Plastid Genomes of the Early Vascular Plant Genus Selaginella Have Unusual Direct Repeat Structures and Drastically Reduced Gene Numbers

 and
and
Abstract
:1. Introduction
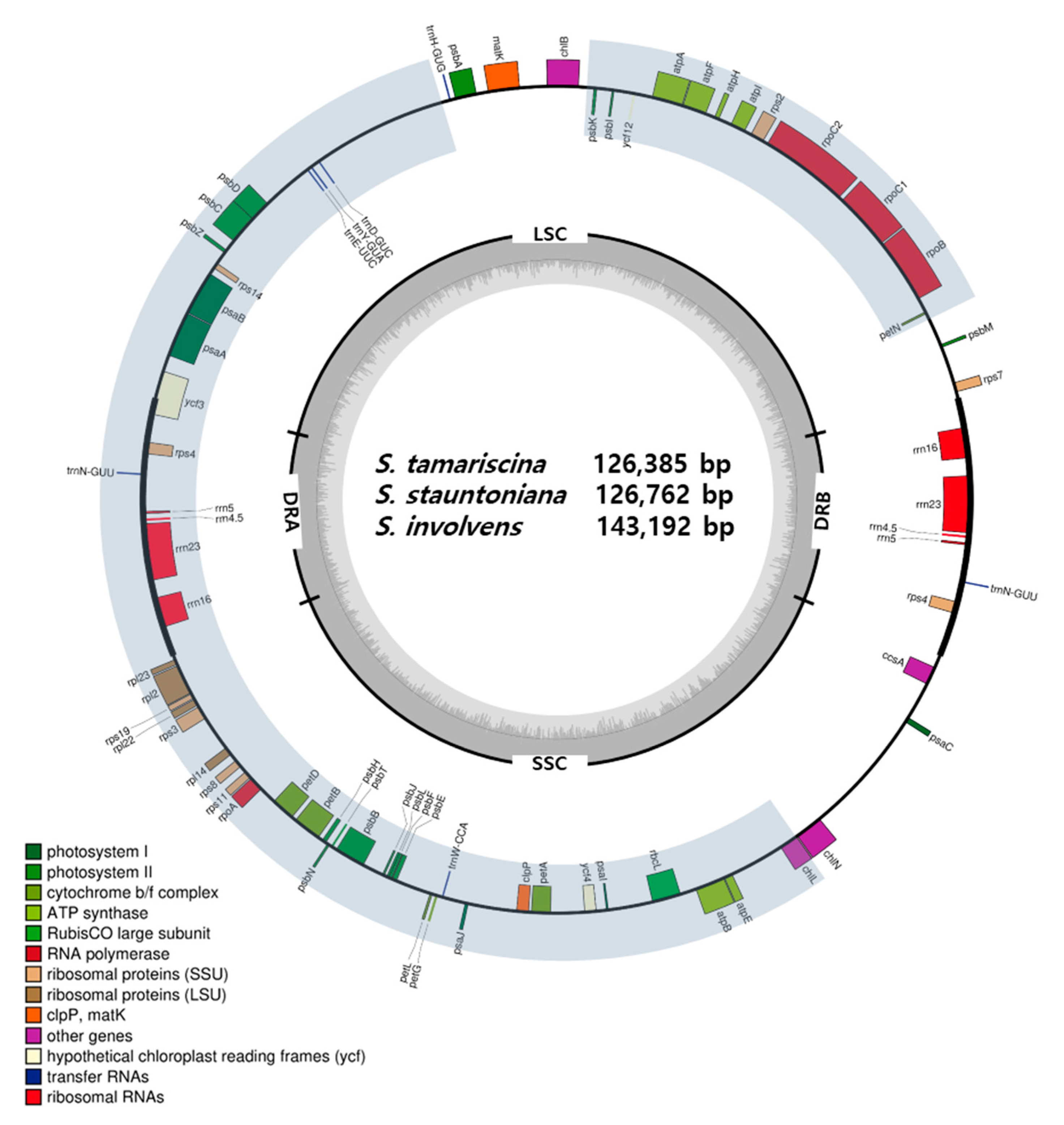
2. Results
2.1. Selaginella Plastomes Contain Reduced LSCs and Unusual DR
2.2. Selaginella Plastomes Contain Fewer Genes than Other Lycophyte Plastomes
2.3. Phylogenetic Analysis Shows Two Main Lineages with Dynamic Structural Variations
2.4. Plastome Diversity among Five S. tamariscina Collections Reflects the Geographical Diversity
2.5. Selaginella Plastomes Exhibit Many Intron Losses and GC Bias
2.6. RNA Editing Is Commonly Found in Selaginella Plastomes
3. Discussion
3.1. Unique Features of Selaginella Plastomes
3.2. One Common and Two Independent Inversions Caused the Appearance of DR and IR Structures during Selaginella Species Evolution
3.3. Gene and Intron Losses
3.4. Intraspecies Diversity
3.5. A High GC Content and Abundant RNA Editing
4. Materials and Methods
4.1. Plant Materials and Publicly Available Plastome Sequences
4.2. DNA Extraction, Sequencing, Plastome Assembly, and Annotation
4.3. Confirmation of Direct Repeat Structures by PCR Amplification
4.4. Gene Contents and Phylogenetic Analysis
4.5. Analysis of Intron Variation and RNA Editing Sites
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| IR | Inverted repeat |
| DR | Direct repeat |
| SC | Single-copy |
| LSC | Long single-copy |
| SSC | Short single-copy |
| NCBI | National Center for Biotechnology Information |
| WGS | Whole genome sequencing |
| PCR | Polymerase Chain Reaction |
| SNP | Single Nucleotide Polymorphisms |
| InDels | Insertions/deletions |
| mya | Million years ago |
| CTAB | Cetyltrimethylammonium bromide |
| dnaLCW | De novo assembly of Low-Coverage Whole genome sequence |
| CDS | Coding sequence |
| tRNA | Transfer RNA |
| rRNA | Ribosomal RNA |
| HPD | Highest Posterior Density |
| GTR | General Time-Reversible |
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence |
|---|---|
| Primer 1 | AGCCGACATTCGTTACGGTT |
| Primer 2 | GGATTCGCATTACCGACAGTG |
| Primer 3 | GCATTTAATCGGCGCGAAGT |
| Primer 4 | CACCACTCCCCTTGAACCTC |
| Primer 5 | TATGCGCCGCGATTAGGC |
| Primer 6 | GTCGGTTCAAAACCCGTGC |
References
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef]
- Ruhlman, T.A.; Jansen, R.K. The Plastid Genomes of Flowering Plants. In Computational Biology; Springer: Berlin/Heidelberg, Germany, 2014; pp. 3–38. [Google Scholar]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 1–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakasugi, T.; Tsudzuki, J.; Ito, S.; Nakashima, K.; Sugiura, M.; Tsudzuki, T. Loss of all ndh genes as determined by sequencing the entire chloroplast genome of the black pine Pinus thunbergii. Proc. Natl. Acad. Sci. USA 1994, 91, 9794–9798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-S.; Wang, Y.-N.; Hsu, C.-Y.; Lin, C.-P.; Chaw, S.-M. Loss of Different Inverted Repeat Copies from the Chloroplast Genomes of Pinaceae and Cupressophytes and Influence of Heterotachy on the Evaluation of Gymnosperm Phylogeny. Genome Biol. Evol. 2011, 3, 1284–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grewe, F.; Guo, W.; Gubbels, E.A.; Hansen, A.K.; Mower, J.P. Complete plastid genomes from Ophioglossum californicum, Psilotum nudum, and Equisetum hyemale reveal an ancestral land plant genome structure and resolve the position of Equisetales among monilophytes. BMC Evol. Biol. 2013, 13, 8. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.D.; Osorio, B.; Aldrich, J.; Thompson, W.F. Chloroplast DNA evolution among legumes: Loss of a large inverted repeat occurred prior to other sequence rearrangements. Curr. Genet. 1987, 11, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Wolf, P.G.; Der, J.P.; Duffy, A.M.; Davidson, J.B.; Grusz, A.L.; Pryer, K.M. The evolution of chloroplast genes and genomes in ferns. Plant Mol. Biol. 2011, 76, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Labiak, P.H.; Karol, K.G.K.G. Plastome sequences of an ancient fern lineage reveal remarkable changes in gene content and architecture. Am. J. Bot. 2017, 104, 1008–1018. [Google Scholar] [CrossRef]
- Cronn, R.C.; Liston, A.; Parks, M.; Gernandt, D.S.; Shen, R.; Mockler, T. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 2008, 36, e122. [Google Scholar] [CrossRef] [Green Version]
- McCoy, S.R.; Kuehl, J.V.; Boore, J.L.; Raubeson, L.A. The complete plastid genome sequence of Welwitschia mirabilis: An unusually compact plastome with accelerated divergence rates. BMC Evol. Biol. 2008, 8, 130. [Google Scholar] [CrossRef] [Green Version]
- Wicke, S.; Müller, K.F.; De Pamphilis, C.W.; Quandt, D.; Wickett, N.J.; Zhang, Y.; Renner, S.S.; Schneeweiss, G.M. Mechanisms of Functional and Physical Genome Reduction in Photosynthetic and Nonphotosynthetic Parasitic Plants of the Broomrape Family. Plant Cell 2013, 25, 3711–3725. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-B.; Tang, M.; Li, H.; Zhang, Z.; Li, D.Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 2013, 13, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Hou, B.-W.; Niu, Z.-T.; Liu, W.; Xue, Q.-Y.; Ding, X.-Y. Comparative Chloroplast Genomes of Photosynthetic Orchids: Insights into Evolution of the Orchidaceae and Development of Molecular Markers for Phylogenetic Applications. PLoS ONE 2014, 9, e99016. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schäferhoff, B.; Depamphilis, C.W.; Müller, K.F. Disproportional Plastome-Wide Increase of Substitution Rates and Relaxed Purifying Selection in Genes of Carnivorous Lentibulariaceae. Mol. Biol. Evol. 2014, 31, 529–545. [Google Scholar] [CrossRef] [Green Version]
- Shmakov, A. A community-derived classification for extant lycophytes and ferns. J. Syst. Evol. 2016, 54, 563–603. [Google Scholar] [CrossRef]
- Banks, J.A.; Nishiyama, T.; Hasebe, M.; Bowman, J.L.; Gribskov, M.; Depamphilis, C.W.; Albert, V.A.; Aono, N.; Aoyama, T.; Ambrose, B.A.; et al. The Selaginella Genome Identifies Genetic Changes Associated with the Evolution of Vascular Plants. Sci. 2011, 332, 960–963. [Google Scholar] [CrossRef] [Green Version]
- Kenrick, P.; Crane, P.R. The origin and early evolution of plants on land. Nat. Cell Biol. 1997, 389, 33–39. [Google Scholar] [CrossRef]
- Zhou, X.-M.; Zhang, L. A classification of Selaginella (Selaginellaceae) based on molecular (chloroplast and nuclear), macromorphological, and spore features. Taxon 2015, 64, 1117–1140. [Google Scholar] [CrossRef]
- Weststrand, S.; Korall, P. Phylogeny of Selaginellaceae: There is value in morphology after all! Am. J. Bot. 2016, 103, 2136–2159. [Google Scholar] [CrossRef] [Green Version]
- Weststrand, S.; Korall, P. A subgeneric classification of Selaginella (Selaginellaceae). Am. J. Bot. 2016, 103, 2160–2169. [Google Scholar] [CrossRef] [Green Version]
- Karol, K.G.; Arumuganathan, K.; Boore, J.L.; Duffy, A.M.; Everett, K.D.; Hall, J.D.; Hansen, S.K.; Kuehl, J.V.; Mandoli, D.F.; Mishler, B.D.; et al. Complete plastome sequences of Equisetum arvense and Isoetes flaccida: Implications for phylogeny and plastid genome evolution of early land plant lineages. BMC Evol. Biol. 2010, 10, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, P.G.; Roper, J.M.; Duffy, A.M. The evolution of chloroplast genome structure in ferns. Genome 2010, 53, 731–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, B.; Fong, R.; Collins, L.J.; McLenachan, P.A.; Penny, D. Two New Fern Chloroplasts and Decelerated Evolution Linked to the Long Generation Time in Tree Ferns. Genome Biol. Evol. 2014, 6, 1166–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [Green Version]
- Turmel, M.; Otis, C.; Lemieux, C. Dynamic Evolution of the Chloroplast Genome in the Green Algal Classes Pedinophyceae and Trebouxiophyceae. Genome Biol. Evol. 2015, 7, 2062–2082. [Google Scholar] [CrossRef] [Green Version]
- Wolf, P.G.; Karol, K.G.; Mandoli, D.F.; Kuehl, J.V.; Arumuganathan, K.; Ellis, M.W.; Mishler, B.D.; Kelch, D.G.; Olmstead, R.G.; Boore, J.L. The first complete chloroplast genome sequence of a lycophyte, Huperzia lucidula (Lycopodiaceae). Gene 2005, 350, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.-Y.; Zhang, H.-R.; Shrestha, N.; Zhang, X.-C. Complete Chloroplast Genome of a Valuable Medicinal Plant, Huperzia serrata (Lycopodiaceae), and Comparison with Its Congener. Appl. Plant Sci. 2016, 4, 1600071. [Google Scholar] [CrossRef]
- Zhang, H.-R.; Kang, J.-S.; Viane, R.L.L.; Zhang, X.-C. The complete chloroplast genome sequence of Huperzia javanica (sw.) C. Y. Yang in Lycopodiaceae. Mitochondrial DNA Part B 2017, 2, 216–218. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, S.; Ueda, K.; Nishiyama, T.; Hasebe, M.; Yoshikawa, S.; Konagaya, A.; Nishiuchi, T.; Yamaguchi, K. The chloroplast genome from a lycophyte (microphyllophyte), Selaginella uncinata, has a unique inversion, transpositions and many gene losses. J. Plant Res. 2007, 120, 281–290. [Google Scholar] [CrossRef]
- Smith, D.R. Unparalleled GC content in the plastid DNA of Selaginella. Plant Mol. Biol. 2009, 71, 627–639. [Google Scholar] [CrossRef]
- Oldenkott, B.; Yamaguchi, K.; Tsuji-Tsukinoki, S.; Knie, N.; Knoop, V. Chloroplast RNA editing going extreme: More than 3400 events of C-to-U editing in the chloroplast transcriptome of the lycophyte Selaginella uncinata. RNA 2014, 20, 1499–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Xin, T.; Bartels, D.; Li, Y.; Gu, W.; Yao, H.; Liu, S.; Yu, H.; Pu, X.; Zhou, J.G.; et al. Genome Analysis of the Ancient Tracheophyte Selaginella tamariscina Reveals Evolutionary Features Relevant to the Acquisition of Desiccation Tolerance. Mol. Plant 2018, 11, 983–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mower, J.P.; Ma, P.F.; Grewe, F.; Taylor, A.; Michael, T.P.; VanBuren, R.; Qiu, Y.L. Lycophyte plastid genomics: Extreme variation in GC, gene and intron content and multiple inversions between a direct and inverted orientation of the rRNA repeat. New Phytol. 2019, 222, 1061–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-R.; Qiao-Ping, X.; Zhang, X.-C. The Unique Evolutionary Trajectory and Dynamic Conformations of DR and IR/DR-Coexisting Plastomes of the Early Vascular Plant Selaginellaceae (Lycophyte). Genome Biol. Evol. 2019, 11, 1258–1274. [Google Scholar] [CrossRef]
- Zhang, H.-R.; Zhang, X.-C.; Xiang, Q.-P. Directed repeats co-occur with few short-dispersed repeats in plastid genome of a Spikemoss, Selaginella vardei (Selaginellaceae, Lycopodiopsida). BMC Genom. 2019, 20, 1–11. [Google Scholar]
- Palmer, J.D. Chloroplast DNA exists in two orientations. Nat. Cell Biol. 1983, 301, 92–93. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Kwon, E.-C.; Kim, J.-H.; Kim, N.-S. Comprehensive genomic analyses with 115 plastomes from algae to seed plants: Structure, gene contents, GC contents, and introns. Genes Genom. 2020, 42, 553–570. [Google Scholar] [CrossRef]
- Park, J.; Kim, Y.; Lee, G.-H.; Park, C.-H. The complete chloroplast genome of Selaginella tamariscina (Beauv.) Spring (Selaginellaceae) isolated in Korea. Mitochondrial DNA Part B 2020, 5, 1654–1656. [Google Scholar] [CrossRef] [Green Version]
- Weng, M.-L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the Ancestral Plastid Genome in Geraniaceae Reveals a Correlation between Genome Rearrangements, Repeats, and Nucleotide Substitution Rates. Mol. Biol. Evol. 2014, 31, 645–659. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.D.; Thompson, W.F. Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell 1982, 29, 537–550. [Google Scholar] [CrossRef]
- Reith, M.; Munholland, J. The ribosomal RNA repeats are non-identical and directly oriented in the chloroplast genome of the red alga Porphyra purpurea. Curr. Genet. 1993, 24, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.E. Chloroplast origins and evolution. In The Molecular Biology of Cyanobacteria; Springer: Berlin/Heidelberg, Germany, 1994; pp. 91–118. [Google Scholar]
- Ruhlman, T.; Chang, W.-J.; Chen, J.J.; Huang, Y.-T.; Chan, M.-T.; Zhang, J.; Liao, D.-C.; Blazier, J.C.; Jin, X.-H.; Shih, M.-C.; et al. NDH expression marks major transitions in plant evolution and reveals coordinate intracellular gene loss. BMC Plant Biol. 2015, 15, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, P.G.; Rowe, C.A.; Sinclair, R.B.; Hasebe, M. Complete Nucleotide Sequence of the Chloroplast Genome from a Leptosporangiate Fern, Adiantum capillus-veneris L. DNA Res. 2003, 10, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Morden, C.W.; Wolfe, K.; DePamphilis, C.; Palmer, J. Plastid translation and transcription genes in a non-photosynthetic plant: Intact, missing and pseudo genes. EMBO J. 1991, 10, 3281–3288. [Google Scholar] [CrossRef]
- Rogalski, M.; Karcher, D.; Bock, R. Superwobbling facilitates translation with reduced tRNA sets. Nat. Struct. Mol. Biol. 2008, 15, 192–198. [Google Scholar] [CrossRef]
- Pfitzinger, H.; Weil, J.; Pillay, D.; Guillemaut, P. Codon recognition mechanisms in plant chloroplasts. Plant Mol. Biol. 1990, 14, 805–814. [Google Scholar] [CrossRef]
- Chorev, M.; Carmel, L. The Function of Introns. Front. Genet. 2012, 3, 55. [Google Scholar] [CrossRef] [Green Version]
- Dabbagh, N.; Bennett, M.S.; Triemer, R.E.; Preisfeld, A. Chloroplast genome expansion by intron multiplication in the basal psychrophilic euglenoid Eutreptiella pomquetensis. PeerJ 2017, 5, e3725. [Google Scholar] [CrossRef] [Green Version]
- Vogel, J.; Börner, T.; Hess, W.R. Comparative analysis of splicing of the complete set of chloroplast group II introns in three higher plant mutants. Nucleic Acids Res. 1999, 27, 3866–3874. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Hipp, A.L.; Gailing, O. Sharing of chloroplast haplotypes among red oak species suggests interspecific gene flow between neighboring populations. Botany 2015, 93, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Norberg, J.; Swaney, D.P.; Dushoff, J.; Lin, J.; Casagrandi, R.; Levin, S.A. Phenotypic diversity and ecosystem functioning in changing environments: A theoretical framework. Proc. Natl. Acad. Sci. USA 2001, 98, 11376–11381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björklund, M.; Ranta, E.; Kaitala, V.; Bach, L.A.; Lundberg, P.; Stenseth, N.C. Quantitative Trait Evolution and Environmental Change. PLoS ONE 2009, 4, e4521. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, J.; Tachida, H. Compositional Properties of Green-Plant Plastid Genomes. J. Mol. Evol. 2005, 60, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006, 46, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Kotera, E.; Tasaka, M.; Shikanai, T. A pentatricopeptide repeat protein is essential for RNA editing in chloroplasts. Nature 2005, 433, 326–330. [Google Scholar] [CrossRef]
- Hecht, J.; Grewe, F.; Knoop, V. Extreme RNA Editing in Coding Islands and Abundant Microsatellites in Repeat Sequences of Selaginella moellendorffii Mitochondria: The Root of Frequent Plant mtDNA Recombination in Early Tracheophytes. Genome Biol. Evol. 2011, 3, 344–358. [Google Scholar] [CrossRef] [Green Version]
- Grewe, F.; Herres, S.; Viehöver, P.; Polsakiewicz, M.; Weisshaar, B.; Knoop, V. A unique transcriptome: 1782 positions of RNA editing alter 1406 codon identities in mitochondrial mRNAs of the lycophyte Isoetes engelmannii. Nucleic Acids Res. 2011, 39, 2890–2902. [Google Scholar] [CrossRef]
- Allen, G.C.; Flores-Vergara, M.A.; Krasynanski, S.; Kumar, S.; Thompson, W.F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 2006, 1, 2320–2325. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.-C.; Lee, J.; Lee, H.O.; Joh, H.J.; Kim, N.-H.; Park, H.-S.; Yang, T.-J. Comprehensive Survey of Genetic Diversity in Chloroplast Genomes and 45S nrDNAs within Panax ginseng Species. PLoS ONE 2015, 10, e0117159. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.-C.; Lee, J.; Yu, Y.; Yang, K.; Choi, B.-S.; Koh, H.-J.; Waminal, N.E.; Choi, H.-I.; Kim, N.-H.; et al. Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci. Rep. 2015, 5, 15655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Phillippy, A.M.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.-A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Wikström, N.; Kenrick, P. Evolution of Lycopodiaceae (Lycopsida): Estimating Divergence Times from rbcL Gene Sequences by Use of Nonparametric Rate Smoothing. Mol. Phylogenetics Evol. 2001, 19, 177–186. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]





| Order | Scientific Name | Structure Size (bp) | Gene Contents (Repeated Genes) | GC Content (%) | GenBank ID | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total Length | LSC | SSC | IRs or DRs | Total | Protein | rRNA | tRNA | Repeat | ||||
| Lycopodiales | Huperzia lucidula | 154,373 | 104,088 | 19,657 | 15,314 | 124 | 85 | 4(4) | 27(4) | IR | 36.25 | NC_006861.1 |
| Huperzia serrata | 154,176 | 104,080 | 19,658 | 15,219 | 130 | 85(2) | 4(4) | 30(5) | IR | 36.28 | NC_033874.1 | |
| Isoetales | Isoetes flaccida | 145,303 | 91,862 | 27,205 | 13,118 | 128 | 82(1) | 4(4) | 32(5) | IR | 37.94 | GU191333.1 |
| Selaginellales | S. lyallii | 110,411 | 44,943 | 45,276 | 10,096 | 83 | 60(1) | 4(4) | 12(2) | DR | 50.75 | NC_041556.1 |
| S. kraussiana | 129,971 | 46,049 | 54,728 | 14,597 | 92 | 70(3) | 4(4) | 10(1) | DR | 52.33 | NC_040926.1 | |
| S. remotifolia | 131,867 | 46,351 | 55,844 | 14,836 | 95 | 70(3) | 4(4) | 12(2) | DR | 56.49 | NC_041644.1 | |
| S. indica | 122,460 | 45,711 | 48,395 | 14,177 | 86 | 61(3) | 4(4) | 12(2) | DR | 53.55 | MK156801.1 | |
| S. vardei | 121,254 | 45,792 | 47,676 | 13,893 | 84 | 61(3) | 4(4) | 10(2) | DR | 53.21 | MG272482.1 | |
| S. lepidophylla | 114,693 | 80,625 | 19,452 | 7308 | 85 | 64 | 4(4) | 12(1) | IR | 51.94 | NC_040927.1 | |
| S. sanguinolenta | 147,148 | 54,436 | 59,650 | 16,531 | 102 | 67(2) | 4(4) | 22(3) | DR | 50.78 | NC_041645.1 | |
| S. stauntoniana | 126,762 | 54,231 | 47,745 | 12,393 | 76 | 60(1) | 4(4) | 6(1) | DR | 54.06 | MK460598 † | |
| S. tamariscina | 126,385 | 53,219 | 47,600 | 12,783 | 75 | 59(1) | 4(4) | 6(1) | DR | 53.98 | MK460597 † | |
| S. doederleinii | 142,752 | 57,841 | 62,865 | 11,023 | 100 | 75(1) | 4(4) | 14(2) | DR | 51.13 | NC_041641.1 | |
| S. involvens | 143,192 | 58,193 | 61,075 | 11,962 | 102 | 75(1) | 4(4) | 14(4) | DR | 50.82 | MK460599 † | |
| S. moellendorffii | 143,525 | 58,198 | 61,129 | 12,099 | 99 | 75(1) | 4(4) | 12(3) | DR | 51.00 | MG272484.1 | |
| S. bisulcata | 140,509 | 55,598 | 59,659 | 12,626 | 85 | 59(3) | 4(4) | 12(3) | DR | 52.77 | NC_041640.1 | |
| S. pennata | 138,024 | 54,979 | 59,847 | 11,599 | 93 | 71(3) | 4(4) | 9(2) | DR | 52.91 | NC_041643.1 | |
| S. hainanensis | 144,201 | 77,780 | 40,819 | 12,801 | 103 | 77(3) | 4(4) | 12(3) | IR | 54.83 | NC_041642.1 | |
| S. uncinata | 144,170 | 77,706 | 40,886 | 12,789 | 101 | 75(3) | 4(4) | 11(4) | IR | 54.85 | AB197035.2 | |
| SNP | ||||||
|---|---|---|---|---|---|---|
| SNU 2014 † | SNU 2018 † | Korea 2020 | China 2018 | China 2019 | ||
| InDel | SNU 2014 | - | 1 | 1 | 151 | 1219 |
| SNU 2018 | 8 | - | 0 | 150 | 1218 | |
| Korea 2020 | 14 | 15 | - | 150 | 1218 | |
| China 2018 | 113 | 113 | 116 | - | 1246 | |
| China 2019 | 396 | 394 | 386 | 401 | - | |
| Function | Gene | H. lu | H. se | I. fl | S. ly | S. kra | S. rem | S. ind | S. va | S. lep | S. san | S. st† | S. ta† | S. doe | S. inv† | S. mo | S. bis | S. pen | S. hai | S. un |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ATP synthase | atpF | + | + | + | + | + | + | – | – | + | + | + | + | + | + | + | + | + | + | + |
| Other | clpP-1 | + | + | + | – | – | – | – | – | – | + | – | – | + | + | + | + | + | + | + |
| clpP-2 | + | + | + | – | – | – | – | – | – | + | – | – | + | – | – | – | – | – | – | |
| NADH dehydrogenase | ndhA | + | + | + | + | + | + | + | + | + | + | + | ||||||||
| ndhB | + | + | + | + | + | + | + | + | + | + | + | |||||||||
| Cytochrome | petB | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| petD | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Large subunit ribosomal protein | rpl16 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ||
| rpl2 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| RNA polymerase | rpoC1 | + | + | + | – | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Small subunit ribosomal protein | rps12 | + | + | + | ||||||||||||||||
| rps16 | + | + | – | |||||||||||||||||
| Unknown | ycf3–1 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| ycf3–2 | + | + | + | – | – | – | – | – | – | + | + | + | + | + | + | + | + | + | + | |
| ycf66 | – | + | + | |||||||||||||||||
| tRNA | trnL | + | + | + | – | – | – | – | – | |||||||||||
| trnG | – | – | + | – | – | – | – | |||||||||||||
| trnI | + | + | – | – | – | |||||||||||||||
| trnA | + | + | + | |||||||||||||||||
| trnK | + | + | ||||||||||||||||||
| trnV | + | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shim, H.; Lee, H.J.; Lee, J.; Lee, H.-O.; Kim, J.-H.; Yang, T.-J.; Kim, N.-S. Plastid Genomes of the Early Vascular Plant Genus Selaginella Have Unusual Direct Repeat Structures and Drastically Reduced Gene Numbers. Int. J. Mol. Sci. 2021, 22, 641. https://doi.org/10.3390/ijms22020641
Shim H, Lee HJ, Lee J, Lee H-O, Kim J-H, Yang T-J, Kim N-S. Plastid Genomes of the Early Vascular Plant Genus Selaginella Have Unusual Direct Repeat Structures and Drastically Reduced Gene Numbers. International Journal of Molecular Sciences. 2021; 22(2):641. https://doi.org/10.3390/ijms22020641
Chicago/Turabian StyleShim, Hyeonah, Hyeon Ju Lee, Junki Lee, Hyun-Oh Lee, Jong-Hwa Kim, Tae-Jin Yang, and Nam-Soo Kim. 2021. "Plastid Genomes of the Early Vascular Plant Genus Selaginella Have Unusual Direct Repeat Structures and Drastically Reduced Gene Numbers" International Journal of Molecular Sciences 22, no. 2: 641. https://doi.org/10.3390/ijms22020641