Anti-Warburg Effect of Melatonin: A Proposed Mechanism to Explain its Inhibition of Multiple Diseases



Abstract
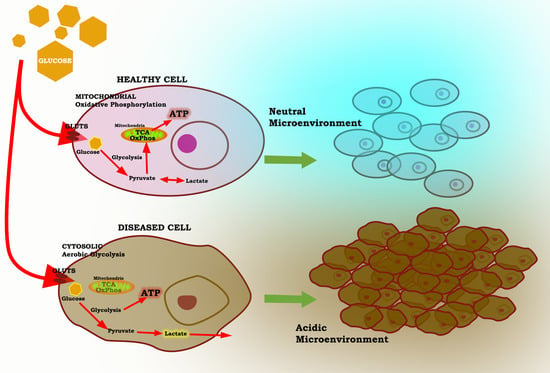
:
1. Introduction
2. Mitochondria, An Ecosystem in Which Melatonin is Produced and Functions
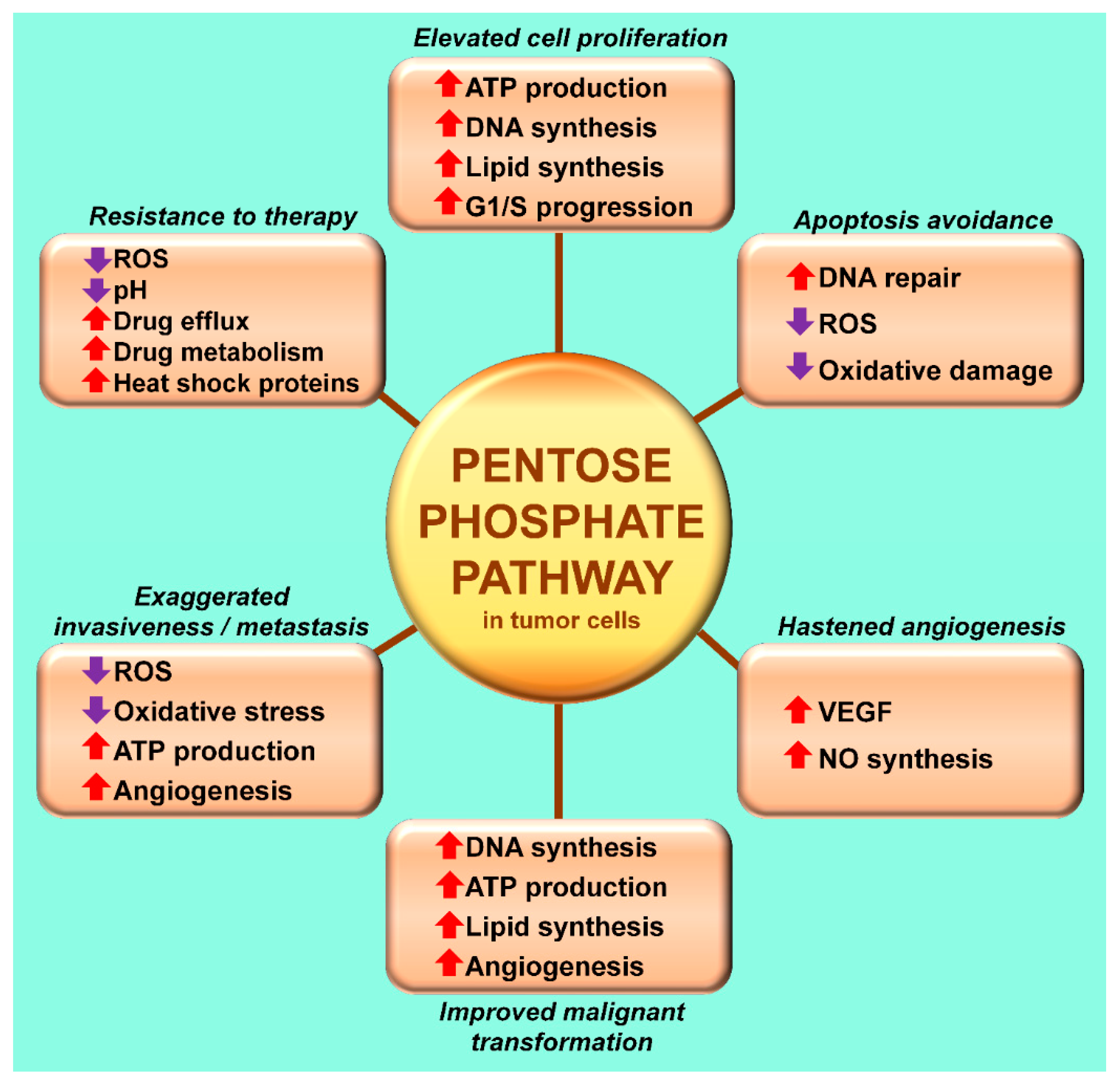
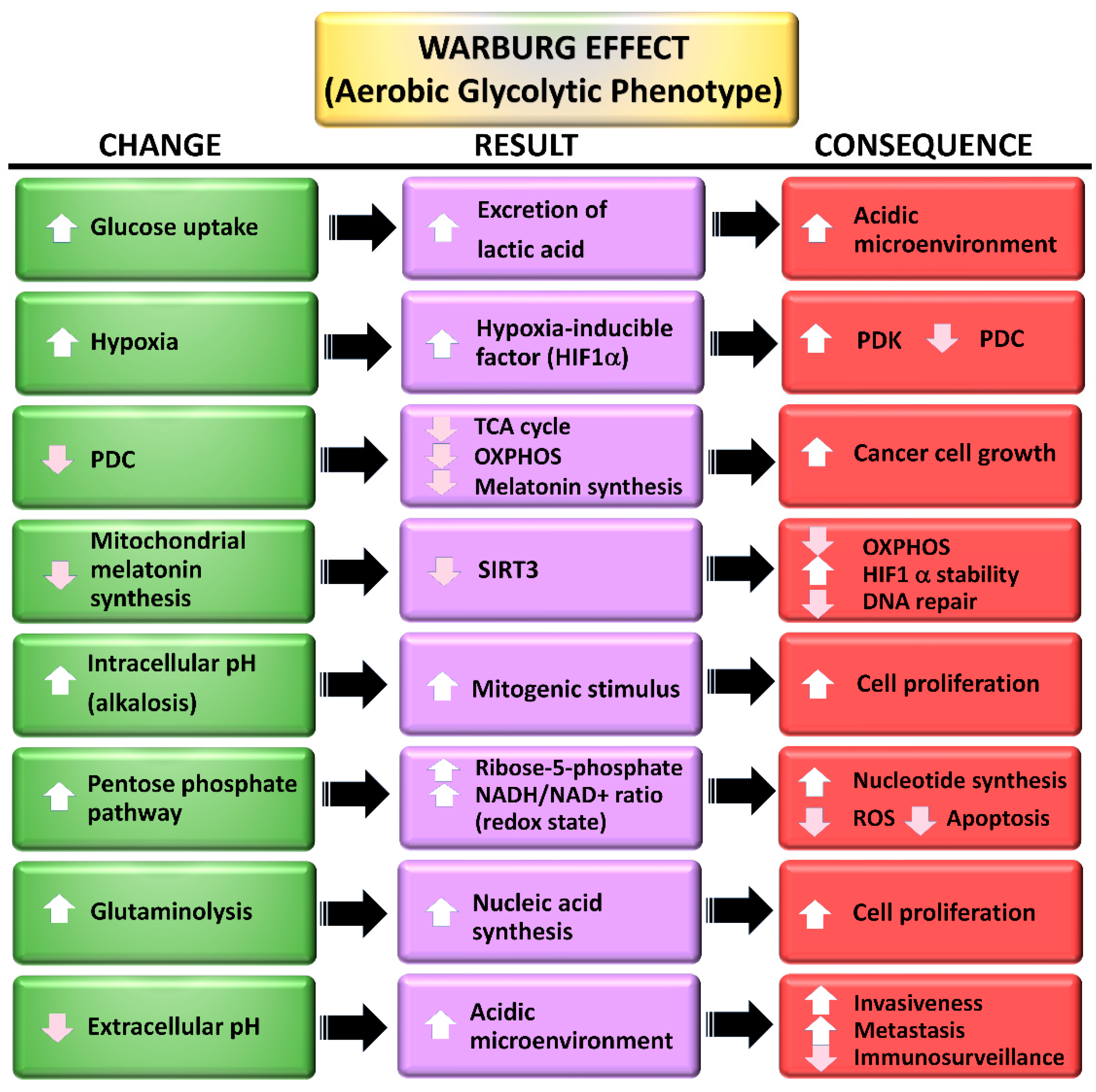
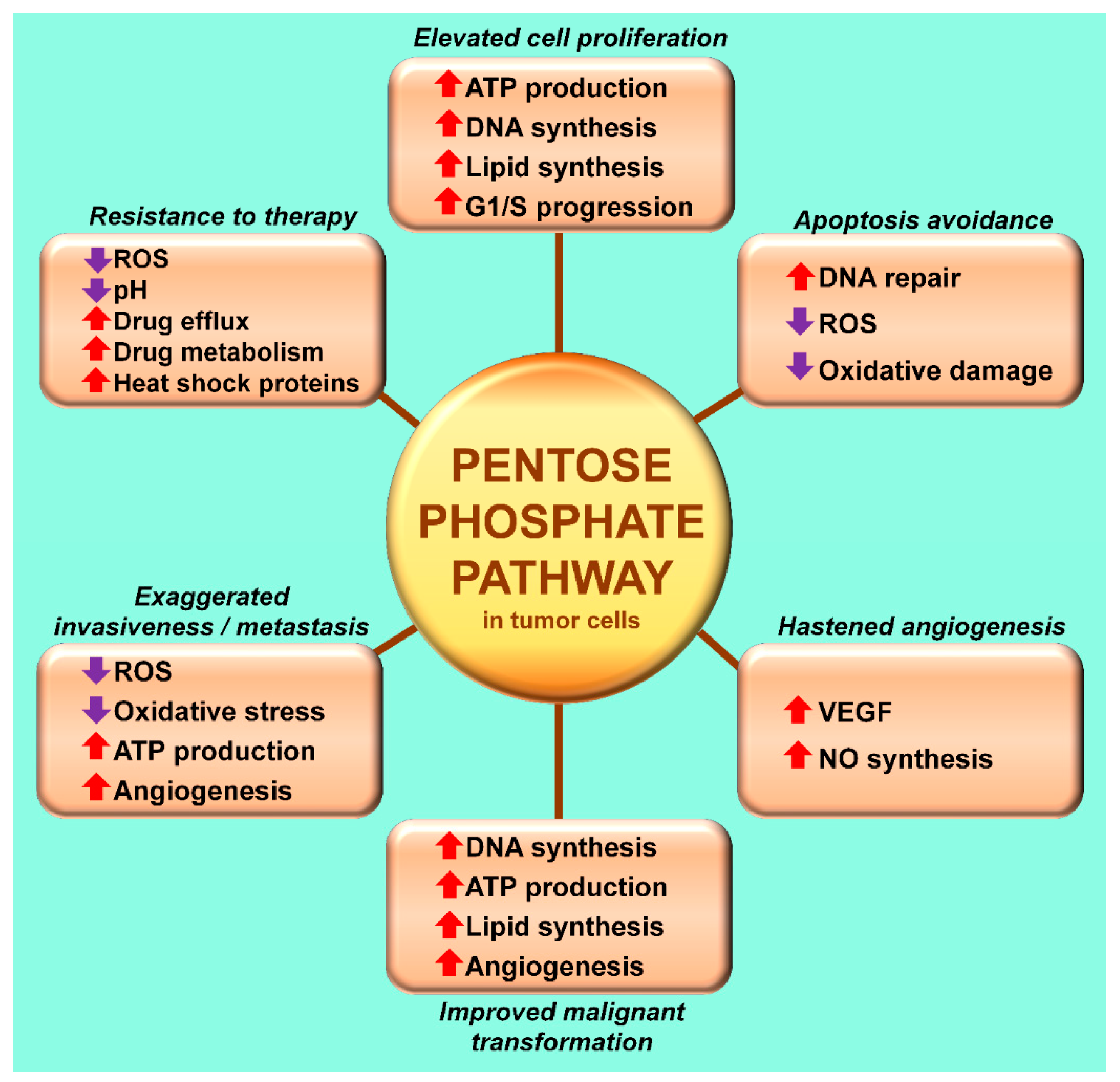
3. The Warburg Effect: A Focal Point for a Number of Diseases
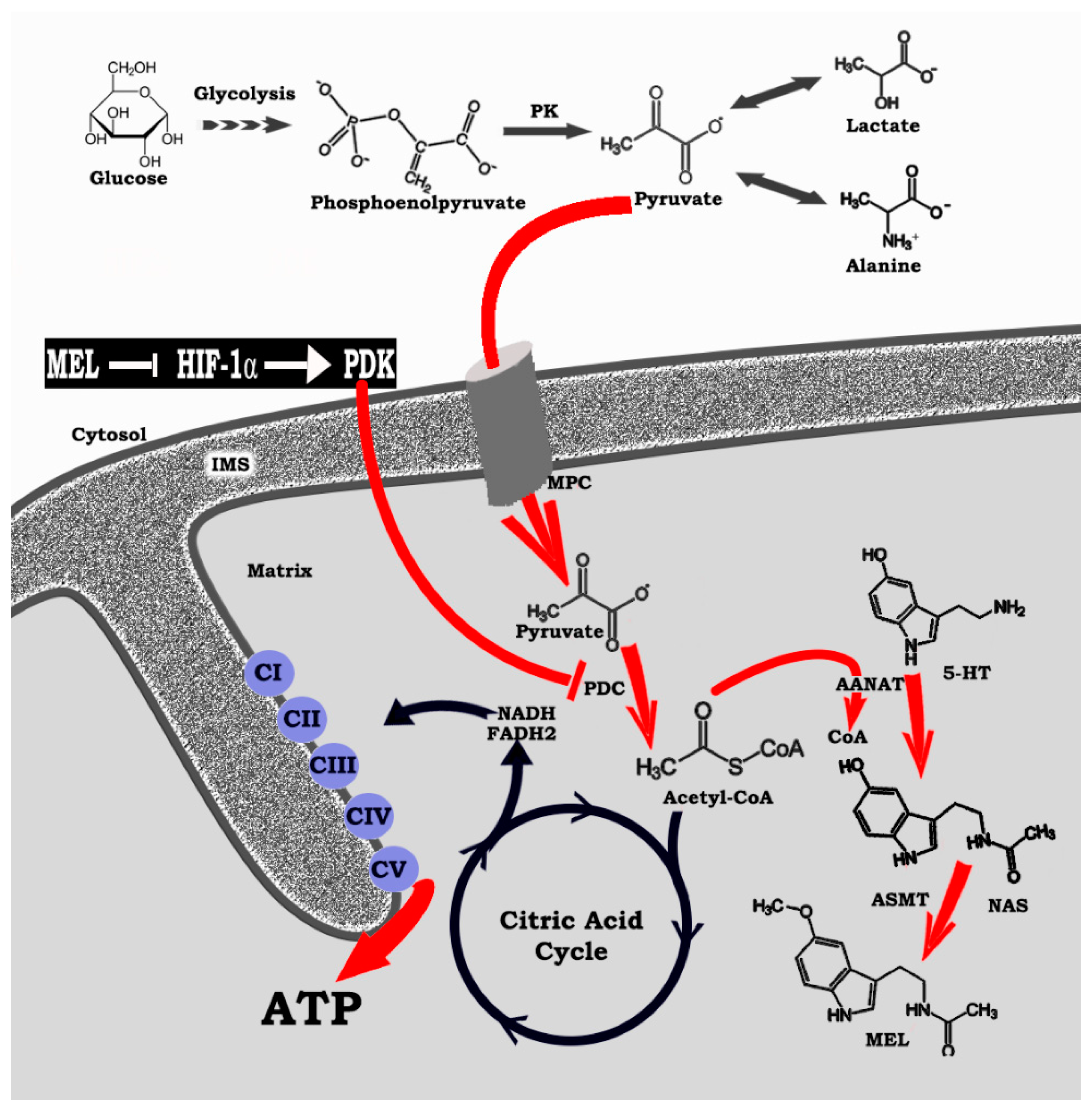
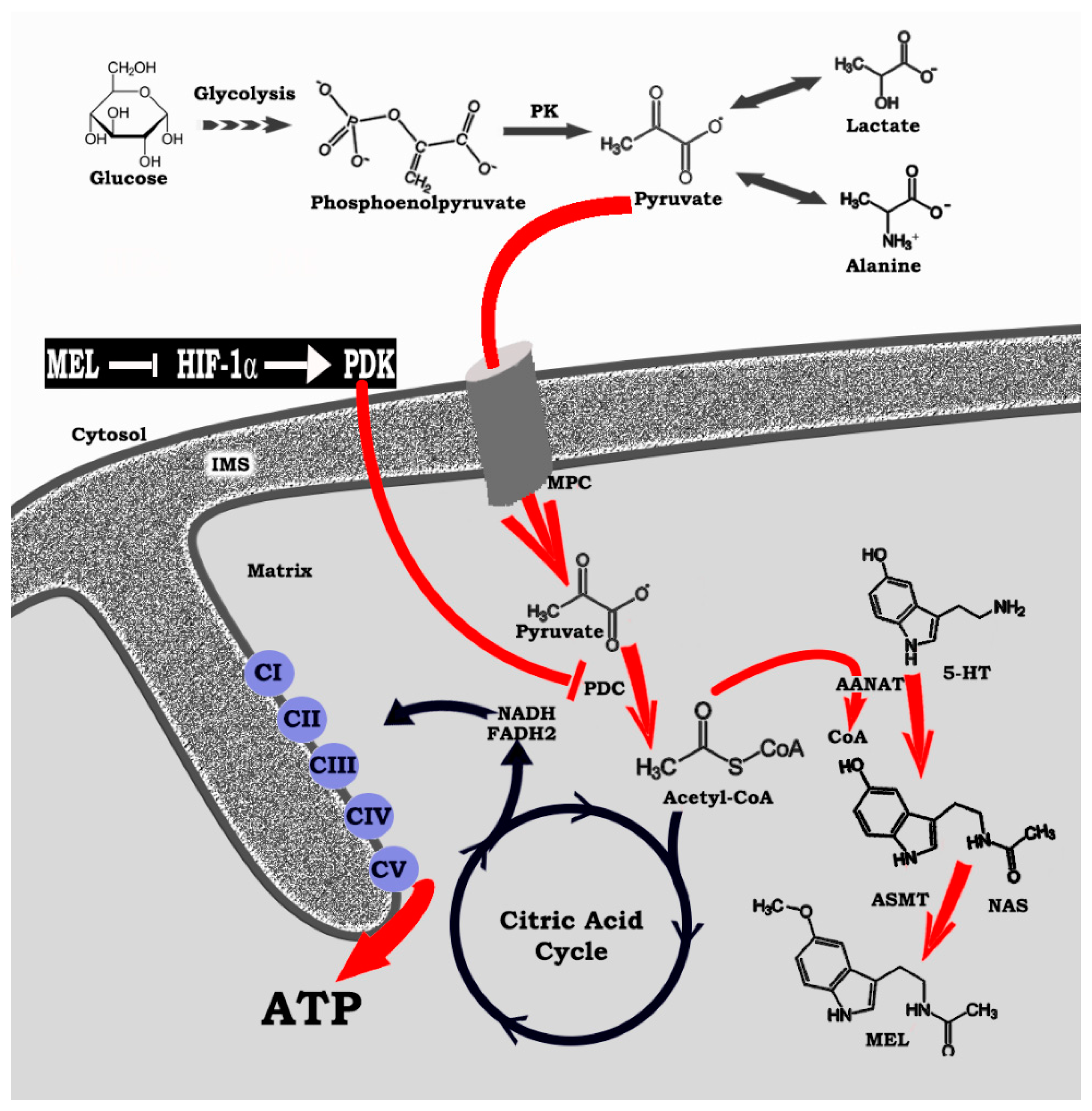
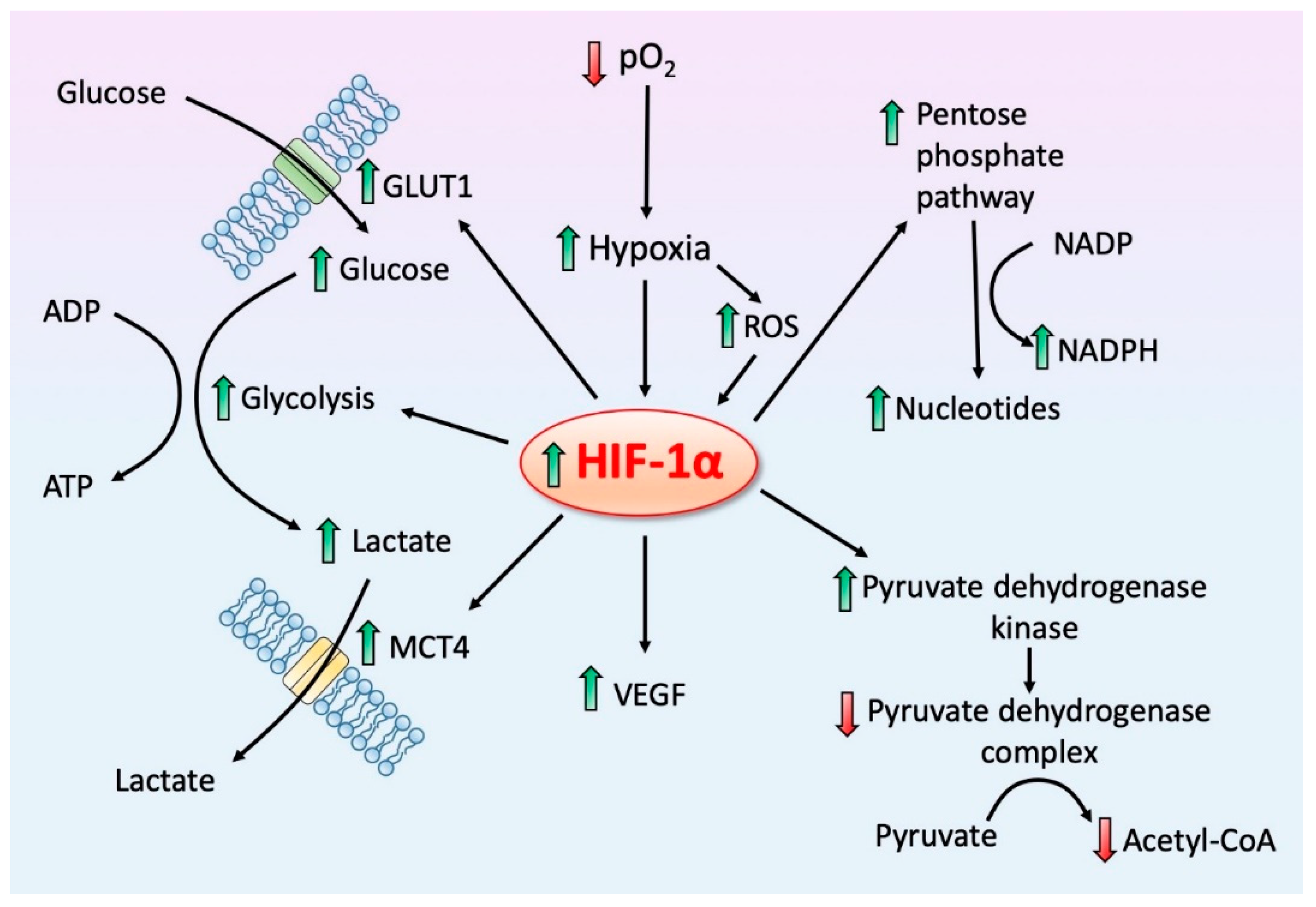
4. HIF-1α, a Regulator of Regulators: Influence on Mitochondrial Melatonin
5. Melatonin Reprograms Glucose Metabolism: Converting Diseased Cells to a Healthier Phenotype
6. Melatonin: A Molecular Peacekeeper during Troubled Metabolic Times
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pascale, R.M.; Calvisi, D.F.; Simile, M.M.; Feo, C.F.; Feo, F. The Warburg effect 97 years after its discovery. Cancers 2020, 12, 2819. [Google Scholar] [CrossRef]
- Urbano, A.M. Otto Warburg: The journey towards the seminal discovery of tumor cell bioenergetic reprogramming. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1867, 165965. [Google Scholar] [CrossRef]
- Li, J.; Zhou, Y.; Pei, M.; Zhang, Y.; Jiang, Y. Berberine inhibits the Warburg effect through TET3/miR-145/HK2 pathways in ovarian cancer cells. J. Cancer. 2021, 12, 207–216. [Google Scholar] [CrossRef]
- Tran, Q.; Lee, H.; Kim, C.; Kong, G.; Gong, N.; Kwon, S.H.; Park, J.; Kim, S.H.; Park, J. Revisiting the Warburg effect: Diet-based strategies for cancer prevention. Biomed. Res. Int. 2020. [Google Scholar] [CrossRef]
- Southan, J.; McHugh, E.; Walker, H.; Ismail, H.M. Metabolic signature of articular cartilage following mechanical injury: An integrated transcriptomics and metabolomics analysis. Front. Mol. Biosci. 2020, 7, 592905. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, S.; Yu, D. Metabolic reprogramming of chemoresistant cancer cells and the potential significance of metabolic regulation in the reversal of cancer chemoresistance. Metabolites 2020, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Malley, C.O.; Pidgeon, G.P. The mTOR pathway in obesity driven gastrointestinal tumors: Potential targets and clinical trials. BBA Clin. 2015, 5, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, S.; Boley, D.; Moller, P.; Stark, H.; Kaleta, C. Mathematical models for explaining the Warburg effect: A review focus on ATP and biomass production. Biochem. Soc. Trans. 2015, 43, 1187–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.C.; Hsu, T.W.; Yeh, C.C.; Huang, H.B. The role of transcription factor caudal-related homeobox transcription factor 2 in colorectal cancer. Tzu Chi Med. J. 2020, 32, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Hou, J.; Cao, Y.; Shan, J.J.; Zhao, J. Spinster homolog 2 in cancers: Its functions and mechanisms. Cell. Signal. 2020, 31, 109821. [Google Scholar] [CrossRef]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The role of the pentose phosphate pathway in diabetes and cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, H.; Zhang, W.; Mu, W. Ribase-5-phosphate isomerases: Characteristics, structural features, and applications. Appl. Microbiol. Biotechnol. 2020, 104, 6429–6441. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Mayernik, J.K.; Moin, L.; Sloan, B.F. Acidosis and proteolysis in the tumor microenvironment. Cancer Metastasis Rev. 2019, 38, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.M.; Blask, D.E.; Xiang, S.; Yuan, L.; Mao, L.; Dauchy, R.T.; Dauchy, E.M.; Frasch, T.; Duplesis, T. Melatonin and associated signaling pathways that control normal breast epithelium and breast cancer. J. Mammary Gland Biol. Neoplasia 2011, 16, 235–245. [Google Scholar] [CrossRef]
- Sanchez-Barcelo, E.; Mediavilla, M.D.; Alonso-Gonzalez, C.; Rueda, N. Breast cancer therapy based on melatonin. Rec. Pat. Endocr. Metab. Immune Drug Discov. 2012, 6, 108–116. [Google Scholar] [CrossRef]
- Slominski, A.T.; Zmijewski, M.A.; Semak, I.; Zbytek, B.; Pisarcik, A.; Li, W.; Zjawiony, J.; Tuckey, R.C. Cytochrome p450 and skin cancer: Role of local endocrine pathways. Anti-Cancer Agents Med. Chem. 2014, 14, 77–96. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Rosales-Corral, S.A.; Tan, D.X.; Acuna-Castroviejo, D.; Qin, L.; Yang, S.F.; Xu, K. Melatonin, a full service anti-cancer agent: Inhibition of initiation, progression and metastasis. Int. J. Mol. Sci. 2017, 18, 843. [Google Scholar] [CrossRef]
- Pourhanifeh, M.H.; Hosseinzadeh, A.; Juybari, K.B.; Mehrzadi, S. Melatonin and urological cancers: A new therapeutic approach. Cancer Cell Int. 2020, 20, 444. [Google Scholar] [CrossRef]
- Chuffa, L.G.A.; Carvalho, R.F.; Justulin, L.A.; Cury, S.S.; Seiva, F.R.F.; Jardim-Perassi, B.V.; Zucarri, D.A.P.C.; Reiter, R.J. A meta-analysis of microRNA networks regulated by melatonin in cancer: Portrait of potential candidates for breast cancer treatment. J. Pineal Res. 2020, 69, e12693. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, W.; Li, L.; Chen, L. Involvement of the Warburg effect in non-tumor diseases processes. J. Cell Physiol. 2018, 233, 2839–2849. [Google Scholar] [CrossRef]
- Schwartz, L.; Peres, S.; Jolicoeur, M.; da Veigo Moreira, J. Cancer and Alzheimer’s disease: Intracellular pH scales and metabolic disorders. Biogerontology 2020, 21, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Dauchy, R.T.; Wren-Dail, M.A.; Dupepe, L.M.; Hill, S.M.; Xiang, S.; Anbalagan, M.; Belancio, V.P.; Dauchy, E.M.; Blask, D.E. Effect of daytime blue-enriched LED light on the nighttime circadian melatonin inhibition of hepatoma 7288CTC Warburg effect and progression. Comp. Med. 2018, 68, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Ma, Q.; Sharma, R. Melatonin in mitochondria: Mitigating clear and present dangers. Physiology 2020, 35, 86–95. [Google Scholar] [CrossRef]
- Lyra-Leite, D.M.; Peterson, A.P.; Ariyasinghe, N.R.; Cho, N.; McCain, M.L. Mitochondrial architecture in cardiac myocytes depends on cell shape and matrix rigidity. J. Mol. Cell. Cardiol. 2021, 150, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Nathan, A.T.; Singer, M. The oxygen trail: Tissue oxygenation. Br. Med. Bull. 1999, 55, 96–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skulochev, V.P. Mitochondrial physiology and pathology; concepts of programmed death of organelles, cells and organisms. Mol. Aspects Med. 1999, 20, 139–184. [Google Scholar] [CrossRef]
- Acuna-Castroviejo, D.; Martin, M.; Macias, M.; Escames, G.; Leon, J.; Khaldy, H.; Reiter, R.J. Melatonin, mitochondria, and cellular bioenergetics. J. Pineal Res. 2001, 30, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Korycka-Dahl, M.B.; Richardson, T. Activated oxygen species and oxidation of food constituents. CRC Crit. Rev. Food Sci. Nutr. 1978, 10, 209–241. [Google Scholar] [CrossRef]
- Finkelstein, E.; Rosen, G.M.; Rauckman, E.J. Spin trapping of superoxide and hydroxyl radical: Practical aspects. Arch. Biochem. Biophys. 1980, 200, 1–16. [Google Scholar] [CrossRef]
- Crow, J.P.; Beckman, J.S. Reactions between nitric oxide, superoxide, and peroxynitrite: Footprints of peroxynitrite in vivo. Adv. Pharmacol. 1995, 34, 17–43. [Google Scholar]
- Radi, R. Kinetic analysis of reactivity of peroxynitrite with biomolecules. Methods Enzymol. 1996, 269, 354–366. [Google Scholar] [PubMed]
- Pryor, W.A. Oxy-radicals and related species: Their formation, lifetimes, and reactions. Annu. Rev. Physiol. 1986, 48, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, G.; Mirabelli, F. Oxidative stress injury studied in isolated intact cells. Mol. Toxicol. 1987, 1, 281–293. [Google Scholar] [PubMed]
- Aust, S.D.; Chignell, C.F.; Bray, T.M.; Kalyanaraman, B.; Mason, R.P. Free radicals in toxicology. Toxicol. Appl. Pharmacol. 1993, 120, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Gilad, E.; Cuzzocrea, B.; Zingarelli, B.; Salzman, A.L.; Szabo, C. Melatonin is a scavenger of peroxynitrite. Life Sci. 1997, 60, PL169–PL174. [Google Scholar] [CrossRef]
- Ramis, M.R.; Esteban, S.; Miralles, A.; Tan, D.X.; Reiter, R.J. Protective effects of melatonin and mitochondria-targeted antioxidants against oxidative stress: A review. Curr. Med. Chem. 2015, 22, 2690–2711. [Google Scholar] [CrossRef] [PubMed]
- Okatani, Y.; Wakatsuki, A.; Reiter, R.J.; Enzan, H.; Miyahara, Y. Protective effect of melatonin against mitochondrial injury induced by ischemia and reperfusion of rat liver. Eur. J. Pharmacol. 2003, 469, 145–152. [Google Scholar] [CrossRef]
- Jou, M.J.; Peng, T.I.; Reiter, R.J.; Jou, S.B.; Wu, H.Y.; Wen, S.T. Visualization of the antioxidative effects of melatonin at the mitochondrial level during oxidative stress-induced apoptosis of rat brain astrocytes. J. Pineal Res. 2004, 37, 55–70. [Google Scholar] [CrossRef]
- Lowes, D.A.; Webster, N.R.; Murphy, M.P.; Galley, H.F. Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br. J. Anaesth. 2013, 110, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Macias, M.; Escames, G.; Reiter, R.J.; Agapito, M.T.; Ortiz, G.G.; Acuna-Castroviejo, D. Melatonin-induced increased activity of the respiratory chain complexes I and IV can prevent mitochondrial damage induced by ruthenium red in vivo. J. Pineal Res. 2000, 28, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Tan, D.X.; Rosales-Corral, S.; Manchester, L.C. The universal nature, unequal distribution and antioxidant functions of melatonin. Mini Rev. Med. Chem. 2013, 13, 373–384. [Google Scholar] [PubMed]
- Wakatsuki, A.; Okatani, Y.; Shinohara, K.; Ikenoue, N.; Fukaya, T. Melatonin protects against ischemia/reperfusion-induced oxidative damage to mitochondria in fetal rat brain. J. Pineal Res. 2001, 31, 167–172. [Google Scholar] [CrossRef]
- Melchiorri, D.; Reiter, R.J.; Sewerynek, E.; Hara, M.; Chen, L.; Nistico, G. Paraquat toxicity and oxidative damage. Reduction by melatonin. Biochem. Pharmacol. 1996, 51, 1095–1099. [Google Scholar] [CrossRef]
- Garcia, J.J.; Lopez-Pingarron, L.; Almeda-Souza, P.; Tres, A.; Escudero, P.; Garcia-Gil, F.A.; Tan, D.X.; Reiter, R.J.; Ramirez, J.M.; Bernal-Perez, M. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: A review. J. Pineal Res. 2014, 56, 225–237. [Google Scholar] [CrossRef]
- Gitto, E.; Tan, D.X.; Reiter, R.J.; Karbownik, M.; Manchester, L.C.; Cuzzocrea, S.; Fulia, F.; Barberi, I. Individual and synergistic antioxidative actions of melatonin: Studies with vitamin E, vitamin C, glutathione and desferrioxamine (deferoxamine) in rat liver homogenates. J. Pharm. Pharmacol. 2001, 53, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Milczarek, R.; Hallmann, A.; Sokolowska, E.; Kaletha, K.; Klimek, J. Melatonin enhances antioxidant action of alpha tocopherol and ascorbate acid against NADPH- and iron-dependent lipid peroxidation in human placenta mitochondria. J. Pineal Res. 2010, 49, 149–155. [Google Scholar] [PubMed]
- Martin, M.; Macias, M.; Escames, G.; Leon, J.; Acuna-Castroviejo, D. Melatonin but not vitamins C and E maintains glutathione homeostasis in t-butyl hydroperoxide-induced mitochondrial oxidative stress. FASEB J. 2000, 14, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Jou, M.J.; Peng, T.I.; Yu, P.Z.; Jou, S.B.; Reiter, R.J.; Chen, J.Y.; Wu, H.Y.; Chen, C.C.; Hsu, L.F. Melatonin protects against common deletion of mitochondrial DNA-augmented mitochondrial oxidative stress and apoptosis. J. Pineal Res. 2007, 43, 389–403. [Google Scholar] [CrossRef]
- Noda, Y.; Mori, A.; Liburdy, R.; Packer, L. Melatonin and its precursors scavenge nitric oxide. J. Pineal Res. 1999, 27, 159–163. [Google Scholar] [CrossRef]
- Tan, D.X.; Hardeland, R.; Manchester, L.C.; Galano, A.; Reiter, R.J. Cyclic-3-hydroxymelatonin (C3HOM), a potent antioxidant scavenges free radicals and suppresses oxidative reactions. Curr. Med. Chem. 2014, 21, 1557–1565. [Google Scholar] [CrossRef]
- Galano, A.; Tan, D.X.; Reiter, R.J. On the free radical scavenging activities of melatonin’s metabolites, AFMK and AMK. J. Pineal Res. 2013, 54, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Tan, D.X.; Reiter, R.J. Kynuramines, metabolites of melatonin and other indoles: The resurrection of an almost forgotten class of biogenic amines. J. Pineal Res. 2009, 47, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Dellegar, S.M.; Murphy, S.A.; Bourne, A.E.; DiCesare, J.C.; Purser, G.H. Identification of the factors affecting the rate of deactivation of hypochlorous acid by melatonin. Biochem. Biophys. Res. Commun. 1999, 257, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Matuszak, Z.; Reszka, K.; Chignell, C.F. Reaction of melatonin and related indoles with hydroxyl radicals: EPR and spin trapping investigations. Free Rad. Biol. Med. 1997, 23, 367–372. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin and the electron transport chain. Cell. Mol. Life Sci. 2017, 74, 3883–3896. [Google Scholar] [CrossRef]
- Mayo, J.C.; Sainz, R.M.; Antoli, I.; Herrera, F.; Martin, V.; Rodriguez, C. Melatonin regulation of antioxidant enzyme gene expression. Cell. Mol. Life Sci. 2002, 59, 1706–1713. [Google Scholar] [CrossRef]
- Zalachoras, I.; Hollis, F.; Ramos-Fernandez, E.; Trovo, L.; Sonnay, S.; Gieser, E.; Preitner, N.; Steiner, P.; Sandi, C.; Morato, L. Therapeutic potential of glutathione-enhancers in stress-related psychopathologies. Neurosci. Biobehav. Rev. 2020, 114, 134–155. [Google Scholar] [CrossRef]
- Winiarska, K.; Fraczyk, T.; Malinska, D.; Drozak, J.; Bryla, J. Melatonin attenuates diabetes-induced oxidative stress in rabbits. J. Pineal Res. 2006, 40, 168–176. [Google Scholar] [CrossRef]
- Urata, Y.; Honma, S.; Goto, S.; Todoroki, S.; Iida, T.; Cho, S.; Honma, K.; Kondo, T. Melatonin induces gamma-glutamylcysteine synthetase mediated by activator protein-1 in human vascular endothelial cells. Free Radic. Biol. Med. 1999, 27, 838–847. [Google Scholar] [CrossRef]
- Okatani, Y.; Wakatsuki, A.; Kanedo, C. Melatonin increases activities of glutathione peroxidase and superoxide dismutase in fetal rat brain. J. Pineal Res. 2000, 28, 89–96. [Google Scholar] [CrossRef]
- Venegas, C.; Garcia, J.A.; Escames, G.; Ortiz, F.; Lopez, A.; Doerrier, C.; Gario-Corzo, L.; Lopez, L.C.; Reiter, R.J.; Acuna-Castroviejo, D. Extrapineal melatonin: Analysis of its subcellular distribution and daily fluctuation. J. Pineal Res. 2016, 52, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Qin, L.; Reiter, R.J. Melatonin: A mitochondrial targeting molecule involving mitochondrial protection and dynamics. Int. J. Mol. Sci. 2016, 17, 2124. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Rosales-Corral, S.; Tan, D.X.; Jou, M.J.; Galano, A.; Xu, B. Melatonin as a mitochondria-targeted antioxidant: One of evolution’s best ideas. Cell. Mol. Life Sci. 2017, 74, 3863–3881. [Google Scholar] [CrossRef]
- Manchester, L.C.; Poeggeler, B.; Alvares, F.L.; Ogden, G.B.; Reiter, R.J. Melatonin immunoreactivity in the photosynthetic prokaryote Rhodospirillum rubrum: Implications for an ancient antioxidant system. Cell Mol. Biol. Res. 1995, 41, 391–395. [Google Scholar]
- Margulis, L.; Bermudes, D. Symbiosis as a mechanism of evolution: Status of cell symbiosis theory. Symbiosis 1985, 1, 101–124. [Google Scholar]
- Martin, W.F. Physiology, anaerobes, and the origin of mitosing cells 50 years on. J. Theor. Biol. 2017, 434, 2–10. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rosales-Corral, S.; Manucha, W. Circadian and non-circadian melatonin: Influences on glucose metabolism in cancer cells. J. Curr. Sci. Technol. 2020, 10, 85–98. [Google Scholar]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rosales-Corral, S.; de Almeida Chuffa, L.G. Melatonin inhibits Warburg-dependent cancer by redirecting glucose oxidation to the mitochondria: A mechanistic hypothesis. Cell. Mol. Life Sci. 2020, 77, 2527–2542. [Google Scholar] [CrossRef]
- Zhao, D.; Wang, H.; Chen, S.; Yu, D.; Reiter, R.J. Phytomelatonin: An emerging regulator of plant biotic stress resistance. Trends Plant Sci. 2020. [Google Scholar] [CrossRef]
- Kerenyi, N.A.; Sotonyi, P.; Somogyi, E. Localizing acetyl-serotonin transferase by electron microscopy. Histochemistry 1975, 46, 77–80. [Google Scholar] [CrossRef]
- Klein, D.C.; Weller, J. Rapid light-induced decrease in pineal serotonin N-acetyltransferase activity. Science 1972, 177, 532–533. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, J.; Weissbach, H. Enzymatic O-methylation of N-acetylserotonin to melatonin. Science 1960, 131, 1312. [Google Scholar] [CrossRef]
- Klein, D.C.; Berg, G.R.; Weller, J.; Glinsmann, W. Pineal gland: Dibutyryl cyclic adenosine monophosphate stimulation of labeled melatonin production. Science 1970, 167, 1738–1740. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Liu, X.; Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Reiter, R.J. Mitochondria and chloroplasts as the original sites of melatonin synthesis: A hypothesis related to melatonin’s primary function and evolution in eukaryotes. J. Pineal Res. 2013, 54, 127–138. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wang, J.; Zhang, Z.; Yang, M.; Li, Y.; Tian, X.; Ma, T.; Tao, J.; Zhu, K.; Song, Y.; et al. Mitochondria synthesize melatonin to ameliorate its function and improve mice oocyte’s quality under in vitro conditions. Int. J. Mol. Sci. 2016, 17, 939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champney, T.H.; Holtorf, A.P.; Steger, R.W.; Reiter, R.J. Concurrent determination of enzymatic activities and substrate concentrations in the melatonin synthetic pathway within the same rat pineal gland. J. Neurosci. Res. 1984, 11, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Suofu, Y.; Li, W.; Jean-Alphonse, F.G.; Jia, J.; Khattar, N.K.; Li, J.; Baranov, S.V.; Leronni, D.; Mihalik, A.L.; He, Y.; et al. Dual role of mitochondria in producing melatonin and during GPCR signaling to block cytochrome c release. Proc. Natl. Acad. Sci. USA 2017, 114, E7997–E8006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troiani, M.E.; Reiter, R.J.; Vaughan, M.K.; Oakin, S.; Vaughan, G.M. Swimming depresses nighttime melatonin content without changing N-acetyltransferase activity in the rat pineal gland. Neuroendocrinology 1988, 47, 55–60. [Google Scholar] [CrossRef]
- Hevia, D.; Gonzalez-Menendez, P.; Quiros-Gonzalez, I.; Miar, A.; Rodriguez-Garcia, A.; Tan, D.X.; Reiter, R.J.; Mayo, J.C.; Sainz, R.M. Melatonin uptake through glucose transporters: New target for melatonin inhibition of cancer. J. Pineal Res. 2015, 58, 234–250. [Google Scholar] [CrossRef]
- Mayo, J.C.; Sainz, R.M.; Gonzalez-Menendez, P.; Hevia, D.; Cernuda-Cernuda, R. Melatonin transport into mitochondria. Cell. Mol. Life Sci. 2017, 74, 3927–3940. [Google Scholar] [CrossRef] [PubMed]
- Hevia, D.; Gonzalez-Menendez, P.; Fernandez-Fernandez, M.; Cueto, S.; Rodriguez-Gonzalez, P.; Garcia-Alonso, J.L.; Mayo, J.C.; Sainz, R.M. Melatonin decreases glucose metabolism in prostate cancer cells: A13C stable isotope–resolved metabolic study. Int. J. Mol. Sci. 2017, 18, 1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, X.; Wang, C.; Yu, Z.; Peng, Y.; Wang, S.; Feng, S.; Zhang, S.; Tian, X.; Sun, C.; Liu, K.; et al. Human transporters, PEPT1/2, facilitate melatonin transportation into mitochondria of cancer cells: An implication of the therapeutic potential. J. Pineal Res. 2017, 62, 12390. [Google Scholar] [CrossRef] [PubMed]
- Blask, D.E.; Dauchy, R.T.; Dauchy, E.M.; Mao, L.; Hill, S.M.; Greene, M.W.; Belanco, V.P.; Sauer, L.A.; Davidson, L. Light exposure at night disrupts host/cancer circadian regulatory dynamics: Impact on the Warburg effect, lipid signaling and tumor growth prevention. PLoS ONE 2014, 9, e102776. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Sharma, R.; Ma, Q. Switching diseased cells from cytosolic aerobic glycolysis to mitochondrial oxidative phosphorylation: A metabolic rhythm regulated by melatonin? J. Pineal Res. 2020, e12677. [Google Scholar] [CrossRef]
- Acuna-Castroviejo, D.; Noguiera-Navarro, M.T.; Reiter, R.J.; Escames, G. Melatonin actions in the heart: More than a hormone. Melatonin Res. 2018, 1, 21–26. [Google Scholar] [CrossRef]
- Martinez, C.A.; Scafoglio, C. Heterogeneity of glucose transport in lung cancer. Biomolecules 2020, 10, 868. [Google Scholar] [CrossRef]
- Heydarzadeh, S.; Moshtaghie, A.A.; Daneshpoor, M.; Hedayati, M. Regulators of glucose uptake in thyroid cancer cell lines. Cell Commun. Signal. 2020, 18, 83. [Google Scholar] [CrossRef]
- Luznik, Z.; Anchouche, S.; Dana, R.; Yin, J. Regulatory T cells in angiogenesis. J. Immunol. 2020, 205, 2557–2565. [Google Scholar] [CrossRef]
- Stein, Y.; Aloni-Grinstein, R.; Rotter, V. Mutant p53 oncogenicity-dominant negative or gain-of-function? Carcinogenesis 2020, 41, 117. [Google Scholar] [CrossRef]
- Bizzarri, M.; Proietti, S.; Cucina, A.; Reiter, R.J. Molecular mechanisms of the pro-apoptotic actions of melatonin in cancer: A review. Exp. Opin. Ther. Targets 2013, 17, 1483–1496. [Google Scholar] [CrossRef] [PubMed]
- Proietti, S.; Cucina, A.; Minini, M.; Bizzarri, M. Melatonin, mitochondria, and the cancer cell. Cell Mol. Life Sci. 2017, 74, 4015–4025. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.F.; Collins, S.E.; Charest, P.G. Ras, PI3K and mTORC2-three’s a crowd? J. Cell Sci. 2020, 133, 234930. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Song, S.; Wang, X.; Hao, J. Small-molecule inhibitors of breast cancer-related targets: Potential therapeutic agents for breast cancer. Eur. J. Med. Chem. 2020, 29, 112954. [Google Scholar] [CrossRef] [PubMed]
- Hinz, N.; Jucker, M. Distinct functions of AKT isoforms in breast cancer: A comprehensive review. Cell. Commun. Signal. 2019, 17, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signaling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Wu, J.; Zhao, Q.; Fu, S.; Jin, J. Emerging roles of aerobic glycolysis in breast cancer. Clin. Transl. Oncol. 2020, 22, 631–646. [Google Scholar] [CrossRef]
- Schito, L.; Rey, S. Hypoxia: Turning vessels into vassals of cancer immunotolerance. Cancer Lett. 2020, 487, 74–84. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Metabolic heterogeneity of cancer cells: An interplay between HIF-1, GLUTs and AMPK. Cancers 2020, 12, 862. [Google Scholar] [CrossRef] [Green Version]
- Masoud, G.N.; Li, W. HIF-1α: Role, regulation and intervention in cancer therapy. Acta Pharm. Sin. B. 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetzman, E.S.; Prochownik, E.V. The role of Myc in coordinating glycolysis, oxidative phosphorylation, glutaminolysis, and fatty acid metabolism in normal and neoplastic tissues. Front. Endocrinol. 2018, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- He, T.L.; Zhang, Y.J.; Jiang, H.; Li, X.H.; Zhu, H.; Zheng, K.L. The c-Myc-LDHA axis positively regulates aerobic glycolysis and promotes tumor progression in pancreatic cancer. Med. Oncol. 2015, 32, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powis, G.; Kirkpatrick, L. Hypoxia- inducible factor-1 alpha as a cancer drug target. Mol. Cancer Ther. 2004, 3, 647–654. [Google Scholar]
- Xia, Y.; Choi, H.K.; Lee, K. Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur. J. Med. Chem. 2012, 49, 24–40. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 and tumor progression: Pathophysiology and therapeutics. Trends Mol. Med. 2002, 8, S62–S67. [Google Scholar] [CrossRef]
- Frattaruolo, L.; Brindisi, M.; Curcio, R.; Marra, F.; Dolce, V.; Cappello, A.R. Targeting the mitochondrial metabolic network: A promising strategy in cancer treatment. Int. J. Mol. Sci. 2020, 21, 6014. [Google Scholar] [CrossRef]
- Ghasemishahrestani, Z.; Melo Mattos, L.M.; Tilli, T.M.; Dos Santos, A.S.; Pereira, M.D. Pieces of the complex puzzle of cancer cell energy metabolism: On overview of energy metabolism and alternatives for targeted cancer therapy. Curr. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Melillo, G. Hypoxia-inducible factor 1 inhibitors. Method. Enzymol. 2007, 435, 385–402. [Google Scholar]
- Cecchini, C.; Tardy, S.; Ceserani, V.; Theurillat, J.P.; Scapozza, L. Exploring the ubiquitin-proteasome system (UPS) through PROTAC technology. Chimia (Aarau) 2020, 74, 274–277. [Google Scholar] [CrossRef]
- Vriend, J.; Reiter, R.J. Melatonin and the von Hippel-Lindau/HIF-1 oxygen sensing mechanism: A review. Biochim. Biophys. Acta. 2016, 1865, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hulver, M.W.; McMillan, R.P.; Cline, M.A.; Gilbert, E.R. The pivotal role of pyruvate dehydrogenase complex in metabolic flexibility. Nutr. Metab. 2014, 11, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacpoole, P.W. Therapeutic targeting of the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molino-Pinelo, S.; Paz-Ares, L. Current challenges in cancer treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [Green Version]
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A convergence-based framework for cancer drug resistance. Cancer Cell. 2018, 33, 801–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Jeon, J.H.; Min, B.K.; Ha, C.M.; Thoudam, T.; Park, B.Y.; Lee, I.K. Role of the pyruvate dehydrogenase complex in metabolic remodeling: Differential pyruvate dehydrogenase complex functions in metabolism. Diabetes Metab. J. 2018, 42, 270–281. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Rajendran, G.; Harris, R.A.; Taylor, J.A., 3rd. Metabolic flexibility in cancer: Targeting the pyruvate dehydrogenase kinase: Pyruvate dehydrogenase axis. Mol. Cancer Ther. 2019, 18, 1673–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeoung, N.H. Pyruvate dehydrogenase kinases: Therapeutic targets for diabetes and cancers. Diabetes Metab. J. 2015, 39, 188–197. [Google Scholar] [CrossRef]
- Sradhanjali, S.; Reddy, M.M. Inhibition of pyruvate dehydrogenase kinase as a therapeutic strategy against cancer. Curr. Top. Med. Chem. 2018, 18, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef]
- Schwartz, L.; Supuran, C.T.; Alfarouk, K.O. The Warburg effect and the hallmarks of cancer. Anticancer Agents Med. Chem. 2017, 17, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Alfarouk, K.O.; Ahmed, S.B.M.; Elliott, R.L.; Benoit, A.; Alqahtani, S.S.; Ibrahim, M.E.; Bashir, A.H.H.; Alhoufie, S.T.S.; Elhassan, G.O.; Wales, C.C.; et al. The pentose phosphate pathway dynamics in cancer and its dependency on intracellular pH. Metabolites 2020, 10, 285. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Dauchy, R.T.; Blask, D.E.; Dauchy, E.M.; Slakey, L.M.; Brimer, S.; Yuan, L.; Xiang, S.; Hauch, A.; Smith, K.; et al. Melatonin suppression of aerobic glycolysis (Warburg effect), survival signaling and metastasis in human leiomyosarcoma. J. Pineal Res. 2016, 60, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Sanchez, A.M.; Antolin, I.; Puente-Moncada, N.; Suarez, S.; Gomez-Lobo, M.; Rodriguez, C.; Martin, V. Melatonin cytotoxicity is associated to Warburg effect inhibition in Ewing sarcoma cells. PLoS ONE 2015, 10, e0135420. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rosales-Corral, S.; Acuna-Castroviejo, D.; Escames, G. Inhibition of mitochondrial pyruvate dehydrogenase kinase: A proposed mechanism by which melatonin causes cancer cells to overcome cytosolic glycolysis, reduce tumor biomass and reverse insensitivity to chemotherapy. Melatonin Res. 2019, 2, 105–119. [Google Scholar] [CrossRef]
- Farhadi, P.; Yarani, R.; Dokaneheifard, S.; Mansouri, K. The emerging role of targeting cancer metabolism for cancer therapy. Tumour Biol. 2020, 42, 1–18. [Google Scholar] [CrossRef]
- Reiter, R.J.; Rosales-Corral, S.; Zhou, X.; Tan, D.X. Role of SIRT3/SOD2 signaling in mediating the antioxidant actions of melatonin in mitochondria. Curr. Trends Endocrinol. 2017, 9, 45–49. [Google Scholar]
- Reiter, R.J.; Tan, D.X.; Rosales-Corral, S.; Galano, A.; Jou, M.J.; Acuna-Castroviejo, D. Melatonin mitigates mitochondrial meltdown: Interactions with SIRT3. Int. J. Mol. Sci. 2018, 19, 2439. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, C.; Puente-Moncada, N.; Reiter, R.J.; Sanchez-Sanchez, A.M.; Herrera, F.; Rodriguez-Blanco, J.; Duarte-Olivenzo, C.; Turos-Cobal, M.; Antolin, I.; Martin, V. Regulation of cancer cell glucose metabolism is determinant for cancer cell fate after melatonin administration. J. Cell Physiol. 2020, 236, 27–40. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Liu, C.; Manucha, W.; Abreu-Gonzalez, P.; Dominquez-Rodriguez, A. Plasticity of glucose metabolism in activated immune cells: Advantages for melatonin inhibition of COVID-19 disease. Melatonin Res. 2020, 3, 362–379. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef]
- Ke, Q.D.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Hwang, M.S.; Suh, S.I.; Baek, W.K. Melatonin down-regulates HIF-1 alpha expression through inhibition of protein translation in prostate cancer cells. J. Pineal Res. 2009, 46, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Ciu, P.; Yu, M.; Han, J.; Li, H.; Xiu, R. Melatonin modulates the expression of VEGF and HIF-1 alpha induced by CoCl2 in cultured cancer cells. J. Pineal Res. 2008, 44, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Jang, W.J.; Yi, E.Y.; Jang, J.Y.; Jung, Y.; Jeong, J.W.; Kim, Y.J. Melatonin suppressed tumor angiogenesis by inhibiting HIF-1alpha stabilization under hypoxia. J. Pineal Res. 2010, 48, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Carbajo-Percador, S.; Ordonez, R.; Benet, M.; Jover, R.; Garcia-Palomo, A.; Mauriz, J.L.; Gonzalez-Gallego, J. Inhibition of VEGF expression through blockade of HIF-1 alpha and STAT3 signaling mediates the anti-angiogenic effect of melatonin in HepG2 liver cancer cells. Br. J. Cancer 2013, 109, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, Q.; Wang, F.; Ling, E.A.; Liu, S.; Wang, L.; Yang, Y.; Yao, L.; Chen, X.; Wang, F.; et al. Melatonin antagonizes hypoxia-mediated glioblastoma cell migration and invasion via inhibition of HIF-1α. J. Pineal Res. 2013, 55, 121–130. [Google Scholar] [CrossRef]
- Sohn, E.J.; Won, G.; Lee, J.; Lee, S.; Kim, S.H. Upregulation of miRNA3195 and miRNA374b mediates the anti-angiogenic properties of melatonin in hypoxic PL-3 prostate cancer cells. J. Cancer 2015, 6, 19–28. [Google Scholar] [CrossRef]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1 alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locasale, J.W.; Cantley, L.C. Altered metabolism in cancer. BMC Biol. 2010, 88, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Dauchy, R.T.; Tirrell, P.C.; Wu, S.S.; Lynch, D.T.; Jitawatanarat, P.; Burrington, C.M.; Dauchy, E.M.; Blask, D.E.; Greene, M.W. Light at night activates IGF-1R/PDK1 signaling and accelerates tumor growth in human breast cancer xenografts. Cancer Res. 2011, 71, 2622–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterniti, I.; Cordaro, M.; Esposito, E.; Cuzzocrea, S. The antioxidant property of melatonin against brain ischemia. Exp. Rev. Neurother. 2016, 16, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Lochner, A.; Marais, E.; Huisamen, B. Melatonin and cardioprotection against ischaemia/reperfusion injury: What’s new? A Review. J. Pineal Res. 2018, 65, e12490. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.J.; Lin, H.; Xiao, F.Y.; Huang, Z.Q.; Chen, Y.H. Melatonin against myocardial ischemia-reperfusion injury: A meta-analysis and mechanism insight from animal studies. Oxid. Med. Cell. Longev. 2020, 2020, 1241065. [Google Scholar] [CrossRef]
- Rosenberger, C.; Eckardt, K.U. Oxygenation of the transplanted kidney. Semin. Nephrol. 2019, 39, 554–566. [Google Scholar] [CrossRef]
- Sethi, K.; Rao, K.; Shulkes, A.; Baldwin, G.; Bolton, D.; Patel, O.; Ichia, J. Targeting HIF-1α to prevent renal ischemia-reperfusion injury: Does it work? Int. J. Cell. Biol. 2018, 9852791. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W.; Lee, H.S.; Sung, M.S.; Kim, S.J. The effect of melatonin on retinal ganglion cell survival in ischemic retina. Chonnam Med. J. 2012, 48, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Kryl‘skii, E.D.; Popova, T.; Safonova, O.A.; Stolyarova, A.O.; Razuvaeu, G.A.; Pinheiro de Carvalho, M.A. Transcriptional regulation of antioxidant enzymes activity and modulation of oxidative stress by melatonin in rats under cerebral ischemia/reperfusion conditions. Neuroscience 2019, 406, 653–666. [Google Scholar] [CrossRef]
- Yeung, C.; Gibson, A.E.; Issaq, S.H.; Oshima, W.; Baumgart, J.T.; Edessa, L.D.; Rai, G.; Urban, D.J.; Johnson, M.S.; Benavides, G.A.; et al. Targeting glycolysis through inhibition of lactate dehydrogenase impairs tumor growth preclinical models of Ewing sarcoma. Cancer Res. 2019, 79, 5060–5073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; Yao, T.; Xu, Y.; Zhang, D.; Fan, S.; Ma, J. CircECE1 activates energy metabolism in osteosarcoma by stabilizing c-Myc. Mol. Cancer 2020, 19, 151. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.H.; Lin, R.C.; Yang, J.S.; Yang, W.E.; Reiter, R.J.; Yang, S.F. Molecular and cellular mechanisms of melatonin in osteosarcoma. Cells 2019, 8, 1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tailor, D.; Going, C.C.; Resendez, A.; Kumar, V.; Nambiar, D.K.; Li, Y.; Dheeraj, A.; La Gory, E.L.; Ghoochani, A.; Birk, A.M.; et al. Novel Aza-podophyllotoxin derivative induces oxidative phosphorylation and cell death via AMPK activation in triple-negative breast cancer. Br. J. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.M.; Belancio, V.P.; Dauchy, R.T.; Xiang, S.; Brimer, S.; Mao, L.; Hauch, A.; Lundberg, P.W.; Summers, W.; Yuan, L.; et al. Melatonin: An inhibitor of breast cancer. Endocr. Relat. Cancer 2015, 22, R183–R204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Liu, Y.; Bai, Y.; Chen, M.; Cheng, D.; Wu, M.; Xia, J. RHOF promotes hepatocellular carcinoma metastasis by altering the metabolic status of cancer cells via RAB3B. Hepatology 2020. [Google Scholar] [CrossRef]
- Elmahallway, E.K.; Mohamed, Y.; Abdo, W.; Yanai, T. Melatonin and mesenchymal stem cells as a key for functional integrity for liver cancer treatment. Int. J. Mol. Sci. 2020, 21, 4521. [Google Scholar] [CrossRef]
- Du, P.; Liao, Y.; Zhao, H.; Zhang, J.; Mu, K. ANXA2P2/miR-9/LDHA axis regulate Warburg effect and affects glioblastoma proliferation and apoptosis. Cell Signal. 2020, 74, 109718. [Google Scholar] [CrossRef]
- Moretti, E.; Favero, G.; Rodella, L.; Rezzani, R. Melatonin’s antineoplastic potential against glioblastoma. Cells 2020, 9, 599. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Huang, R.; Wei, X.; Yu, W.; Min, Z.; Ye, M. The SIRT6-autophagy-Warburg effect axis in papillary thyroid cancer. Font. Oncol. 2020, 10, 1265. [Google Scholar] [CrossRef]
- Liao, Y.; Gao, Y.; Chang, A.; Li, Z.; Wang, H.; Cao, J.; Gu, W.; Tang, R. Melatonin synergizes BRAF-targeting agent dabrafenib for the treatment of anaplastic thyroid cancer by inhibiting AKT/hTERT signaling. J. Cell. Mol. Med. 2020, 24, 12119–12130. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, V.; Iommelli, F.; Morti, M.; Fonti, R.; Votta, G.; Stoppelli, M.P.; Del Vecchio, S. Reversal of Warburg effect and reactivation of oxidative phosphorylation by differential inhibition of EGFR Signaling pathways in non-small cell lung cancer. Clin. Cancer Res. 2015, 21, 5110–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Yang, Y.; Fan, C.; Han, J.; Wang, D.; Di, S.; Hu, W.; Liu, D.; Li, X.; Reiter, R.J.; et al. Melatonin as a potential anticarcinogen for non-small-cell lung cancer. Oncotarget 2016, 7, 46768–46784. [Google Scholar] [CrossRef] [Green Version]
- Schoepke, E.; Billon, C.; Haynes, K.M.; Avdagic, A.; Sitaula, S.; Sanders, R.; Adeyemi, C.M.; Walker, J.K.; Burns, T.P. A selective ERR alpha/gamma inverse agonist, SLU-PP-1072, inhibits the Warburg effect and induces apoptosis in prostate cancer cells. ACS Chem. Biol. 2020, 15, 2338–2345. [Google Scholar] [CrossRef] [PubMed]
- De Almeida Chuffa, L.G.; Seiva, F.R.F.; Cucielo, M.S.; Silveira, H.S.; Reiter, R.J.; Lupi, L.A. Mitochondrial functions and melatonin: A tour of the reproductive cancers. Cell. Mol. Life Sci. 2019, 76, 837–863. [Google Scholar] [CrossRef]
- Freidus, L.G.; Kumar, P.; Marimuthu, T.; Pradeep, P.; Pillay, V.; Choonara, Y.E. Synthesis and properties of CurNQ for theranostic application in ovarian cancer intervention. Molecules 2020, 25, 4471. [Google Scholar] [CrossRef]
- Bu, S.; Wang, Q.; Sun, J.; Li, X.; Gu, T.; Lai, D. Melatonin suppresses chronic restraint stress-mediated metastasis of epithelial ovarian cancer via NE/AKT/beta-catenin/SLUG axis. Cell. Death Dis. 2020, 11, 644. [Google Scholar] [CrossRef]
- Fu, R.; Yang, P.; Amin, S.; Li, Z. A novel miR-206/huRNPA1/PKM2 axis reshapes the Warburg effect to suppress colon cancer growth. Biochem. Biophys. Res. Commun. 2020, 531, 465–471. [Google Scholar] [CrossRef]
- Gil-Martin, E.; Lopez-Munoz, F.; Reiter, R.J.; Romero, A. Understanding the oncostatic actions displayed by melatonin by melatonin in colorectal cancer therapy. Future Med. Chem. 2020, 12, 1201–1204. [Google Scholar] [CrossRef]
- Annas, D.; Cheon, S.Y.; Yusuf, M.; Bae, S.J.; Ha, K.T.; Park, K.H. Synthesis and initial screening of lactate dehydrogenase inhibitor activity of 1,3-benzodioxole derivatives. Sci. Rep. 2020, 10, 19889. [Google Scholar] [CrossRef]
- Tamtaji, O.R.; Mirhosseini, N.; Reiter, R.J.; Behnamfar, M.; Asemi, Z. Melatonin and pancreatic cancer: Current knowledge and future perspectives. J. Cell Physiol. 2019, 234, 5372–5378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, F.; Li, S.; Guo, H.; Xi, H.; Deng, J.; Han, Q.; Zhang, W. ERG the modulates Warburg effect and tumor progression in cervical cancer. Biochem. Biophys. Res. Commun. 2020, 522, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xue, Y.; Shen, L.; Qin, P.; Sang, X.; Tao, Z.; Yi, J.; Wang, J.; Liu, P.; Cheng, H. Inhibition of SGK1 confers vulnerability to redox dysregulation in cervical cancer. Redox. Biol. 2019, 24, 101225. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, Y.; Zhu, H.; Cai, R.; Wang, K.F.; Song, J.; Wang, R.X.; Zhou, R.X. Role of transforming growth factor β1 in the inhibition of gastric cancer cell proliferation by melatonin in vitro and in vivo. Oncol. Rep. 2019, 42, 753–762. [Google Scholar] [CrossRef]
- Liu, H.; Chen, K.; Wang, L.; Zeng, X.; Huang, Z.; Li, M.; Dong, P.; Chen, X. miR-613 inhibits Warburg effect in gastric cancer by targeting PFKFB2. Biochem. Biophys. Res. Commun. 2019, 515, 37–43. [Google Scholar] [CrossRef]
- Kumar, P.R.; Moore, J.A.; Bowles, K.M.; Rushworth, S.A.; Moncrieff, M.D. Mitochondrial oxidative phosphorylation in cutaneous melanoma. Br. J. Cancer 2020. [Google Scholar] [CrossRef]
- Alvarez-Artime, A.; Cernuda-Cernuda, R.; Artime-Naveda, F.; Cepas, V.; Gonzalez-Menendez, P.; Fernandez-Vega, S.; Quiros-Gonzalez, I.; Sainz, R.M.; Mayo, J.C. Melatonin-induced cytoskeletal reorganization leads to inhibition of melanoma cancer cell proliferation. Int. J. Mol. Sci. 2020, 21, 548. [Google Scholar] [CrossRef] [Green Version]
- Baumeister, J.; Chatain, N.; Hubrich, A.; Maie, T.; Costa, I.G.; Denecke, B.; Han, L.; Kustermann, C.; Santag, S.; Sere, K.; et al. Hypoxia-inducible factor 1 (HIF-1) is a new therapeutic target in JAK2V617F-positive myeloproliferative neoplasms. Leukemia 2020, 34, 1062–1074. [Google Scholar] [CrossRef]
- Shafabakhsh, R.; Mirzaei, H.; Asemi, Z. Melatonin: A promising agent targeting leukemia. J. Cell. Biochem. 2020, 121, 2730–2738. [Google Scholar] [CrossRef]
- Alfonso, J.; Santos, L.L.; Longatto-Filho, A.; Baltazar, F. Competitive glucose metabolism as a target to boost bladder cancer immunotherapy. Nat. Rev. Urol. 2020, 17, 77–106. [Google Scholar] [CrossRef]
- Chen, Y.T.; Huang, C.R.; Chang, C.L.; Chiang, J.Y.; Luo, C.W.; Chen, H.H.; Yip, H.K. Jagged2 progressively increased expression from Stage I to III of bladder cancer and melatonin-mediated downregulation of NOTCH/Jagged 2 suppresses the bladder tumorigenesis, via inhibiting PI3K/AKT/mTOR/MMPs signaling. Int. J. Biol. Sci. 2020, 16, 2648–2662. [Google Scholar] [CrossRef] [PubMed]
- Salama, S.A.; Mahammad, M.A.; Diaz-Arrastia, C.R.; Kamel, M.W.; Kilic, G.S.; Ndofor, B.T.; Abel-Baki, M.S.; Theiler, S.K. Estradiol 17-β upregulates pyruvate kinase M2 expression to coactivate estrogen receptor-α and to integrate metabolic reprogramming with the mitogenic response in endometrial cells. J. Clin. Endocrinol. Metab. 2014, 99, 3790–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, C.; Yang, H.; Chang, K.; Zhang, B.; Xie, F.; Ye, J.; Chang, R.; Qui, X.; Wang, Y.; Qu, Y.; et al. Melatonin alleviates progression of uterine endometrial cancer by suppressing estrogen/ubiquitin C/SDHB-mediated succinate accumulation. Cancer Lett. 2020, 476, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Lessi, F.; Mazzanti, C.M.; Tomei, S.; Di Cristofano, C.; Minervini, A.; Menicagli, M.; Apollo, A.; Masieri, L.; Collecchi, P.; Minervini, R.; et al. VHL and HIF-1α: Gene variations and prognosis in early-stage clear cell renal cell carcinoma. Med. Oncol. 2014, 31, 840. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.C.; Lin, Y.W.; Chu, C.Y.; Yang, Y.C.; Yang, S.F.; Liu, Y.F.; Hsiao, M.; Lee, W.J.; Chien, M.H. Melatonin-triggered post-transcriptional and post-translational modifications of ADAMTS1 coordinately retard tumorigenesis and metastasis of renal cell carcinoma. J. Pineal Res. 2020, 69, e12668. [Google Scholar] [CrossRef]
- Kornberg, M.D. The immunologic Warburg effect: Evidence and therapeutic opportunities in autoimmunity. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1486. [Google Scholar]
- Lopez-Gonzalez, A.; Alvarez-Sanchez, N.; Lardone, P.J.; Cruz-Chamorro, I.; Martinez-Lopez, A.; Guerrero, J.M.; Reiter, R.J.; Carillo-Vico, A. Melatonin treatment improves primary progressive multiple sclerosis: A case report. J. Pineal Res. 2015, 58, 173–177. [Google Scholar] [CrossRef]
- Altante, A.; de Bari, L.; Bobba, A.; Amadoro, G. A disease with a sweet tooth: Exploring the Warburg effect in Alzheimer’s disease. Biogerontology 2017, 18, 301–319. [Google Scholar]
- Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Coto-Montes, A.; Boga, J.A.; Manchester, L.C.; Fuentes-Broto, L.; Korkmaz, A.; Ma, S.; Tan, D.X.; Reiter, R.J. Alzheimer’s disease: Pathological mechanisms and the beneficial role of melatonin. J. Pineal Res. 2012, 52, 167–202. [Google Scholar] [CrossRef]
- Damiano, M.; Galvan, L.; Deglon, N.; Brouillet, E. Mitochondria in Huntington’s disease. Biochim. Biophys. Acta. 2010, 1802, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Wongprayoon, P.; Govitrapong, P. Melatonin as a mitochondrial protector in neurodegenerative disease. Cell. Mol. Life Sci. 2017, 74, 3999–4014. [Google Scholar] [CrossRef] [PubMed]
- Vallee, A.; Lecarpentier, Y.; Guillevin, R.; Vallee, J.N. Aerobic glycolysis in amyotrophic lateral sclerosis and Huntington’s disease. Rev. Neurosci. 2018, 29, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Sandhu, A.F.; Rungratanawanich, W.; Williams, G.E.; Akbar, M.; Zhou, S.; Song, B.J.; Wang, X. Melatonin and autophagy in aging-related neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 7174. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Targeting the mitochondrial pyruvate carrier for neuroprotection. Brain Sci. 2019, 9, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Zhang, T.; Lee, T.H. Cellular mechanisms of melatonin: Insight from neurodegenerative diseases. Biomolecules 2020, 10, 1158. [Google Scholar] [CrossRef]
- Podrini, C.; Cassina, L.; Boletta, A. Metabolic reprogramming and the role of mitochondria in polycystic kidney disease. Cell Signal. 2020, 67, 109495. [Google Scholar] [CrossRef]
- Millet-Boureima, C.; Rozencwaig, R.; Polyak, F.; Gamberi, C. Cyst reduction by melatonin in a novel Drosophila model of polycystic kidney disease. Molecules 2020, 25, 5477. [Google Scholar] [CrossRef]
- Morita, M.; Kanasaki, K. Sodium-glucose cotransporter-2 inhibitors for diabetic kidney disease: Targeting Warburg effects in proximal tubular cells. Diabetes Metab. 2020, 46, 353–361. [Google Scholar] [CrossRef]
- Promsan, S.; Lungkaphin, A. The roles of melatonin on kidney injury in obese and diabetic conditions. Biofactors 2020, 46, 531–549. [Google Scholar] [CrossRef]
- Del Valle Bessone, C.; Fajreldines, H.D.; de Barboza, G.E.D.; de Talamoni, N.G.; Allemandi, D.A.; Carpentieri, A.R.; Quinteros, D.A. Protective role of melatonin in retinal ganglionar cell: In vitro and in vivo evidence. Life Sci. 2019, 218, 233–240. [Google Scholar] [CrossRef]
- Yu, Z.; Yanxia, H.; Limin, G.; Yun, Z.; Mingxuan, Z.; Fuyao, X.; Cheng, T.; Jufang, H.; Dan, D. Melatonin alleviates pyroptosis of retinal neurons following acute intraocular hypertension. CNS Neurol. Disord. Drug Targets 2020. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Wu, D.; Dasgupta, A.; Chen, K.H.; Mewburn, J.; Potus, F.; Lima, P.D.A.; Hong, Z.; Zhao, Y.Y.; Hindmarch, C.C.T.; et al. Epigenetic metabolic reprogramming of right ventricular fibroblasts in pulmonary arterial hypertension: A pyruvate dehydrogenase kinase-dependent shift in mitochondrial metabolism promotes right ventricular fibrosis. Circ. Res. 2020, 126, 1723–1735. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Liang, S.; Zhang, J.; Du, Z.; Xu, Q.; Duan, J.; Sun, Z. Melatonin ameliorates PM 2.5-induced cardiac perivascular fibrosis through regulating mitochondrial redox homeostasis. J. Pineal Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cottrill, K.A.; Chan, S.Y. Metabolic dysfunction in pulmonary hypertension: The expanding relevance of the Warburg effect. Eur. J. Clin. Investig. 2013, 43, 855–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLean, M.R. Melatonin: Shining some light on pulmonary hypertension. Cardiovasc. Res. 2020, 116, 2036–2037. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiid, I.; Hoal-van Helden, E.; Hon, D.; Lombard, C.; van Helden, P. Potentiation of isoniazid activity against mycobacterium tuberculosis by melatonin. Antimicrob. Agents Chemother. 1999, 43, 975–977. [Google Scholar] [CrossRef] [Green Version]
- Ji, R.; Chen, W.; Wang, Y.; Gong, F.; Huang, S.; Zhong, M.; Liu, Z.; Chen, Y.; Ma, L.; Yang, Z.; et al. The Warburg effect promotes mitochondrial injury regulated by uncoupling protein-2 in septic acute kidney injury. Shock 2020. [Google Scholar] [CrossRef]
- Biancatelli, R.M.L.C.; Berill, M.; Mohammed, Y.H.; Marik, P.E. Melatonin for the treatment of sepsis: The scientific rationale. J. Thoracic Dis. 2020, 12, S54–S65. [Google Scholar] [CrossRef]
- Ma, W.Q.; Sun, X.J.; Zhu, Y.; Liu, N.E. PDK4 promotes vascular calcification by interfering with autophagic activity and metabolic reprogramming. Cell Death Dis. 2020, 11, 991. [Google Scholar] [CrossRef]
- Sezgin, D.; Aslan, G.; Sahin, K.; Tuzcu, M.; Llhan, N.; Sahna, E. The effects of melatonin against atherosclerosis-induced endothelial dysfunction and inflammation in hypercholesterolemic rats. Arch. Physiol. Biochem. 2020, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ramirez, I.; Carillo-Garcia, A.; Contreras-Paredes, A.; Ortiz-Sanchez, E.; Cruz-Gregorio, A.; Lizano, M. Regulation of cellular metabolism by high-risk human papillomaviruses. Int. J. Mol. Sci. 2018, 19, 1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Huang, Y.; Song, S. Inhibiting the HPV16 oncogene-mediated glycolysis sensitizes human cervical carcinoma cells to 5-flourouracil. Onco Targets Ther. 2019, 12, 6711–6720. [Google Scholar] [CrossRef] [PubMed]
- Di Sotto, A.; di Giacomo, S.; Amatore, D.; Locatelli, M.; Vitalone, A.; Toniolo, C.; Rotino, G.L.; Scalzo, R.L.; Palamara, A.T.; Marcocci, M.E.; et al. A polyphenol rich extract from Solanum melongena L. DR2 peel exhibits antioxidant properties and anti-herpes simplex virus type 1 activity in vitro. Molecules 2018, 23, 2066. [Google Scholar] [CrossRef] [Green Version]
- da Silva Nunez, O.; de Souza Pereira, R. Regression of herpes viral infection symptoms using melatonin and SB-73: Comparison with Acyclovir. J. Pineal Res. 2008, 44, 373–378. [Google Scholar]
- Icard, P.; Lincet, H.; Wu, Z.; Coquerel, A.; Forgez, P.; Alifano, M.; Fournel, L. The key role of Warburg effect in SARS-CoV-2 replication and associated inflammatory response. Biochimie 2020, 180, 169–177. [Google Scholar] [CrossRef]
- Ramlall, V.; Zuker, J.; Tatonetti, N. Melatonin significantly associated with survival of intubated COVID-19 patients. medRxiv. 2020. [Google Scholar] [CrossRef]
- Tarasenko, T.N.; Jestin, M.; Matsumoto, S.; Saito, K.; Hwang, S.; Gavrilova, O.; Trivedi, N.; Zerfas, P.M.; Barca, E.; DiMauro, S.; et al. Macrophage derived TNFα promotes hepatic reprogramming to Warburg-like metabolism. J. Mol. Med. 2019, 97, 1231–1243. [Google Scholar] [CrossRef]
- Crespo, I.; Fernandez-Palanca, P.; San Miguel, B.; Alvarez, M.; Gonzalez-Gallego, J.; Tunon, M.J. Melatonin modulates mitophagy, innate immunity and circadian clocks in a model of viral-induced fulminant hepatic failure. J. Cell. Mol. Med. 2020, 24, 7625–7636. [Google Scholar] [CrossRef]
- Dagenais-Lussier, X.; Beji, C.; Telittchenko, R.; Routy, J.P.; van Grevenynghe, J. Plasticity of T-cell mitochondrial metabolism: A necessary peacekeeper during the troubled times of persistent HIV-1 infection. Cytokine Growth Factor Rev. 2020, 55, 26–36. [Google Scholar]
- Lissoni, P.; Vigore, L.; Rescaldani, R.; Rovelli, F.; Brivio, F.; Giani, L.; Barni, S.; Tancini, G.; Ardizzoia, A.; Vigano, M.G. Neuroimmunotherapy with low-dose subcutaneous interleukin-2 plus melatonin in AIDS patients with CD4 cell number below 200/mm3: A biological phase-II study. J. Biol. Regul. Homeost. Agents 1995, 9, 155–158. [Google Scholar] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, A.M.; Smith, C.O.; Shum, L.C.; Awad, H.; Eliseev, R.A. Lactate Dehydrogenase Inhibition with Oxamate Exerts Bone Anabolic Effect. J. Bone Miner. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S. Melatonin: An endogenous miraculous indolamine, fights against cancer progression. J. Cancer Res. Clin. Oncol. 2020, 146, 1893–1922. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Tumour microenvironment: Roles of the aryl hydrocarbon receptor, O-GlcNAcylation, acetyl-CoA and melatonergic pathway in regulating dynamic metabolic interactions across cell types-Tumor microenvironment and metabolism. Int. J. Mol. Sci. 2020, 22, 141. [Google Scholar] [CrossRef]
- Huang, W.Y.; Jou, M.J.; Peng, T.I. mtDNA T8993G mutation-induced F1F0-ATP synthase defect augments mitochondrial dysfunction associated with hypoxia/reoxygenation: The protective role of melatonin. PLoS ONE 2013, 8, e81546. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Rosales-Corral, S.A. Melatonin reprograms glucose metabolism in cancer cell mitochondria. Endocrinol. Diabet. Metab. 2019, 1, 52–60. [Google Scholar]
- Reiter, R.J.; Richardson, B.A.; Johnson, L.Y.; Ferguson, B.N.; Dinh, D.T. Pineal melatonin rhythm: Reduction in aging Syrian hamsters. Science 1980, 210, 1372–1373. [Google Scholar] [CrossRef]
- Reiter, R.J.; Craft, C.M.; Johnson, J.E., Jr.; King, T.S.; Richardson, B.A.; Vaughan, G.M.; Vaughan, M.K. Age-associated reduction in nocturnal pineal melatonin levels in female rats. Endocrinology 1981, 109, 1295–1297. [Google Scholar] [CrossRef]
- Sack, R.L.; Lewy, A.J.; Erb, D.L.; Vollmer, W.M.; Singer, C.M. Human melatonin production decreases with age. J. Pineal Res. 1986, 3, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Scholtens, R.M.; van Munster, B.C.; van Kempen, M.F.; de Rooij, S.E. Physiological melatonin levels in healthy older people: A systematic review. J. Psychosom Res 2016, 86, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Jauhari, A.; Baranov, S.V.; Suofu, Y.; Kim, J.; Singh, T.; Yablonska, S.; Li, F.; Wang, X.; Oberly, P.; Minnigh, M.B.; et al. Melatonin inhibits cytosolic mitochondrial DNA-induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J. Clin. Investig. 2020, 130, 3124–3136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhdanova, I.V.; Wurtman, R.J.; Balcioglu, A.; Kartashov, A.I.; Lynch, H.J. Endogenous melatonin levels and the fate of exogenous melatonin: Age effects. J. Gerontol. A Biol. Sci. Med. Sci. 1998, 53, B293-8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimenez, V.M.; Prado, N.; Diez, E.; Manucha, W.; Reiter, R.J. New proposal involving nanoformulated melatonin targeted to the mitochondria as a potential COVID-19 treatment. Nanomedicine 2020. [Google Scholar] [CrossRef]
- Andersen, L.P.; Gogenur, I.; Rosenberg, J.; Reiter, R.J. The Safety of Melatonin in Humans. Clin. Drug Investig. 2016, 36, 169–175. [Google Scholar] [CrossRef]
- Zetner, D.; Andersen, L.P.K.; Alder, R.; Jessen, M.L.; Tolstrup, A.; Rosenberg, J. Pharmacokinetics and Safety of Intravenous, Intravesical, Rectal, Transdermal, and Vaginal Melatonin in Healthy Female Volunteers: A Cross-Over Study. Pharmacology 2020, 1–8. [Google Scholar] [CrossRef]
- De Bleecker, J.L.; Lamont, B.H.; Verstraete, A.G.; Schelfhout, V.J. Melatonin and painful gynecomastia. Neurology 1999, 53, 435–436. [Google Scholar] [CrossRef]
- Grilo-Bensusan, I.; Gomez-Delgado, E.; Gomez-Regife, L. Melatonin as a possible cause of diarrhoea. Rev. Esp. Enferm. Dig. 2015, 107, 119–120. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Types that Display Warburg Effect | References Reporting Warburg Metabolism | References Indicating Melatonin Inhibits these Cell Types |
|---|---|---|
| Cancer | ||
| Ewing sarcoma | Yeung et al. [151] | Sanchez-Sanchez et al. [124] |
| Osteosarcoma | Shen et al. [152] | Lu et al. [153] |
| Breast | Tailor et al. [154] | Hill et al. [155] |
| Hepatocellular carcinoma | Li et al. [156] | Elmahallway et al. [157] |
| Glioblastoma | Du et al. [158] | Moretti et al. [159] |
| Thyroid cancer | Yang et al. [160] | Liao et al. [161] |
| Non-small cell lung | De Rosa et al. [162] | Ma et al. [163] |
| Prostate | Schoepke et al. [164] | De Almeida Chuffa et al. [165] |
| Ovarian | Freidus et al. [166] | Bu et al. [167] |
| Colorectal | Fu et al. [168] | Gil-Martin et al. [169] |
| Pancreatic | Annas et al. [170] | Tamtaji et al. [171] |
| Cervical | Zhang et al. [172] | Wang et al. [173] |
| Stomach | Liu et al. [174] | Liu et al. [175] |
| Melanoma (cutaneous) | Kumar et al. [176] | Alvarez-Artime et al. [177] |
| Myeloproliferative | Baumeister et al. [178] | Shafabakhseh et al. [179] |
| Bladder | Alfonso et al. [180] | Chen et al. [181] |
| Endometrial | Salama et al. [182] | Gu et al. [183] |
| Renal Cell Carcinoma | Lessi et al. [184] | Wen et al. [185] |
| Non-Cancer Diseases | ||
| Multiple sclerosis | Kornberg et al. [186] | Lopez-Gonzalez et al. [187] |
| Alzheimer disease | Altante et al. [188] | Rosales-Corral et al. [189] |
| Huntington disease | Damiano et al. [190] | Wongprayoon and Govitrapons et al. [191] |
| Amyotrophic lateral sclerosis | Vallee et al. [192] | Luo et al. [193] |
| Parkinson disease | Tang et al. [194] | Chen et al. [195] |
| Polycystic kidney disease | Podrini et al. [196] | Millet-Boureima et al. [197] |
| Diabetic kidney disease | Morita and Kawaski et al. [198] | Promsan and Lungkaphin et al. [199] |
| Glaucoma | Del Valle et al. [200] | Yu et al. [201] |
| Fibrosis | Tian et al. [202] | Jiang et al. [203] |
| Pulmonary hypertension | Cottrill and Chan [204] | MacLean [205] |
| Myocbacterium tuberculosis | Krawczyk et al. [206] | Wiid et al. [207] |
| Septic shock | Ji et al. [208] | Colunga-Biancatelli et al. [209] |
| Atherosclerosis | Ma et al. [210] | Sezgin et al. [211] |
| Human papilloma virus | Martinez-Ramirez et al. [212] | Ma et al. [213] |
| Herpes simplex | Di Sotto et al. [214] | Nunez-Oda and Pereira -Rdo [215] |
| SARS-CoV-2 | Icard et al. [216] | Ramlall et al. [217] |
| Viral infected hepatocytes | Tarasenko et al. [218] | Crespo et al. [219] |
| HIV/AIDS | Dagenais-Lussier et al. [220] | Lissoni et al. [221] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reiter, R.J.; Sharma, R.; Rosales-Corral, S. Anti-Warburg Effect of Melatonin: A Proposed Mechanism to Explain its Inhibition of Multiple Diseases. Int. J. Mol. Sci. 2021, 22, 764. https://doi.org/10.3390/ijms22020764
Reiter RJ, Sharma R, Rosales-Corral S. Anti-Warburg Effect of Melatonin: A Proposed Mechanism to Explain its Inhibition of Multiple Diseases. International Journal of Molecular Sciences. 2021; 22(2):764. https://doi.org/10.3390/ijms22020764
Chicago/Turabian StyleReiter, Russel J., Ramaswamy Sharma, and Sergio Rosales-Corral. 2021. "Anti-Warburg Effect of Melatonin: A Proposed Mechanism to Explain its Inhibition of Multiple Diseases" International Journal of Molecular Sciences 22, no. 2: 764. https://doi.org/10.3390/ijms22020764
APA StyleReiter, R. J., Sharma, R., & Rosales-Corral, S. (2021). Anti-Warburg Effect of Melatonin: A Proposed Mechanism to Explain its Inhibition of Multiple Diseases. International Journal of Molecular Sciences, 22(2), 764. https://doi.org/10.3390/ijms22020764