, S.; Abou El Ella, D.A. Recent Advances in 4-Aminoquinazoline Based Scaffold Derivatives Targeting EGFR Kinases as Anticancer Agents. Future J. Pharm. Sci. 2016, 2, 9–19. [Google Scholar] [CrossRef]
- Rewcastle, G.W.; Denny, W.A.; Bridges, A.J.; Zhou, H.; Cody, D.R.; McMichael, A.; Fry, D.W. Tyrosine Kinase Inhibitors. 5. Synthesis and Structure-Activity Relationships for 4-[(Phenylmethyl)Amino]- and 4-(Phenylamino)Quinazolines as Potent Adenosine 5’-Triphosphate Binding Site Inhibitors of the Tyrosine Kinase Domain of the Epidermal Growth Factor Receptor. J. Med. Chem. 1995, 38, 3482–3487. [Google Scholar] [CrossRef]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An Overview of Halogen Bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef] [PubMed]
- Mérour, J.-Y.; Buron, F.; Plé, K.; Bonnet, P.; Routier, S. The Azaindole Framework in the Design of Kinase Inhibitors. Molecules 2014, 19, 19935–19979. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, C.; Simpson, C.; DeRyckere, D.; Van Deusen, A.; Miley, M.J.; Kireev, D.; Norris-Drouin, J.; Sather, S.; Hunter, D.; et al. Discovery of Small Molecule Mer Kinase Inhibitors for the Treatment of Pediatric Acute Lymphoblastic Leukemia. ACS Med. Chem. Lett. 2012, 3, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Gucký, T.; Řezníčková, E.; Radošová Muchová, T.; Jorda, R.; Klejová, Z.; Malínková, V.; Berka, K.; Bazgier, V.; Ajani, H.; Lepšík, M.; et al. Discovery of N2-(4-Amino-Cyclohexyl)-9-Cyclopentyl-N6-(4-Morpholin-4-Ylmethyl-Phenyl)-9H-Purine-2,6-Diamine as a Potent FLT3 Kinase Inhibitor for Acute Myeloid Leukemia with FLT3 Mutations. J. Med. Chem. 2018, 61, 3855–3869. [Google Scholar] [CrossRef]
- Eurofins DiscoverX. KINOMEscan® Assay. Available online: https://www.discoverx.com/services/drug-discovery-development-services/kinase-profiling/kinomescan (accessed on 10 April 2020).
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- The Cancer Cell Line Encyclopedia. Available online: www.broadinstitute.org/ccle. (accessed on 17 December 2020).
- Eurofins, KinaseProfiler, NEK4 Human Other Protein Kinase Enzymatic Radiometric Assay [Km ATP]. Available online: https://www.eurofinsdiscoveryservices.com/catalogmanagement/viewitem/NEK4-Human-Other-Protein-Kinase-Enzymatic-Radiometric-Assay-Km-ATP-KinaseProfiler/15-033KP (accessed on 10 April 2020).
- Mohamed, T.; Rao, P.P.N. 2,4-Disubstituted Quinazolines as Amyloid-β Aggregation Inhibitors with Dual Cholinesterase Inhibition and Antioxidant Properties: Development and Structure-Activity Relationship (SAR) Studies. Eur. J. Med. Chem. 2017, 126, 823–843. [Google Scholar] [CrossRef]
- Tobe, M.; Isobe, Y.; Tomizawa, H.; Nagasaki, T.; Takahashi, H.; Fukazawa, T.; Hayashi, H. Discovery of Quinazolines as a Novel Structural Class of Potent Inhibitors of NF-ΚB Activation. Bioorg. Med. Chem. 2003, 11, 383–391. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tm (°C) | ΔTm (°C) |
|---|---|---|
| USP13 in buffer | 44.56 | N.D. |
| USP13 in 5% DMSO | 45.18 | 0.00 |
| USP13 in 10% DMSO | 44.80 | 0.00 |
| USP13 + 50 μM of 5aa in 5% DMSO | 45.79 | 0.61 |
| USP13 + 100 μM of 5aa in 10% DMSO | 45.86 | 1.06 |
| USP13 + 100 μM of 5aa in 10% DMSO | 44.44 | −0.36 |
| USP13 + 50 μM of 5bc in 5% DMSO | 46.58 | 1.40 |
| USP13 + 100 μM of 5bc in 10% DMSO | 43.27 | −1.53 |
| USP13 + 100 μM of 5bc in 10% DMSO | 44.44 | −0.36 |
 |  |  |  | |
|---|---|---|---|---|
| Kinase | Residual Activity (%) | |||
| AAK1 (h) | 95 | 53 | 81 | 72 |
| Abl (m) | 90 | 56 | 83 | 99 |
| ACK1 (h) | 85 | 56 | 84 | 71 |
| ALK (h) | 82 | 72 | 59 | 86 |
| ALK2 (h) | 60 | 50 | 45 | 58 |
| Aurora-B (h) | 85 | 59 | 69 | 48 |
| BrSK1 (h) | 85 | 30 | 89 | 112 |
| BrSK2 (h) | 79 | 42 | 65 | 71 |
| CaMKIIα (h) | 94 | 58 | 88 | 89 |
| CaMKIIγ (h) | 83 | 41 | 74 | 75 |
| CaMKIIδ (h) | 90 | 41 | 79 | 77 |
| CDK2/cyclinE (h) | 69 | 89 | 103 | 54 |
| CDK7/cyclinH/ MAT1 (h) | 62 | 58 | 49 | 59 |
| CDK13/cyclinK(h) | 75 | 105 | 35 | 114 |
| CDKL3 (h) | 77 | 29 | 65 | 71 |
| CDKL4 (h) | 73 | 38 | 94 | 92 |
| CLK1 (h) | 23 | 14 | 28 | 28 |
| CLK2 (h) | 47 | 43 | 53 | 60 |
| CLK4 (h) | 19 | 6 | 12 | 16 |
| DCAMKL2 (h) | 81 | 80 | 64 | 57 |
| DDR1 (h) | 65 | 7 | 29 | 39 |
| DRAK1 (h) | 98 | 57 | 79 | 85 |
| DYRK1A (h) | 74 | 29 | 68 | 63 |
| DYRK1B (h) | 88 | 53 | 82 | 92 |
| DYRK3 (h) | 117 | 57 | 98 | 98 |
| EGFR (h) | 27 | 1 | 46 | 72 |
| EGFR (L858R) (h) | 23 | 3 | 37 | 60 |
| EGFR (L861Q) (h) | 36 | 1 | 51 | 73 |
| EphB4 (h) | 68 | 63 | 59 | 68 |
| ErbB2 (h) | 74 | 20 | 79 | 65 |
| Flt3 (D835Y) (h) | 91 | 49 | 87 | 80 |
| Haspin (h) | 71 | 36 | 72 | 78 |
| Hck (h) activated | 85 | 46 | 85 | 108 |
| HIPK4 (h) | 103 | 42 | 92 | 93 |
| LOK (h) | 91 | 35 | 81 | 87 |
| LRRK2 (h) | 91 | 44 | 91 | 101 |
| Met (h) | 82 | 72 | 57 | 112 |
| Mnk2 (h) | 93 | 19 | 83 | 80 |
| MST4 (h) | 58 | 64 | 81 | 72 |
| NEK4 (h) | 71 | 30 | 27 | 12 |
| NEK11 (h) | 41 | 53 | 71 | 45 |
| PASK (h) | 66 | 65 | 44 | 30 |
| PDGFRα (D842V) (h) | 114 | 50 | 92 | 86 |
| Pim-1 (h) | 66 | 39 | 73 | 70 |
| Ret (h) | 69 | 43 | 99 | 62 |
| RIPK2 (h) | 82 | 22 | 86 | 86 |
| TAF1L (h) | 80 | 32 | 66 | 70 |
| TRB2 (h) | 69 | 31 | 65 | 60 |
| PI3KC2g (h) | 70 | 40 | 57 | 34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsocht, M.; Giron, P.; Maes, L.; Versées, W.; Gutierrez, G.J.; De Grève, J.; Ballet, S. Structure–Activity Relationship (SAR) Study of Spautin-1 to Entail the Discovery of Novel NEK4 Inhibitors. Int. J. Mol. Sci. 2021, 22, 635. https://doi.org/10.3390/ijms22020635
Elsocht M, Giron P, Maes L, Versées W, Gutierrez GJ, De Grève J, Ballet S. Structure–Activity Relationship (SAR) Study of Spautin-1 to Entail the Discovery of Novel NEK4 Inhibitors. International Journal of Molecular Sciences. 2021; 22(2):635. https://doi.org/10.3390/ijms22020635
Chicago/Turabian StyleElsocht, Mathias, Philippe Giron, Laila Maes, Wim Versées, Gustavo J. Gutierrez, Jacques De Grève, and Steven Ballet. 2021. "Structure–Activity Relationship (SAR) Study of Spautin-1 to Entail the Discovery of Novel NEK4 Inhibitors" International Journal of Molecular Sciences 22, no. 2: 635. https://doi.org/10.3390/ijms22020635
APA StyleElsocht, M., Giron, P., Maes, L., Versées, W., Gutierrez, G. J., De Grève, J., & Ballet, S. (2021). Structure–Activity Relationship (SAR) Study of Spautin-1 to Entail the Discovery of Novel NEK4 Inhibitors. International Journal of Molecular Sciences, 22(2), 635. https://doi.org/10.3390/ijms22020635