The Involvement of Innate and Adaptive Immunity in the Initiation and Perpetuation of Sjögren’s Syndrome


 , and
, and 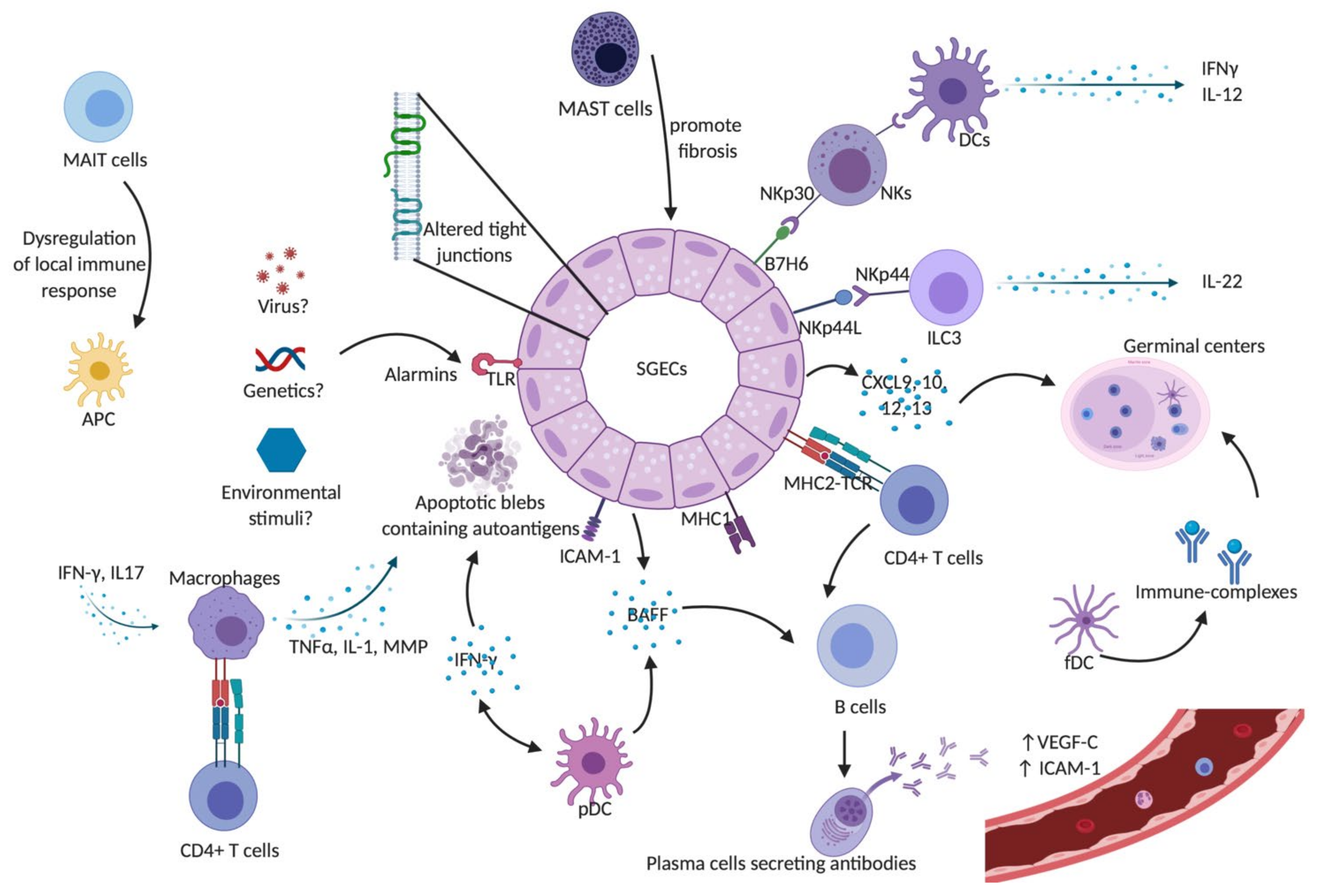{kind=link}
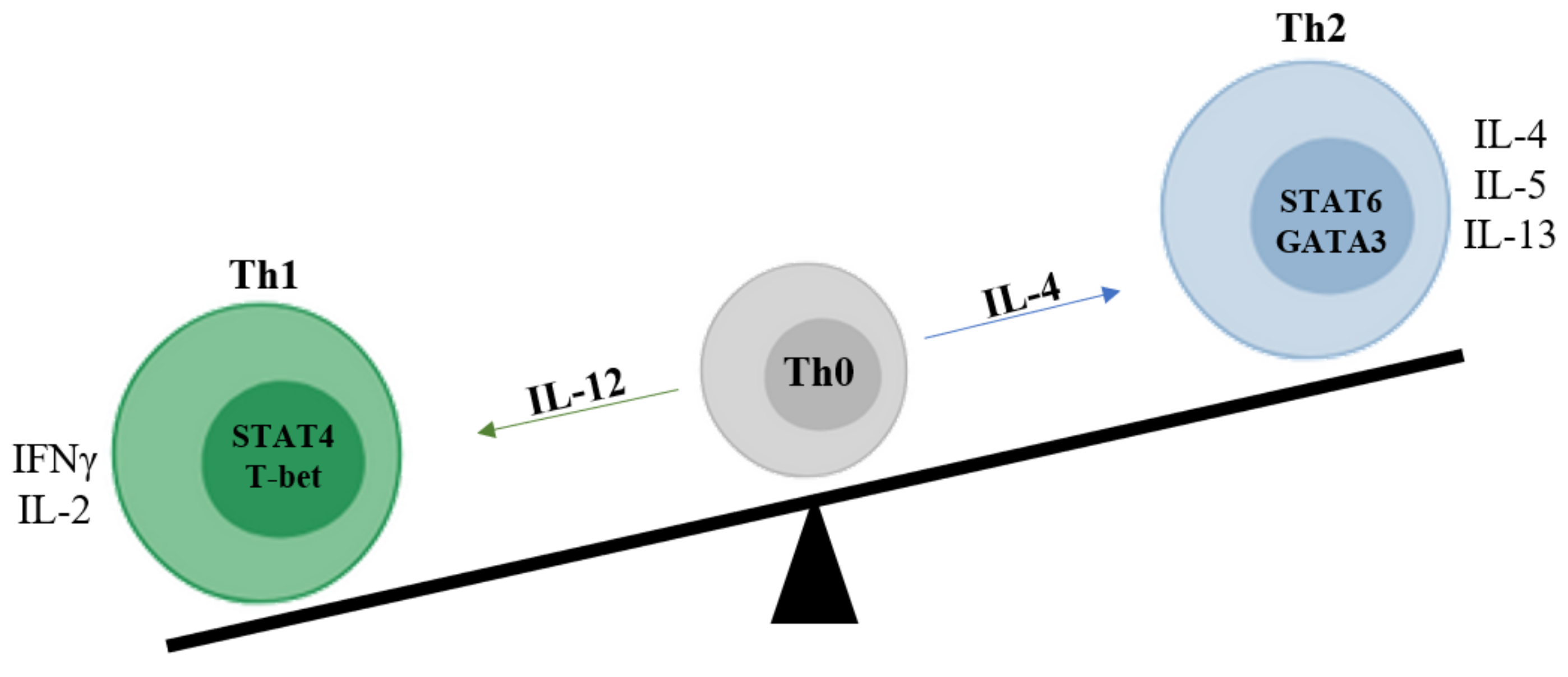{kind=link}
{kind=link}
Abstract
1. Sjögren’s Syndrome
2. Innate Immune Cells Involved in Sjögren’s Syndrome
2.1. Dendritic Cells
2.2. Macrophages
2.3. Mast Cells
2.4. Salivary Gland Epithelial Cells (SGECs)
2.5. Endothelial Cells
2.6. Mucosal-Associated Invariant T (MAIT) Cells
2.7. Natural Killer (NK) Cells
2.8. Natural Killer T (NKT) Cells
2.9. Innate Lymphoid Cells (ILCs)
3. Adaptive Immunity: The Insidious Role of T Cell Subsets in SS
3.1. CD4+ T Cells
3.1.1. Th1-Th2 Cells
3.1.2. Th17
3.1.3. T Follicular Helper Cells
3.1.4. T Regulatory Cells
3.1.5. Follicular Regulatory Cells (Tfr)
3.2. CD8+ T Cells
Innate T Cells
3.3. B Cells
3.3.1. B Cell Hyperactivity
3.3.2. B Cell Subpopulations
3.3.3. Marginal Zone B Cells
3.3.4. Regulatory B Cells
3.3.5. BAFF
3.3.6. Germinal Center-Like Structures
4. Cross Talk between the Innate and Adaptive Immunity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APRIL | a proliferation-inducing ligand |
| BAFF | B-cell activating factor |
| Bcl-6 | B-cell lymphoma 6 |
| Blimp-1 | B lymphocyte-induced maturation protein-1 |
| BTK | Bruton tyrosine kinase t |
| CCL3 | C-C chemokine ligand type 3 |
| CCL4 | C-C chemokine ligand type 4 |
| CCR5 | C-C chemokine receptor type 5 |
| CCR7 | C-C chemokine receptor type 7 |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| CXCL1 | C-X-C motif chemokine type 1 |
| CXCL2 | C-X-C motif chemokine type 2 |
| CXCL9 | C-X-C motif chemokine type 9 |
| CXCL10 | C-X-C motif chemokine type 10 |
| CXCL12 | C-X-C motif chemokine type 12 |
| CXCL13 | C-X-C motif chemokine type 13 |
| CXCR3 | C-X-C motif chemokine receptor type 3 |
| CXCR5 | C-X-C motif chemokine receptor type 5 |
| CCL3 | chemokine (C-C motif) ligand 3 |
| CCL4 | chemokine (C-C motif) ligand 4 |
| CCL19 | chemokine (C-C motif) ligand 19 |
| CCL21 | chemokine (C-C motif) ligand 21 |
| DCs | dendritic cells |
| ECM | extracellular matrix |
| ESSDAI | EULAR Sjögren’s syndrome disease activity index |
| fDCs | follicular dendritic cells |
| FoxP3 | forkhead box protein transcription factor P3 |
| GATA-3 | GATA Binding Protein 3 |
| GITR | glucocorticoid-induced TNFR-related protein |
| HLA | human leukocyte antigen |
| ICAM-1 | intercellular adhesion molecule 1 |
| IC | immune-complexes |
| ICOS | Inducible T-cell COStimulator |
| Id3 | inhibitor of DNA binding 3 |
| Baff | Interferon |
| IFN-γ | interferon gamma |
| IL | Interleukin |
| IL1R1 | interleukin-1 receptor type 1 |
| IL-2Rα | IL-2 receptor α |
| ILC | innate Lymphoid Cells |
| iNKT | invariant natural killer T cells |
| LNs | lymph nodes |
| MAIT | mucosal-associated invariant T cells |
| MALT | mucosa-associated lymphoid tissue |
| MSGs | minor salivary glands |
| MHC-I | major histocompatibility complex class I |
| MHC-II | major histocompatibility complex class II |
| MMPs | Metalloproteases |
| MR1 | MHC-I-related molecule 1 |
| MZ | marginal zone |
| NFAT-2 | nuclear Factor of activated T cells 2 |
| NK | natural killer cells |
| NKT | natural killer T cells |
| NOD | non-obese diabetic |
| PD-1 | programmed death-ligand 1 |
| pDCs | plasmacytoid dendritic cells |
| pSS | primary Sjögren’s syndrome |
| RA | rheumatoid arthritis |
| RORγt | retinoic acid-related orphan receptor gamma t |
| SGs | salivary glands |
| SGECs | salivary glands epithelial cells |
| SLE | systemic lupus erythematosus |
| SS | Sjögren’s syndrome |
| sSS | secondary Sjögren’s syndrome |
| ST2 | suppression of tumorigenicity 2 |
| STAT | signal transducer and activator of transcription |
| TBX21 or T-bet | T-Box Transcription Factor 21 |
| TCR | T cell receptor |
| Tfh | T follicular helper |
| Tfr | follicular regulatory T cells |
| TGFβ | transforming growth factor beta |
| Th1 | type 1 helper cells |
| Th17 | type 17 helper cells |
| TLR | toll-like receptor |
| TNFα | tumor necrosis factor alpha |
| Tregs | T-regulatory cells |
| VCAM-1 | vascular cell adhesion molecule |
| VEGF-C | vascular endothelial growth factor C |
| VEGFR-3 | vascular endothelial growth factor receptor 3 |
| ZO-1 | zonula occludens 1 |
References
- Fox, R.I.; Kang, H.I. Pathogenesis of Sjogren’s syndrome. Rheum. Dis. Clin. N. Am. 1992, 18, 517–538. [Google Scholar]
- Fox, R.I.; Michelson, P. Approaches to the treatment of Sjogren’s syndrome. J. Rheumatol. Suppl. 2000, 61, 15–21. [Google Scholar] [PubMed]
- Basbaum, A.I. Opioid regulation of nociceptive and neuropathic pain. Clin. Neuropharmacol. 1992, 15 Pt A (Suppl. S1), 372A. [Google Scholar] [CrossRef]
- Sullivan, D.A. Sex hormones and Sjogren’s syndrome. J. Rheumatol. Suppl. 1997, 50, 17–32. [Google Scholar] [PubMed]
- Nguyen, C.Q.; Peck, A.B. Unraveling the pathophysiology of Sjogren syndrome-associated dry eye disease. Ocul. Surf. 2009, 7, 11–27. [Google Scholar] [CrossRef]
- Vitali, C.; Bombardieri, S.; Jonsson, R.; Moutsopoulos, H.M.; Alexander, E.L.; Carsons, S.E.; Daniels, T.E.; Fox, P.C.; Fox, R.I.; Kassan, S.S.; et al. Classification criteria for Sjogren’s syndrome: A revised version of the European criteria proposed by the American-European Consensus Group. Ann. Rheum Dis. 2002, 61, 554–558. [Google Scholar] [CrossRef]
- Parisis, D.; Chivasso, C.; Perret, J.; Soyfoo, M.S.; Delporte, C. Current State of Knowledge on Primary Sjogren’s Syndrome, an Autoimmune Exocrinopathy. J. Clin. Med. 2020, 9, 2299. [Google Scholar] [CrossRef]
- Kiripolsky, J.; McCabe, L.G.; Kramer, J.M. Innate immunity in Sjogren’s syndrome. Clin. Immunol. 2017, 182, 4–13. [Google Scholar] [CrossRef]
- Steinman, R.M.; Hemmi, H. Dendritic cells: Translating innate to adaptive immunity. Curr. Top. Microbiol. Immunol. 2006, 311, 17–58. [Google Scholar]
- Gottenberg, J.E.; Cagnard, N.; Lucchesi, C.; Letourneur, F.; Mistou, S.; Lazure, T.; Jacques, S.; Ba, N.; Ittah, M.; Lepajolec, C.; et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjogren’s syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 2770–2775. [Google Scholar] [CrossRef]
- Ozaki, Y.; Ito, T.; Son, Y.; Amuro, H.; Shimamoto, K.; Sugimoto, H.; Katashiba, Y.; Ogata, M.; Miyamoto, R.; Murakami, N.; et al. Decrease of blood dendritic cells and increase of tissue-infiltrating dendritic cells are involved in the induction of Sjogren’s syndrome but not in the maintenance. Clin. Exp. Immunol. 2010, 159, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Wildenberg, M.E.; Welzen-Coppens, J.M.; van Helden-Meeuwsen, C.G.; Bootsma, H.; Vissink, A.; van Rooijen, N.; van de Merwe, J.P.; Drexhage, H.A.; Versnel, M.A. Increased frequency of CD16+ monocytes and the presence of activated dendritic cells in salivary glands in primary Sjogren syndrome. Ann. Rheum. Dis. 2009, 68, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Hillen, M.R.; Pandit, A.; Blokland, S.L.M.; Hartgring, S.A.Y.; Bekker, C.P.J.; van der Heijden, E.H.M.; Servaas, N.H.; Rossato, M.; Kruize, A.A.; van Roon, J.A.G.; et al. Plasmacytoid DCs From Patients With Sjogren’s Syndrome Are Transcriptionally Primed for Enhanced Pro-inflammatory Cytokine Production. Front. Immunol. 2019, 10, 2096. [Google Scholar] [CrossRef]
- Ainola, M.; Porola, P.; Takakubo, Y.; Przybyla, B.; Kouri, V.P.; Tolvanen, T.A.; Hanninen, A.; Nordstrom, D.C. Activation of plasmacytoid dendritic cells by apoptotic particles-mechanism for the loss of immunological tolerance in Sjogren’s syndrome. Clin. Exp. Immunol. 2018, 191, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Colonna, M. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol. Rev. 2010, 234, 142–162. [Google Scholar] [CrossRef]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Vakaloglou, K.M.; Mavragani, C.P. Activation of the type I interferon pathway in primary Sjogren’s syndrome: An update. Curr. Opin. Rheumatol. 2011, 23, 459–464. [Google Scholar] [CrossRef]
- Aloisi, F.; Pujol-Borrell, R. Lymphoid neogenesis in chronic inflammatory diseases. Nat. Rev. Immunol. 2006, 6, 205–217. [Google Scholar] [CrossRef]
- Frank, G.B.; Kokate, T.G. Blockade of high K+ contractures and Ca(+)+-dependent slow action potentials in frog skeletal muscle by opioid drugs. Prog. Clin. Biol. Res. 1990, 328, 283–286. [Google Scholar]
- Stout, R.D.; Jiang, C.; Matta, B.; Tietzel, I.; Watkins, S.K.; Suttles, J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 2005, 175, 342–349. [Google Scholar] [CrossRef]
- Morell, M.; Varela, N.; Maranon, C. Myeloid Populations in Systemic Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2017, 53, 198–218. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, M.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. Characteristics of the minor salivary gland infiltrates in Sjogren’s syndrome. J. Autoimmun. 2010, 34, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.H. Induction, function and regulation of IL-17-producing T cells. Eur. J. Immunol. 2008, 38, 2636–2649. [Google Scholar] [CrossRef] [PubMed]
- Gliozzi, M.; Greenwell-Wild, T.; Jin, W.; Moutsopoulos, N.M.; Kapsogeorgou, E.; Moutsopoulos, H.M.; Wahl, S.M. A link between interferon and augmented plasmin generation in exocrine gland damage in Sjogren’s syndrome. J. Autoimmun. 2013, 40, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.; Grasso, G.; Destro Castaniti, G.M.; Ciccia, F.; Guggino, G. Primary Sjogren Syndrome: Focus on Innate Immune Cells and Inflammation. Vaccines 2020, 8, 272. [Google Scholar] [CrossRef] [PubMed]
- Manoussakis, M.N.; Boiu, S.; Korkolopoulou, P.; Kapsogeorgou, E.K.; Kavantzas, N.; Ziakas, P.; Patsouris, E.; Moutsopoulos, H.M. Rates of infiltration by macrophages and dendritic cells and expression of interleukin-18 and interleukin-12 in the chronic inflammatory lesions of Sjogren’s syndrome: Correlation with certain features of immune hyperactivity and factors associated with high risk of lymphoma development. Arthritis Rheum. 2007, 56, 3977–3988. [Google Scholar] [PubMed]
- Kinoshita, S.; Nakamura, T.; Nishida, K. Pathological keratinization of ocular surface epithelium. Adv. Exp. Med. Biol. 2002, 506 Pt. A, 641–646. [Google Scholar]
- McNamara, N.A. Molecular mechanisms of keratinizing ocular surface disease. Optom Vis. Sci. 2010, 87, 233–238. [Google Scholar] [CrossRef]
- Conti, P.; Stellin, L.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Ronconi, G. Advances in Mast Cell Activation by IL-1 and IL-33 in Sjogren’s Syndrome: Promising Inhibitory Effect of IL-37. Int. J. Mol. Sci. 2020, 21, 4297. [Google Scholar] [CrossRef]
- Leehan, K.M.; Pezant, N.P.; Rasmussen, A.; Grundahl, K.; Moore, J.S.; Radfar, L.; Lewis, D.M.; Stone, D.U.; Lessard, C.J.; Rhodus, N.L.; et al. Minor salivary gland fibrosis in Sjogren’s syndrome is elevated, associated with focus score and not solely a consequence of aging. Clin. Exp. Rheumatol. 2018, 36 (Suppl. S112), 80–88. [Google Scholar]
- Skopouli, F.N.; Li, L.; Boumba, D.; Stefanaki, S.; Hanel, K.; Moutsopoulos, H.M.; Krilis, S.A. Association of mast cells with fibrosis and fatty infiltration in the minor salivary glands of patients with Sjogren’s syndrome. Clin. Exp. Rheumatol. 1998, 16, 63–65. [Google Scholar] [PubMed]
- Xu, D.; Jiang, H.R.; Kewin, P.; Li, Y.; Mu, R.; Fraser, A.R.; Pitman, N.; Kurowska-Stolarska, M.; McKenzie, A.N.; McInnes, I.B.; et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc. Natl. Acad. Sci. USA 2008, 105, 10913–10918. [Google Scholar] [CrossRef] [PubMed]
- de Paula, F.; Teshima, T.H.N.; Hsieh, R.; Souza, M.M.; Nico, M.M.S.; Lourenco, S.V. Overview of Human Salivary Glands: Highlights of Morphology and Developing Processes. Anat. Rec. 2017, 300, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Manoussakis, M.N.; Moutsopoulos, H.M. Sjogren’s syndrome: Autoimmune epithelitis. Baillieres Best Pract Res. Clin. Rheumatol. 2000, 14, 73–95. [Google Scholar] [CrossRef]
- Ewert, P.; Aguilera, S.; Alliende, C.; Kwon, Y.J.; Albornoz, A.; Molina, C.; Urzua, U.; Quest, A.F.; Olea, N.; Perez, P.; et al. Disruption of tight junction structure in salivary glands from Sjogren’s syndrome patients is linked to proinflammatory cytokine exposure. Arthritis Rheum. 2010, 62, 1280–1289. [Google Scholar] [CrossRef]
- Barrera, M.J.; Bahamondes, V.; Sepulveda, D.; Quest, A.F.; Castro, I.; Cortes, J.; Aguilera, S.; Urzua, U.; Molina, C.; Perez, P.; et al. Sjogren’s syndrome and the epithelial target: A comprehensive review. J. Autoimmun. 2013, 42, 7–18. [Google Scholar] [CrossRef]
- Manoussakis, M.N.; Kapsogeorgou, E.K. The role of intrinsic epithelial activation in the pathogenesis of Sjogren’s syndrome. J. Autoimmun. 2010, 35, 219–224. [Google Scholar] [CrossRef]
- Goules, A.V.; Kapsogeorgou, E.K.; Tzioufas, A.G. Insight into pathogenesis of Sjogren’s syndrome: Dissection on autoimmune infiltrates and epithelial cells. Clin. Immunol. 2017, 182, 30–40. [Google Scholar] [CrossRef]
- Hashimoto, A.; Nishikawa, T.; Hayashi, T.; Fujii, N.; Harada, K.; Oka, T.; Takahashi, K. The presence of free D-serine in rat brain. FEBS Lett. 1992, 296, 33–36. [Google Scholar] [CrossRef]
- Manoussakis, M.N.; Spachidou, M.P.; Maratheftis, C.I. Salivary epithelial cells from Sjogren’s syndrome patients are highly sensitive to anoikis induced by TLR-3 ligation. J. Autoimmun. 2010, 35, 212–218. [Google Scholar] [CrossRef]
- Kiripolsky, J.; Kramer, J.M. Current and Emerging Evidence for Toll-Like Receptor Activation in Sjogren’s Syndrome. J. Immunol. Res. 2018, 2018, 1246818. [Google Scholar] [CrossRef] [PubMed]
- Kyriakidis, N.C.; Kapsogeorgou, E.K.; Gourzi, V.C.; Konsta, O.D.; Baltatzis, G.E.; Tzioufas, A.G. Toll-like receptor 3 stimulation promotes Ro52/TRIM21 synthesis and nuclear redistribution in salivary gland epithelial cells, partially via type I interferon pathway. Clin. Exp. Immunol. 2014, 178, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Ping, L.; Zhenjun, L.; Takada, Y.; Sugai, S. Involvement of the interferon-gamma-induced T cell-attracting chemokines, interferon-gamma-inducible 10-kd protein (CXCL10) and monokine induced by interferon-gamma (CXCL9), in the salivary gland lesions of patients with Sjogren’s syndrome. Arthritis Rheum. 2002, 46, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Manoussakis, M.N.; Kapsogeorgou, E.K. The role of epithelial cells in the pathogenesis of Sjogren’s syndrome. Clin. Rev. Allergy Immunol. 2007, 32, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Boumba, D.; Skopouli, F.N.; Moutsopoulos, H.M. Cytokine mRNA expression in the labial salivary gland tissues from patients with primary Sjogren’s syndrome. Br. J. Rheumatol. 1995, 34, 326–333. [Google Scholar] [CrossRef]
- Tsunawaki, S.; Nakamura, S.; Ohyama, Y.; Sasaki, M.; Ikebe-Hiroki, A.; Hiraki, A.; Kadena, T.; Kawamura, E.; Kumamaru, W.; Shinohara, M.; et al. Possible function of salivary gland epithelial cells as nonprofessional antigen-presenting cells in the development of Sjogren’s syndrome. J. Rheumatol. 2002, 29, 1884–1896. [Google Scholar]
- Matsumura, R.; Umemiya, K.; Goto, T.; Nakazawa, T.; Kagami, M.; Tomioka, H.; Tanabe, E.; Sugiyama, T.; Sueishi, M. Glandular and extraglandular expression of costimulatory molecules in patients with Sjogren’s syndrome. Ann. Rheum Dis. 2001, 60, 473–482. [Google Scholar] [CrossRef]
- Kapsogeorgou, E.K.; Dimitriou, I.D.; Abu-Helu, R.F.; Moutsopoulos, H.M.; Manoussakis, M.N. Activation of epithelial and myoepithelial cells in the salivary glands of patients with Sjogren’s syndrome: High expression of intercellular adhesion molecule-1 (ICAM.1) in biopsy specimens and cultured cells. Clin. Exp. Immunol. 2001, 124, 126–133. [Google Scholar] [CrossRef]
- Mitsias, D.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. The role of epithelial cells in the initiation and perpetuation of autoimmune lesions: Lessons from Sjogren’s syndrome (autoimmune epithelitis). Lupus 2006, 15, 255–261. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Mikulowska-Mennis, A.; Xu, B.; Berberian, J.M.; Michie, S.A. Lymphocyte migration to inflamed lacrimal glands is mediated by vascular cell adhesion molecule-1/alpha(4)beta(1) integrin, peripheral node addressin/l-selectin, and lymphocyte function-associated antigen-1 adhesion pathways. Am. J. Pathol. 2001, 159, 671–681. [Google Scholar] [CrossRef]
- Turkcapar, N.; Sak, S.D.; Saatci, M.; Duman, M.; Olmez, U. Vasculitis and expression of vascular cell adhesion molecule-1, intercellular adhesion molecule-1, and E-selectin in salivary glands of patients with Sjogren’s syndrome. J. Rheumatol. 2005, 32, 1063–1070. [Google Scholar]
- Alunno, A.; Ibba-Manneschi, L.; Bistoni, O.; Rosa, I.; Caterbi, S.; Gerli, R.; Manetti, M. Mobilization of lymphatic endothelial precursor cells and lymphatic neovascularization in primary Sjogren’s syndrome. J. Cell Mol. Med. 2016, 20, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Gunn, M.D.; Tangemann, K.; Tam, C.; Cyster, J.G.; Rosen, S.D.; Williams, L.T. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc. Natl. Acad. Sci. USA 1998, 95, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Toubal, A.; Nel, I.; Lotersztajn, S.; Lehuen, A. Mucosal-associated invariant T cells and disease. Nat. Rev. Immunol. 2019, 19, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.C.; McLaren, J.E.; Reistetter, J.A.; Smyk-Pearson, S.; Ladell, K.; Swarbrick, G.M.; Yu, Y.Y.; Hansen, T.H.; Lund, O.; Nielsen, M.; et al. MR1-restricted MAIT cells display ligand discrimination and pathogen selectivity through distinct T cell receptor usage. J. Exp. Med. 2014, 211, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Macardle, C.; Weedon, H.; Beroukas, D.; Banovic, T. Mucosal-associated invariant T cells are reduced and functionally immature in the peripheral blood of primary Sjogren’s syndrome patients. Eur. J. Immunol. 2016, 46, 2444–2453. [Google Scholar] [CrossRef]
- Izumi, Y.; Ida, H.; Huang, M.; Iwanaga, N.; Tanaka, F.; Aratake, K.; Arima, K.; Tamai, M.; Kamachi, M.; Nakamura, H.; et al. Characterization of peripheral natural killer cells in primary Sjogren’s syndrome: Impaired NK cell activity and low NK cell number. J. Lab. Clin. Med. 2006, 147, 242–249. [Google Scholar] [CrossRef]
- Ferlazzo, G.; Tsang, M.L.; Moretta, L.; Melioli, G.; Steinman, R.M.; Munz, C. Human dendritic cells activate resting natural killer (NK) cells and are recognized via the NKp30 receptor by activated NK cells. J. Exp. Med. 2002, 195, 343–351. [Google Scholar] [CrossRef]
- Rusakiewicz, S.; Nocturne, G.; Lazure, T.; Semeraro, M.; Flament, C.; Caillat-Zucman, S.; Sene, D.; Delahaye, N.; Vivier, E.; Chaba, K.; et al. NCR3/NKp30 contributes to pathogenesis in primary Sjogren’s syndrome. Sci. Transl. Med. 2013, 5, 195ra96. [Google Scholar] [CrossRef]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Ferrante, A.; Raimondo, S.; Giardina, A.; Dieli, F.; Campisi, G.; Alessandro, R.; Triolo, G. Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with Sjogren’s syndrome. Ann. Rheum Dis. 2012, 71, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Bendelac, A.; Savage, P.B.; Teyton, L. The biology of NKT cells. Annu. Rev. Immunol. 2007, 25, 297–336. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T. The Hunt for the Source of Primary Interleukin-4: How We Discovered That Natural Killer T Cells and Basophils Determine T Helper Type 2 Cell Differentiation In Vivo. Front. Immunol. 2018, 9, 716. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.; La Barbera, L.; Lo Pizzo, M.; Ciccia, F.; Sireci, G.; Guggino, G. Invariant NKT Cells and Rheumatic Disease: Focus on Primary Sjogren Syndrome. Int. J. Mol. Sci. 2019, 20, 5435. [Google Scholar] [CrossRef] [PubMed]
- Sudzius, G.; Mieliauskaite, D.; Siaurys, A.; Viliene, R.; Butrimiene, I.; Characiejus, D.; Dumalakiene, I. Distribution of Peripheral Lymphocyte Populations in Primary Sjogren’s Syndrome Patients. J. Immunol. Res. 2015, 2015, 854706. [Google Scholar] [CrossRef] [PubMed]
- Guggino, G.; Ciccia, F.; Raimondo, S.; Giardina, G.; Alessandro, R.; Dieli, F.; Sireci, G.; Triolo, G. Invariant NKT cells are expanded in peripheral blood but are undetectable in salivary glands of patients with primary Sjogren’s syndrome. Clin. Exp. Rheumatol. 2016, 34, 25–31. [Google Scholar]
- Yang, J.Q.; Wen, X.; Kim, P.J.; Singh, R.R. Invariant NKT cells inhibit autoreactive B cells in a contact- and CD1d-dependent manner. J. Immunol. 2011, 186, 1512–1520. [Google Scholar] [CrossRef]
- Zook, E.C.; Kee, B.L. Development of innate lymphoid cells. Nat. Immunol. 2016, 17, 775–782. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef]
- Ebbo, M.; Crinier, A.; Vely, F.; Vivier, E. Innate lymphoid cells: Major players in inflammatory diseases. Nat. Rev. Immunol. 2017, 17, 665–678. [Google Scholar] [CrossRef]
- Shikhagaie, M.M.; Germar, K.; Bal, S.M.; Ros, X.R.; Spits, H. Innate lymphoid cells in autoimmunity: Emerging regulators in rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Wenink, M.H.; Leijten, E.F.A.; Cupedo, T.; Radstake, T. Review: Innate Lymphoid Cells: Sparking Inflammatory Rheumatic Disease? Arthritis Rheumatol. 2017, 69, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Blokland, S.L.M.; van den Hoogen, L.L.; Leijten, E.F.A.; Hartgring, S.A.Y.; Fritsch, R.; Kruize, A.A.; van Roon, J.A.G.; Radstake, T. Increased expression of Fas on group 2 and 3 innate lymphoid cells is associated with an interferon signature in systemic lupus erythematosus and Sjogren’s syndrome. Rheumatology 2019, 58, 1740–1745. [Google Scholar] [CrossRef] [PubMed]
- Maazi, H.; Banie, H.; Aleman Muench, G.R.; Patel, N.; Wang, B.; Sankaranarayanan, I.; Bhargava, V.; Sato, T.; Lewis, G.; Cesaroni, M.; et al. Activated plasmacytoid dendritic cells regulate type 2 innate lymphoid cell-mediated airway hyperreactivity. J. Allergy Clin. Immunol. 2018, 141, 893–905 e6. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, L.; Zhao, J.; Li, G.; Zhang, L.; Chen, W.; Nie, W. Plasmacytoid dendritic cells promote HIV-1-induced group 3 innate lymphoid cell depletion. J. Clin. Invest. 2015, 125, 3692–3703. [Google Scholar] [CrossRef]
- Duerr, C.U.; McCarthy, C.D.; Mindt, B.C.; Rubio, M.; Meli, A.P.; Pothlichet, J.; Eva, M.M.; Gauchat, J.F.; Qureshi, S.T.; Mazer, B.D.; et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat. Immunol. 2016, 17, 65–75. [Google Scholar] [CrossRef]
- Skopouli, F.N.; Fox, P.C.; Galanopoulou, V.; Atkinson, J.C.; Jaffe, E.S.; Moutsopoulos, H.M. T cell subpopulations in the labial minor salivary gland histopathologic lesion of Sjogren’s syndrome. J. Rheumatol. 1991, 18, 210–214. [Google Scholar]
- Hooks, J.J.; Moutsopoulos, H.M.; Geis, S.A.; Stahl, N.I.; Decker, J.L.; Notkins, A.L. Immune interferon in the circulation of patients with autoimmune disease. N. Engl. J. Med. 1979, 301, 5–8. [Google Scholar] [CrossRef]
- Youinou, P.; Pers, J.O. Disturbance of cytokine networks in Sjogren’s syndrome. Arthritis Res. Ther. 2011, 13, 227. [Google Scholar] [CrossRef]
- van Woerkom, J.M.; Kruize, A.A.; Wenting-van Wijk, M.J.; Knol, E.; Bihari, I.C.; Jacobs, J.W.; Bijlsma, J.W.; Lafeber, F.P.; van Roon, J.A. Salivary gland and peripheral blood T helper 1 and 2 cell activity in Sjogren’s syndrome compared with non-Sjogren’s sicca syndrome. Ann. Rheum Dis. 2005, 64, 1474–1479. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Coffman, R.L. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 1989, 7, 145–173. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Dudakov, J.A.; Hanash, A.M.; van den Brink, M.R. Interleukin-22: Immunobiology and pathology. Annu. Rev. Immunol. 2015, 33, 747–785. [Google Scholar] [CrossRef]
- Abusleme, L.; Moutsopoulos, N.M. IL-17: Overview and role in oral immunity and microbiome. Oral Dis. 2017, 23, 854–865. [Google Scholar] [CrossRef]
- Curtis, M.M.; Way, S.S. Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology 2009, 126, 177–185. [Google Scholar] [CrossRef]
- Khader, S.A.; Gaffen, S.L.; Kolls, J.K. Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol. 2009, 2, 403–411. [Google Scholar] [CrossRef]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef]
- Dong, C. Genetic controls of Th17 cell differentiation and plasticity. Exp. Mol. Med. 2011, 43, 1–6. [Google Scholar] [CrossRef]
- Yang, X.O.; Pappu, B.P.; Nurieva, R.; Akimzhanov, A.; Kang, H.S.; Chung, Y.; Ma, L.; Shah, B.; Panopoulos, A.D.; Schluns, K.S.; et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 2008, 28, 29–39. [Google Scholar] [CrossRef]
- Kuestner, R.E.; Taft, D.W.; Haran, A.; Brandt, C.S.; Brender, T.; Lum, K.; Harder, B.; Okada, S.; Ostrander, C.D.; Kreindler, J.L.; et al. Identification of the IL-17 receptor related molecule IL-17RC as the receptor for IL-17F. J. Immunol. 2007, 179, 5462–5473. [Google Scholar] [CrossRef] [PubMed]
- Katsifis, G.E.; Rekka, S.; Moutsopoulos, N.M.; Pillemer, S.; Wahl, S.M. Systemic and local interleukin-17 and linked cytokines associated with Sjogren’s syndrome immunopathogenesis. Am. J. Pathol. 2009, 175, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gao, C.; Chen, H.; Li, Y.; Jin, Y.; Qi, H. Analysis of Th17-associated cytokines and clinical correlations in patients with dry eye disease. PLoS ONE 2017, 12, e0173301. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.Q.; Hu, M.H.; Li, Y.; Stewart, C.; Peck, A.B. Salivary gland tissue expression of interleukin-23 and interleukin-17 in Sjogren’s syndrome: Findings in humans and mice. Arthritis Rheum. 2008, 58, 734–743. [Google Scholar] [CrossRef]
- Sakai, A.; Sugawara, Y.; Kuroishi, T.; Sasano, T.; Sugawara, S. Identification of IL-18 and Th17 cells in salivary glands of patients with Sjogren’s syndrome, and amplification of IL-17-mediated secretion of inflammatory cytokines from salivary gland cells by IL-18. J. Immunol. 2008, 181, 2898–2906. [Google Scholar] [CrossRef]
- Goenka, R.; Barnett, L.G.; Silver, J.S.; O’Neill, P.J.; Hunter, C.A.; Cancro, M.P.; Laufer, T.M. Cutting edge: Dendritic cell-restricted antigen presentation initiates the follicular helper T cell program but cannot complete ultimate effector differentiation. J. Immunol. 2011, 187, 1091–1095. [Google Scholar] [CrossRef]
- Chen, M.; Guo, Z.; Ju, W.; Ryffel, B.; He, X.; Zheng, S.G. The development and function of follicular helper T cells in immune responses. Cell. Mol. Immunol. 2012, 9, 375–379. [Google Scholar] [CrossRef]
- Kim, C.H.; Rott, L.S.; Clark-Lewis, I.; Campbell, D.J.; Wu, L.; Butcher, E.C. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center-localized subset of CXCR5+ T cells. J. Exp. Med. 2001, 193, 1373–1381. [Google Scholar] [CrossRef]
- Arnold, C.N.; Campbell, D.J.; Lipp, M.; Butcher, E.C. The germinal center response is impaired in the absence of T cell-expressed CXCR5. Eur. J. Immunol. 2007, 37, 100–109. [Google Scholar] [CrossRef]
- Hardtke, S.; Ohl, L.; Forster, R. Balanced expression of CXCR5 and CCR7 on follicular T helper cells determines their transient positioning to lymph node follicles and is essential for efficient B-cell help. Blood 2005, 106, 1924–1931. [Google Scholar] [CrossRef]
- Haynes, N.M.; Allen, C.D.; Lesley, R.; Ansel, K.M.; Killeen, N.; Cyster, J.G. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J. Immunol. 2007, 179, 5099–5108. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 2011, 29, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Szabo, K.; Papp, G.; Barath, S.; Gyimesi, E.; Szanto, A.; Zeher, M. Follicular helper T cells may play an important role in the severity of primary Sjogren’s syndrome. Clin. Immunol. 2013, 147, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Yu, D.; Li, X.; Yu, N.; Li, X.; Wang, Y.; Wang, Y. CD4+CXCR5+ follicular helper T cells in salivary gland promote B cells maturation in patients with primary Sjogren’s syndrome. Int. J. Clin. Exp. Pathol. 2014, 7, 1988–1996. [Google Scholar] [PubMed]
- Verstappen, G.M.; Meiners, P.M.; Corneth, O.B.J.; Visser, A.; Arends, S.; Abdulahad, W.H.; Hendriks, R.W.; Vissink, A.; Kroese, F.G.M.; Bootsma, H. Attenuation of Follicular Helper T Cell-Dependent B Cell Hyperactivity by Abatacept Treatment in Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2017, 69, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Cosorich, I.; McGuire, H.M.; Warren, J.; Danta, M.; King, C. CCR9 Expressing T Helper and T Follicular Helper Cells Exhibit Site-Specific Identities During Inflammatory Disease. Front. Immunol. 2018, 9, 2899. [Google Scholar] [CrossRef]
- McGuire, H.M.; Vogelzang, A.; Ma, C.S.; Hughes, W.E.; Silveira, P.A.; Tangye, S.G.; Christ, D.; Fulcher, D.; Falcone, M.; King, C. A subset of interleukin-21+ chemokine receptor CCR9+ T helper cells target accessory organs of the digestive system in autoimmunity. Immunity 2011, 34, 602–615. [Google Scholar] [CrossRef]
- Blokland, S.L.M.; Hillen, M.R.; Kruize, A.A.; Meller, S.; Homey, B.; Smithson, G.M.; Radstake, T.; van Roon, J.A.G. Increased CCL25 and T Helper Cells Expressing CCR9 in the Salivary Glands of Patients with Primary Sjogren’s Syndrome: Potential New Axis in Lymphoid Neogenesis. Arthritis Rheumatol. 2017, 69, 2038–2051. [Google Scholar] [CrossRef]
- Schwartz, R.H. Natural regulatory T cells and self-tolerance. Nat. Immunol. 2005, 6, 327–330. [Google Scholar] [CrossRef]
- von Boehmer, H. Mechanisms of suppression by suppressor T cells. Nat. Immunol. 2005, 6, 338–344. [Google Scholar] [CrossRef]
- Sakaguchi, S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat. Immunol. 2005, 6, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Lan, R.Y.; Ansari, A.A.; Lian, Z.X.; Gershwin, M.E. Regulatory T cells: Development, function and role in autoimmunity. Autoimmun. Rev. 2005, 4, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Wraith, D.C.; Nicolson, K.S.; Whitley, N.T. Regulatory CD4+ T cells and the control of autoimmune disease. Curr. Opin. Immunol. 2004, 16, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Rasmussen, J.P.; Rudensky, A.Y. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 2007, 8, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Noack, M.; Miossec, P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev. 2014, 13, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Gottenberg, J.E.; Lavie, F.; Abbed, K.; Gasnault, J.; Le Nevot, E.; Delfraissy, J.F.; Taoufik, Y.; Mariette, X. CD4 CD25high regulatory T cells are not impaired in patients with primary Sjogren’s syndrome. J. Autoimmun. 2005, 24, 235–242. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Qian, L.; Wang, G.; Zhang, H.; Wang, X.; Chen, K.; Zhai, Z.; Li, Q.; Wang, Y.; et al. T regulatory cells are markedly diminished in diseased salivary glands of patients with primary Sjogren’s syndrome. J. Rheumatol. 2007, 34, 2438–2445. [Google Scholar]
- Liu, M.F.; Lin, L.H.; Weng, C.T.; Weng, M.Y. Decreased CD4+CD25+bright T cells in peripheral blood of patients with primary Sjogren’s syndrome. Lupus 2008, 17, 34–39. [Google Scholar] [CrossRef]
- Christodoulou, M.I.; Kapsogeorgou, E.K.; Moutsopoulos, N.M.; Moutsopoulos, H.M. Foxp3+ T-regulatory cells in Sjogren’s syndrome: Correlation with the grade of the autoimmune lesion and certain adverse prognostic factors. Am. J. Pathol. 2008, 173, 1389–1396. [Google Scholar] [CrossRef]
- Szodoray, P.; Papp, G.; Horvath, I.F.; Barath, S.; Sipka, S.; Nakken, B.; Zeher, M. Cells with regulatory function of the innate and adaptive immune system in primary Sjogren’s syndrome. Clin. Exp. Immunol. 2009, 157, 343–349. [Google Scholar] [CrossRef]
- Sarigul, M.; Yazisiz, V.; Bassorgun, C.I.; Ulker, M.; Avci, A.B.; Erbasan, F.; Gelen, T.; Gorczynski, R.M.; Terzioglu, E. The numbers of Foxp3 + Treg cells are positively correlated with higher grade of infiltration at the salivary glands in primary Sjogren’s syndrome. Lupus 2010, 19, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Furuzawa-Carballeda, J.; Hernandez-Molina, G.; Lima, G.; Rivera-Vicencio, Y.; Ferez-Blando, K.; Llorente, L. Peripheral regulatory cells immunophenotyping in primary Sjogren’s syndrome: A cross-sectional study. Arthritis Res. Ther. 2013, 15, R68. [Google Scholar] [CrossRef] [PubMed]
- Alunno, A.; Petrillo, M.G.; Nocentini, G.; Bistoni, O.; Bartoloni, E.; Caterbi, S.; Bianchini, R.; Baldini, C.; Nicoletti, I.; Riccardi, C.; et al. Characterization of a new regulatory CD4+ T cell subset in primary Sjogren’s syndrome. Rheumatology 2013, 52, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Alunno, A.; Nocentini, G.; Bistoni, O.; Petrillo, M.G.; Bartoloni Bocci, E.; Ronchetti, S.; Lo Vaglio, E.; Riccardi, C.; Gerli, R. Expansion of CD4+CD25-GITR+ regulatory T-cell subset in the peripheral blood of patients with primary Sjogren’s syndrome: Correlation with disease activity. Reumatismo 2012, 64, 293–298. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Linterman, M.A.; Pierson, W.; Lee, S.K.; Kallies, A.; Kawamoto, S.; Rayner, T.F.; Srivastava, M.; Divekar, D.P.; Beaton, L.; Hogan, J.J.; et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011, 17, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Wollenberg, I.; Agua-Doce, A.; Hernandez, A.; Almeida, C.; Oliveira, V.G.; Faro, J.; Graca, L. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J. Immunol. 2011, 187, 4553–4560. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.; Tanaka, S.; Chu, F.; Nurieva, R.I.; Martinez, G.J.; Rawal, S.; Wang, Y.H.; Lim, H.; Reynolds, J.M.; Zhou, X.H.; et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 2011, 17, 983–988. [Google Scholar] [CrossRef]
- Gerner, M.Y.; Torabi-Parizi, P.; Germain, R.N. Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity 2015, 42, 172–185. [Google Scholar] [CrossRef]
- Kerfoot, S.M.; Yaari, G.; Patel, J.R.; Johnson, K.L.; Gonzalez, D.G.; Kleinstein, S.H.; Haberman, A.M. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity 2011, 34, 947–960. [Google Scholar] [CrossRef]
- Sage, P.T.; Paterson, A.M.; Lovitch, S.B.; Sharpe, A.H. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity 2014, 41, 1026–1039. [Google Scholar] [CrossRef]
- Vaeth, M.; Eckstein, M.; Shaw, P.J.; Kozhaya, L.; Yang, J.; Berberich-Siebelt, F.; Clancy, R.; Unutmaz, D.; Feske, S. Store-Operated Ca(2+) Entry in Follicular T Cells Controls Humoral Immune Responses and Autoimmunity. Immunity 2016, 44, 1350–1364. [Google Scholar] [CrossRef] [PubMed]
- Sage, P.T.; Alvarez, D.; Godec, J.; von Andrian, U.H.; Sharpe, A.H. Circulating T follicular regulatory and helper cells have memory-like properties. J. Clin. Invest. 2014, 124, 5191–5204. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Liu, X.; Lin, X.; Feng, H.; Sun, L.; Li, S.; Chen, H.; Tang, H.; Lu, L.; Jin, W.; et al. Deficiency in T follicular regulatory cells promotes autoimmunity. J. Exp. Med. 2018, 215, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Verstappen, G.M.; Nakshbandi, U.; Mossel, E.; Haacke, E.A.; van der Vegt, B.; Vissink, A.; Bootsma, H.; Kroese, F.G.M. Is the T Follicular Regulatory:Follicular Helper T Cell Ratio in Blood a Biomarker for Ectopic Lymphoid Structure Formation in Sjogren’s Syndrome? Comment on the Article by Fonseca et al. Arthritis Rheumatol. 2018, 70, 1354–1355. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, V.R.; Romao, V.C.; Agua-Doce, A.; Santos, M.; Lopez-Presa, D.; Ferreira, A.C.; Fonseca, J.E.; Graca, L. The Ratio of Blood T Follicular Regulatory Cells to T Follicular Helper Cells Marks Ectopic Lymphoid Structure Formation While Activated Follicular Helper T Cells Indicate Disease Activity in Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2018, 70, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, S.; Suzuki, K.; Nishikawa, A.; Kassai, Y.; Takiguchi, M.; Kurisu, R.; Okuzono, Y.; Miyazaki, T.; Takeshita, M.; Yoshimoto, K.; et al. Multiomic disease signatures converge to cytotoxic CD8 T cells in primary Sjogren’s syndrome. Ann. Rheum Dis. 2017, 76, 1458–1466. [Google Scholar] [CrossRef]
- Zhou, J.; Yu, Q. Disruption of CXCR3 function impedes the development of Sjogren’s syndrome-like xerostomia in non-obese diabetic mice. Lab. Invest. 2018, 98, 620–628. [Google Scholar] [CrossRef]
- Barr, J.Y.; Wang, X.; Meyerholz, D.K.; Lieberman, S.M. CD8 T cells contribute to lacrimal gland pathology in the nonobese diabetic mouse model of Sjogren syndrome. Immunol. Cell Biol. 2017, 95, 684–694. [Google Scholar] [CrossRef]
- Brandt, D.; Hedrich, C.M. TCRalphabeta(+)CD3(+)CD4(-)CD8(-) (double negative) T cells in autoimmunity. Autoimmun. Rev. 2018, 17, 422–430. [Google Scholar] [CrossRef]
- Gao, Y.; Williams, A.P. Role of Innate T Cells in Anti-Bacterial Immunity. Front Immunol. 2015, 6, 302. [Google Scholar] [CrossRef]
- Hansen, A.; Lipsky, P.E.; Dorner, T. B cells in Sjogren’s syndrome: Indications for disturbed selection and differentiation in ectopic lymphoid tissue. Arthritis Res. Ther. 2007, 9, 218. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.; Reiter, K.; Ziprian, T.; Jacobi, A.; Hoffmann, A.; Gosemann, M.; Scholze, J.; Lipsky, P.E.; Dorner, T. Dysregulation of chemokine receptor expression and function by B cells of patients with primary Sjogren’s syndrome. Arthritis Rheum. 2005, 52, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Kroese, F.G.; Abdulahad, W.H.; Haacke, E.; Bos, N.A.; Vissink, A.; Bootsma, H. B-cell hyperactivity in primary Sjogren’s syndrome. Expert Rev. Clin. Immunol. 2014, 10, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Scheid, J.F.; Mouquet, H.; Kofer, J.; Yurasov, S.; Nussenzweig, M.C.; Wardemann, H. Differential regulation of self-reactivity discriminates between IgG+ human circulating memory B cells and bone marrow plasma cells. Proc. Natl. Acad. Sci. USA 2011, 108, 18044–18048. [Google Scholar] [CrossRef]
- Mouquet, H.; Nussenzweig, M.C. Polyreactive antibodies in adaptive immune responses to viruses. Cell. Mol. Life Sci. 2012, 69, 1435–1445. [Google Scholar] [CrossRef]
- Hayakawa, I.; Tedder, T.F.; Zhuang, Y. B-lymphocyte depletion ameliorates Sjogren’s syndrome in Id3 knockout mice. Immunology 2007, 122, 73–79. [Google Scholar] [CrossRef]
- Nocturne, G.; Mariette, X. Advances in understanding the pathogenesis of primary Sjogren’s syndrome. Nat. Rev. Rheumatol. 2013, 9, 544–556. [Google Scholar] [CrossRef]
- Puga, I.; Cols, M.; Barra, C.M.; He, B.; Cassis, L.; Gentile, M.; Comerma, L.; Chorny, A.; Shan, M.; Xu, W.; et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat. Immunol. 2011, 13, 170–180. [Google Scholar] [CrossRef]
- Szabo, K.; Papp, G.; Szanto, A.; Tarr, T.; Zeher, M. A comprehensive investigation on the distribution of circulating follicular T helper cells and B cell subsets in primary Sjogren’s syndrome and systemic lupus erythematosus. Clin. Exp. Immunol. 2016, 183, 76–89. [Google Scholar] [CrossRef]
- Corneth, O.B.J.; Verstappen, G.M.P.; Paulissen, S.M.J.; de Bruijn, M.J.W.; Rip, J.; Lukkes, M.; van Hamburg, J.P.; Lubberts, E.; Bootsma, H.; Kroese, F.G.M.; et al. Enhanced Bruton’s Tyrosine Kinase Activity in Peripheral Blood B Lymphocytes from Patients with Autoimmune Disease. Arthritis Rheumatol. 2017, 69, 1313–1324. [Google Scholar] [CrossRef]
- Hansen, A.; Odendahl, M.; Reiter, K.; Jacobi, A.M.; Feist, E.; Scholze, J.; Burmester, G.R.; Lipsky, P.E.; Dorner, T. Diminished peripheral blood memory B cells and accumulation of memory B cells in the salivary glands of patients with Sjogren’s syndrome. Arthritis Rheum. 2002, 46, 2160–2171. [Google Scholar] [CrossRef] [PubMed]
- Mingueneau, M.; Boudaoud, S.; Haskett, S.; Reynolds, T.L.; Nocturne, G.; Norton, E.; Zhang, X.; Constant, M.; Park, D.; Wang, W.; et al. Cytometry by time-of-flight immunophenotyping identifies a blood Sjogren’s signature correlating with disease activity and glandular inflammation. J. Allergy Clin. Immunol. 2016, 137, 1809–1821.e12. [Google Scholar] [CrossRef] [PubMed]
- Daridon, C.; Pers, J.O.; Devauchelle, V.; Martins-Carvalho, C.; Hutin, P.; Pennec, Y.L.; Saraux, A.; Youinou, P. Identification of transitional type II B cells in the salivary glands of patients with Sjogren’s syndrome. Arthritis Rheum. 2006, 54, 2280–2288. [Google Scholar] [CrossRef]
- Fletcher, C.A.; Sutherland, A.P.; Groom, J.R.; Batten, M.L.; Ng, L.G.; Gommerman, J.; Mackay, F. Development of nephritis but not sialadenitis in autoimmune-prone BAFF transgenic mice lacking marginal zone B cells. Eur. J. Immunol. 2006, 36, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.Q.; Kim, H.; Cornelius, J.G.; Peck, A.B. Development of Sjogren’s syndrome in nonobese diabetic-derived autoimmune-prone C57BL/6.NOD-Aec1Aec2 mice is dependent on complement component-3. J. Immunol. 2007, 179, 2318–2329. [Google Scholar] [CrossRef]
- Shen, L.; Zhang, C.; Wang, T.; Brooks, S.; Ford, R.J.; Lin-Lee, Y.C.; Kasianowicz, A.; Kumar, V.; Martin, L.; Liang, P.; et al. Development of autoimmunity in IL-14alpha-transgenic mice. J. Immunol. 2006, 177, 5676–5686. [Google Scholar] [CrossRef]
- Shen, L.; Gao, C.; Suresh, L.; Xian, Z.; Song, N.; Chaves, L.D.; Yu, M.; Ambrus, J.L., Jr. Central role for marginal zone B cells in an animal model of Sjogren’s syndrome. Clin. Immunol. 2016, 168, 30–36. [Google Scholar] [CrossRef]
- Nocturne, G.; Mariette, X. Sjogren Syndrome-associated lymphomas: An update on pathogenesis and management. Br. J. Haematol. 2015, 168, 317–327. [Google Scholar] [CrossRef]
- Fogel, O.; Riviere, E.; Seror, R.; Nocturne, G.; Boudaoud, S.; Ly, B.; Gottenberg, J.E.; Le Guern, V.; Dubost, J.J.; Nititham, J.; et al. Role of the IL-12/IL-35 balance in patients with Sjogren syndrome. J. Allergy Clin. Immunol. 2018, 142, 258–268.e5. [Google Scholar] [CrossRef]
- Mariette, X.; Roux, S.; Zhang, J.; Bengoufa, D.; Lavie, F.; Zhou, T.; Kimberly, R. The level of BLyS (BAFF) correlates with the titre of autoantibodies in human Sjogren’s syndrome. Ann. Rheum. Dis. 2003, 62, 168–171. [Google Scholar] [CrossRef]
- Goenka, R.; Scholz, J.L.; Sindhava, V.J.; Cancro, M.P. New roles for the BLyS/BAFF family in antigen-experienced B cell niches. Cytokine Growth Factor Rev. 2014, 25, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Zhang, W.; Haskett, S.; Pellerin, A.; Xu, S.; Petersen, B.; Jandreski, L.; Hamann, S.; Reynolds, T.L.; Zheng, T.S.; et al. BAFF overexpression increases lymphocytic infiltration in Sjogren’s target tissue, but only inefficiently promotes ectopic B-cell differentiation. Clin. Immunol. 2016, 169, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Risselada, A.P.; Looije, M.F.; Kruize, A.A.; Bijlsma, J.W.; van Roon, J.A. The role of ectopic germinal centers in the immunopathology of primary Sjogren’s syndrome: A systematic review. Semin. Arthritis Rheum. 2013, 42, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Bombardieri, M.; Lewis, M.; Pitzalis, C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat. Rev. Rheumatol. 2017, 13, 141–154. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chivasso, C.; Sarrand, J.; Perret, J.; Delporte, C.; Soyfoo, M.S. The Involvement of Innate and Adaptive Immunity in the Initiation and Perpetuation of Sjögren’s Syndrome. Int. J. Mol. Sci. 2021, 22, 658. https://doi.org/10.3390/ijms22020658
Chivasso C, Sarrand J, Perret J, Delporte C, Soyfoo MS. The Involvement of Innate and Adaptive Immunity in the Initiation and Perpetuation of Sjögren’s Syndrome. International Journal of Molecular Sciences. 2021; 22(2):658. https://doi.org/10.3390/ijms22020658
Chicago/Turabian StyleChivasso, Clara, Julie Sarrand, Jason Perret, Christine Delporte, and Muhammad Shahnawaz Soyfoo. 2021. "The Involvement of Innate and Adaptive Immunity in the Initiation and Perpetuation of Sjögren’s Syndrome" International Journal of Molecular Sciences 22, no. 2: 658. https://doi.org/10.3390/ijms22020658
APA StyleChivasso, C., Sarrand, J., Perret, J., Delporte, C., & Soyfoo, M. S. (2021). The Involvement of Innate and Adaptive Immunity in the Initiation and Perpetuation of Sjögren’s Syndrome. International Journal of Molecular Sciences, 22(2), 658. https://doi.org/10.3390/ijms22020658