
Identification of the Primary Factors Determining the Specificity of Human VKORC1 Recognition by Thioredoxin-Fold Proteins


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The Trx-Fold Proteins as Possible Partners of VKORC1
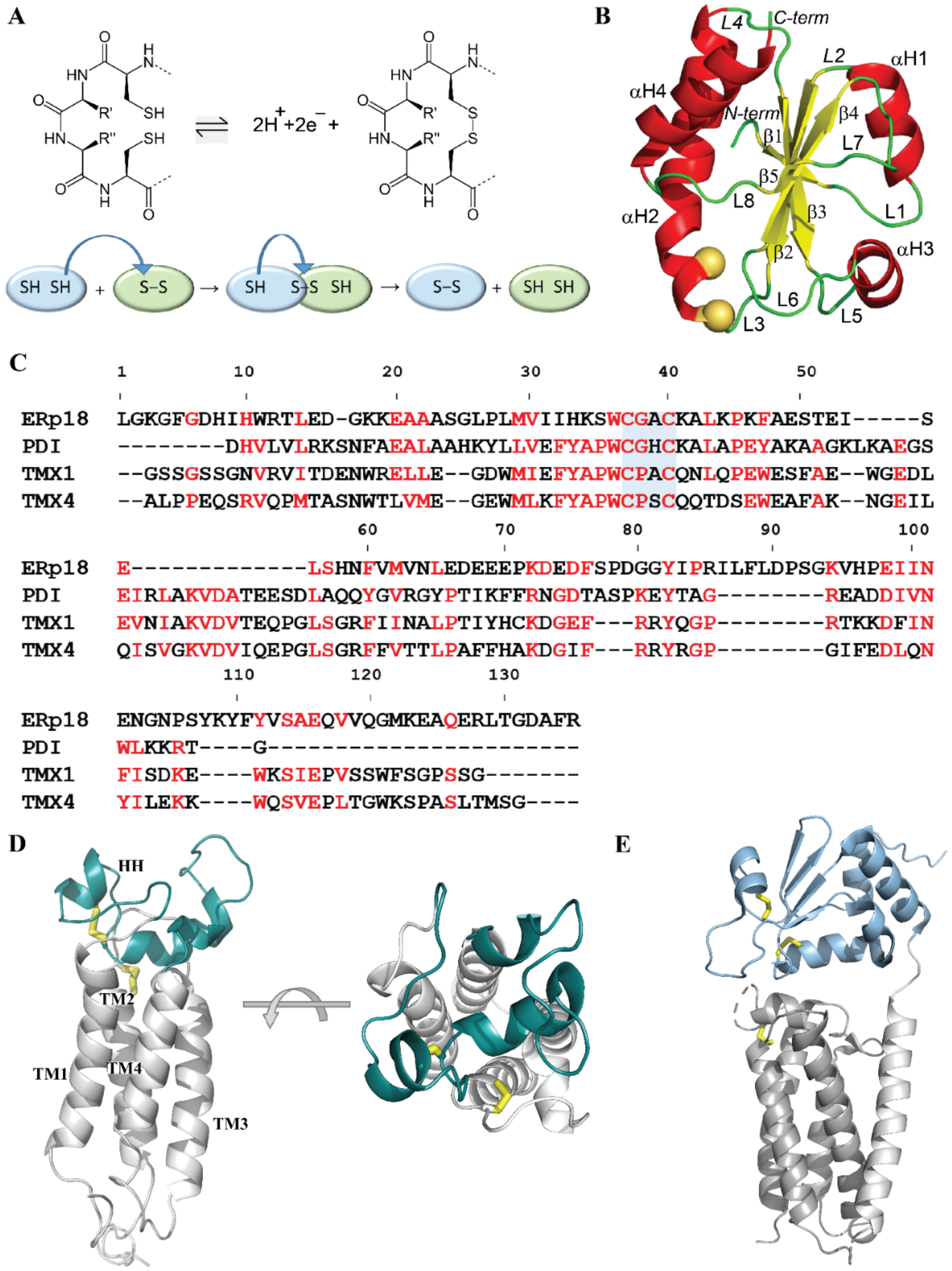
2.1.1. Sequences and Structural Data
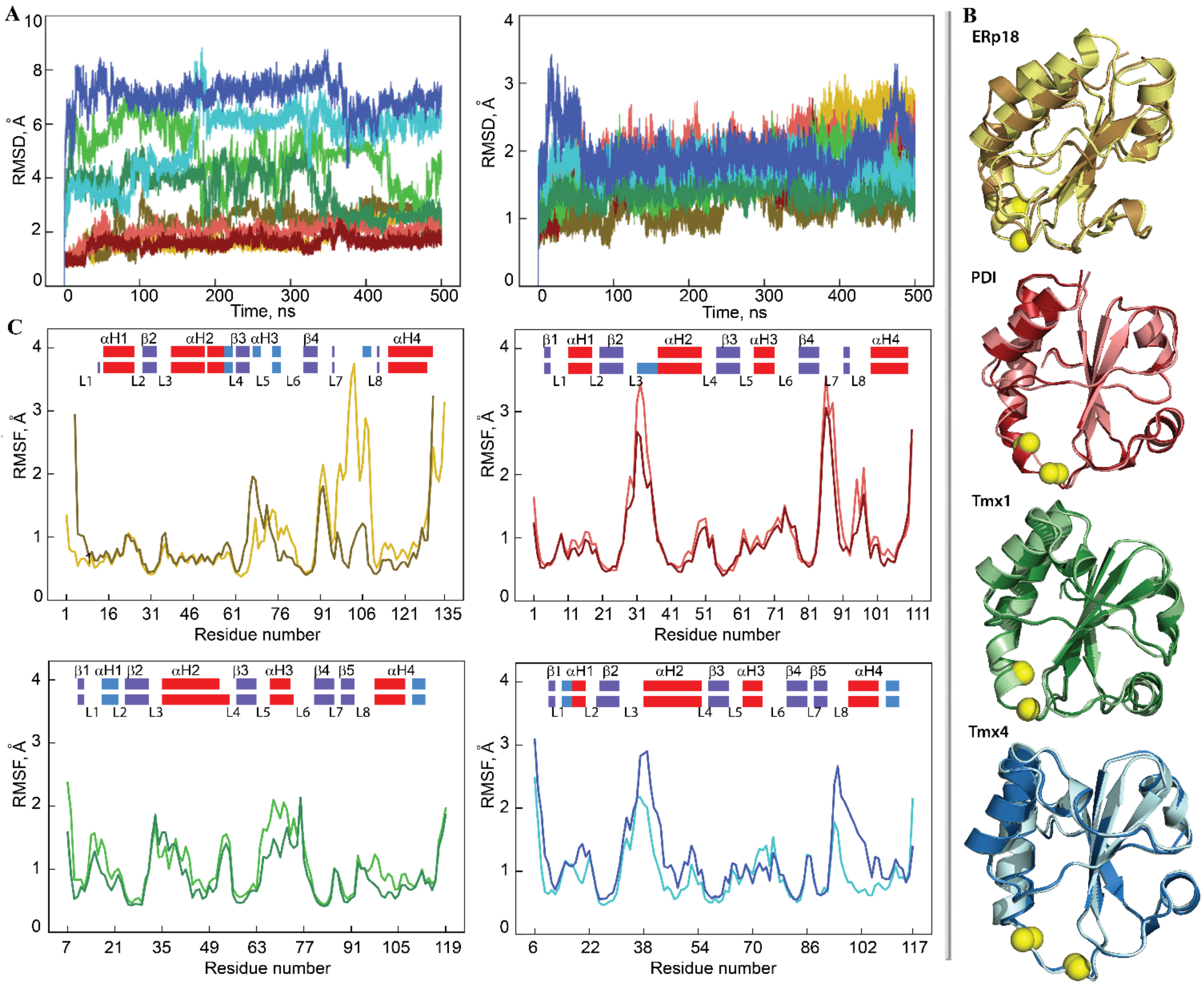
2.1.2. General Characterisation of Trx-Fold Proteins Using MD Simulations
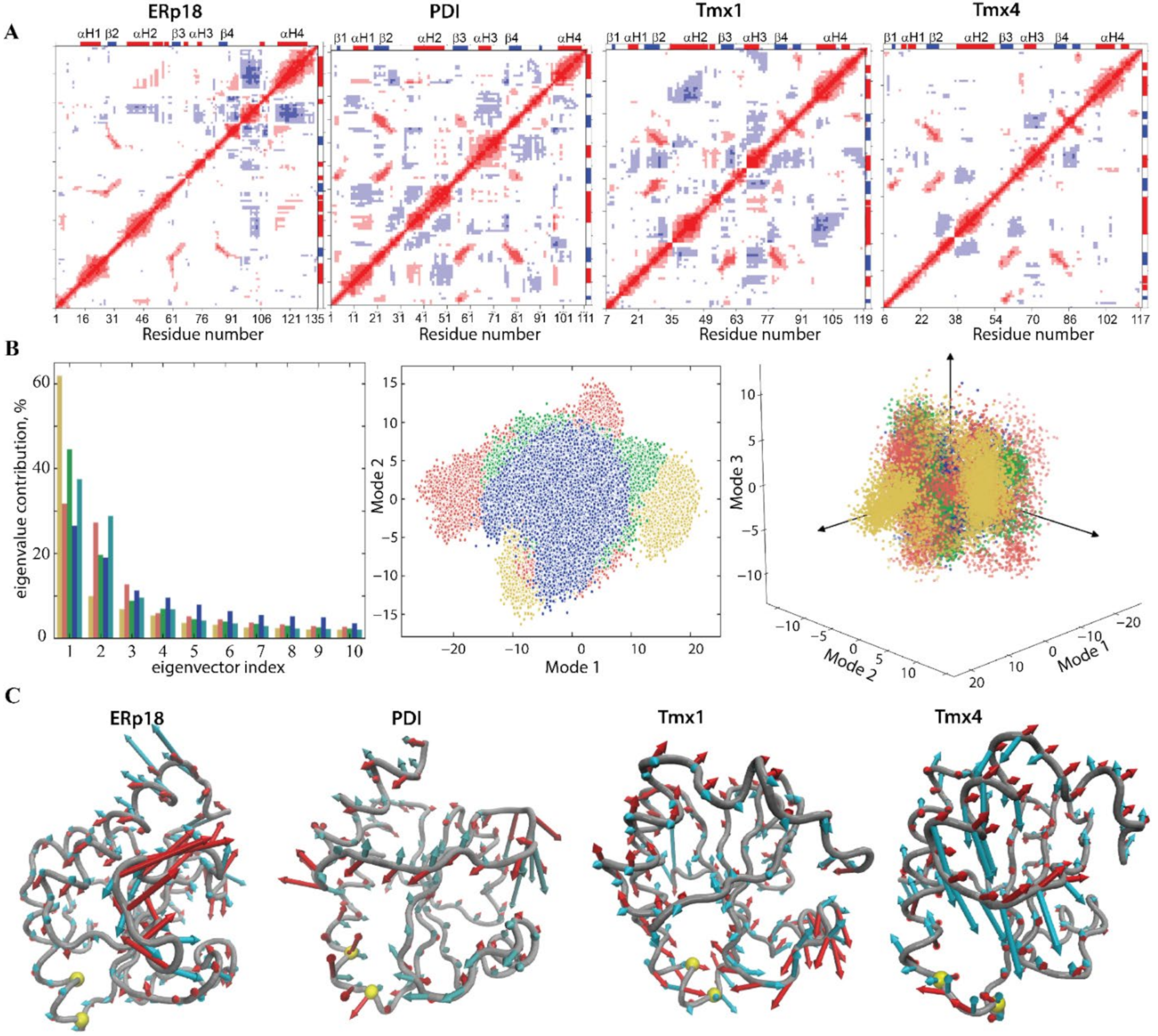
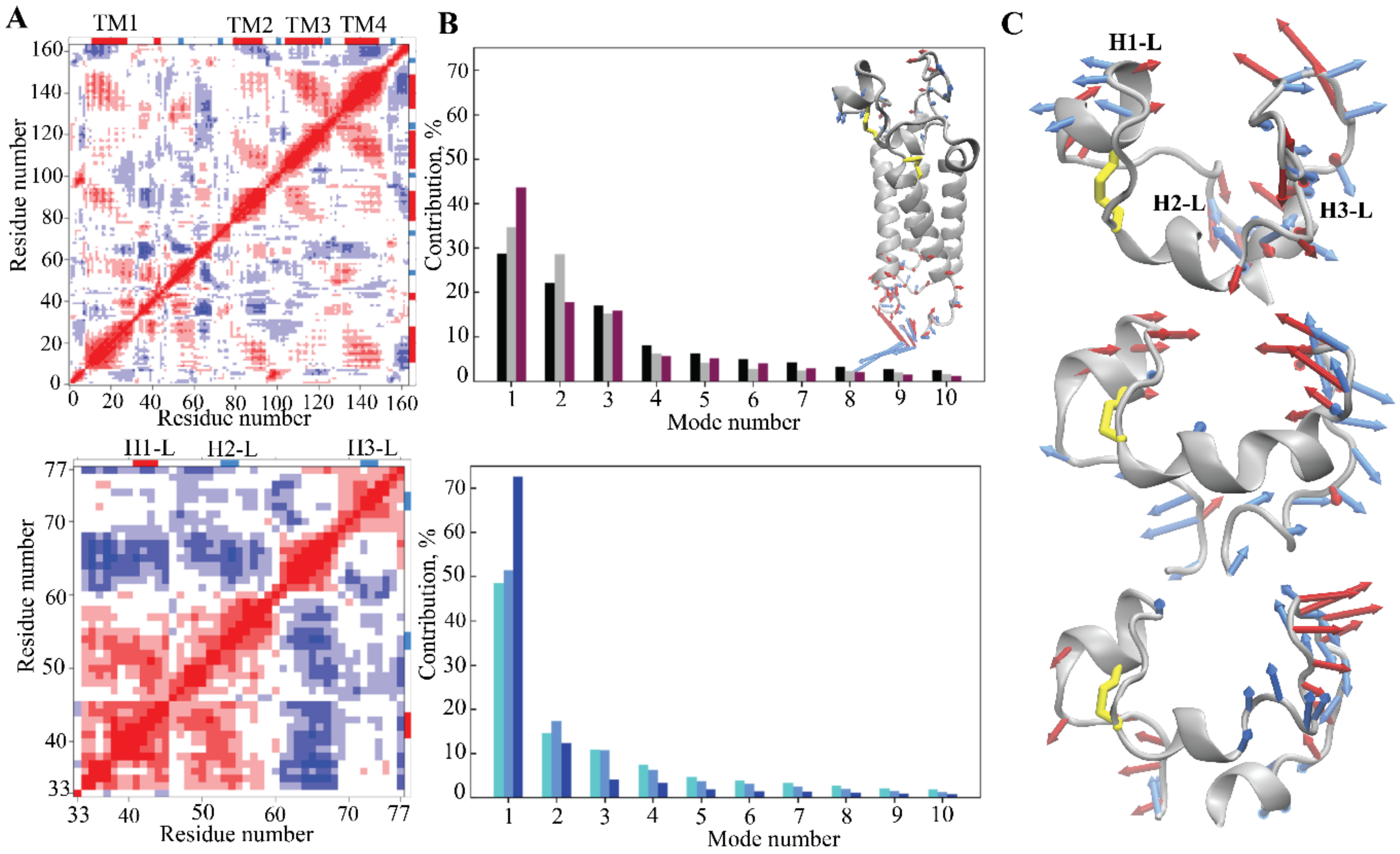
2.1.3. Intrinsic Motion and Its Interdependence on Trx-Folded Proteins
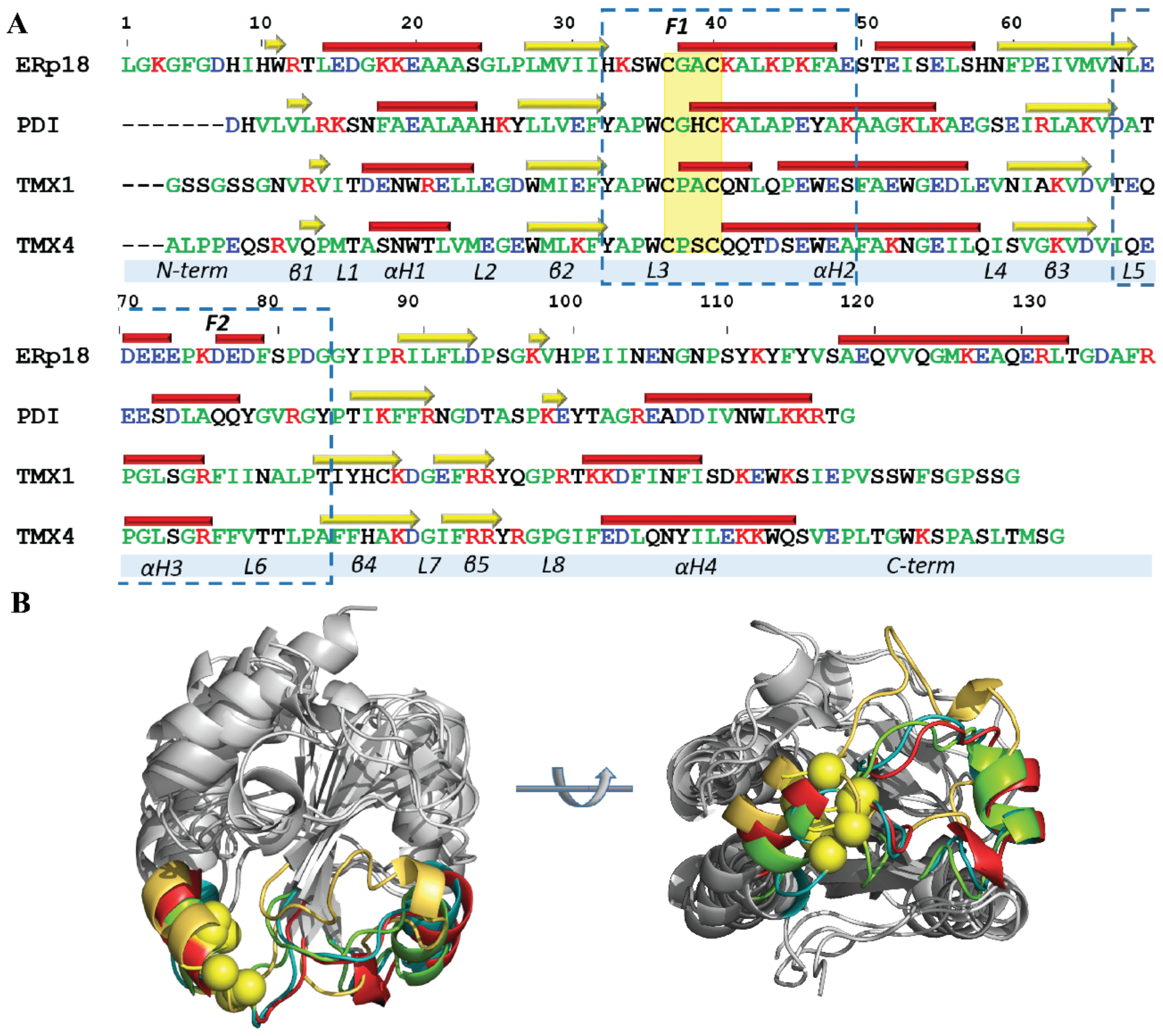
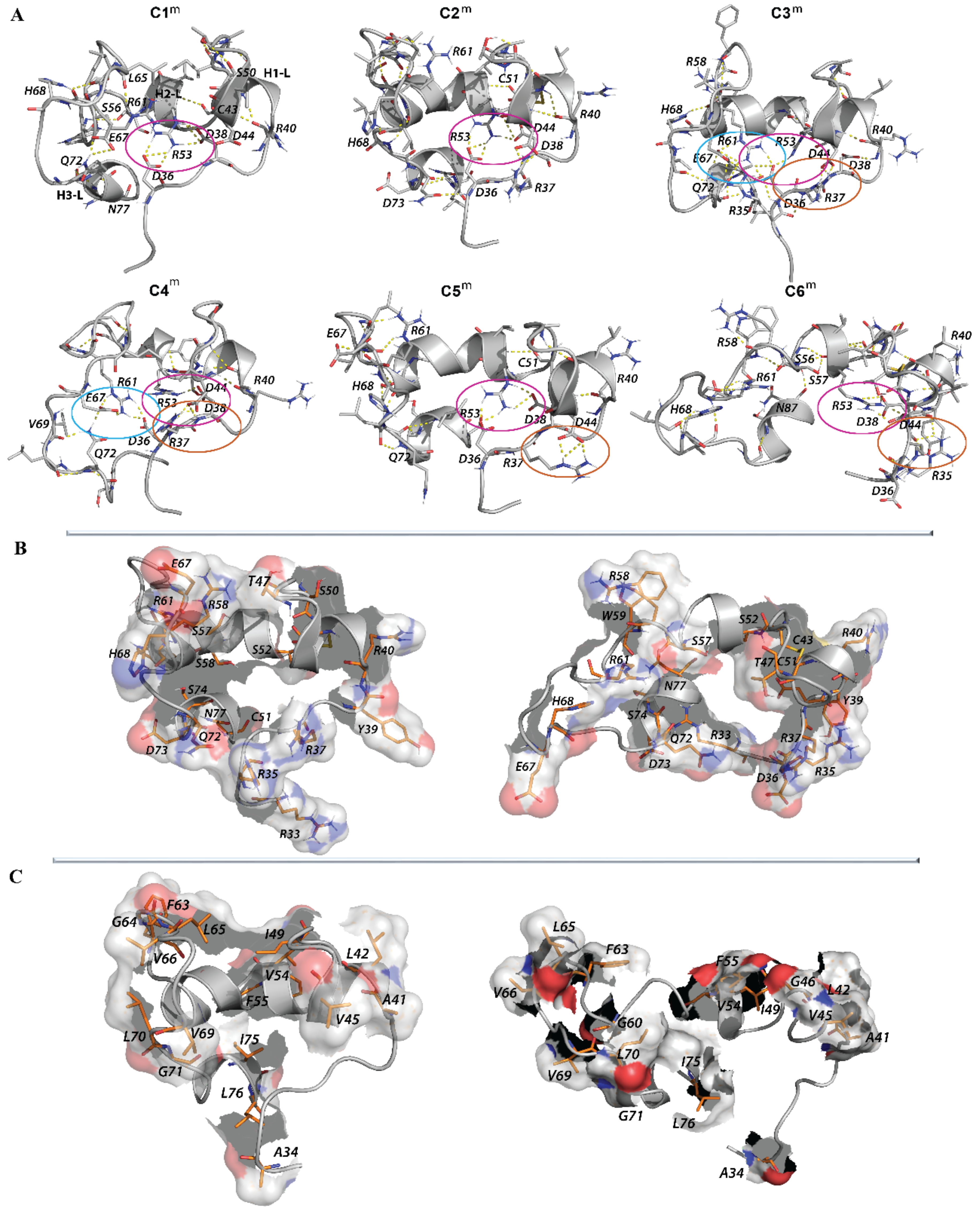
2.1.4. Focus on the Region of Trx-Fold Proteins Potentially Involved in Target Recognition and/or Electron Transfer Reaction
2.1.5. Geometry of the CX1X2C Motif
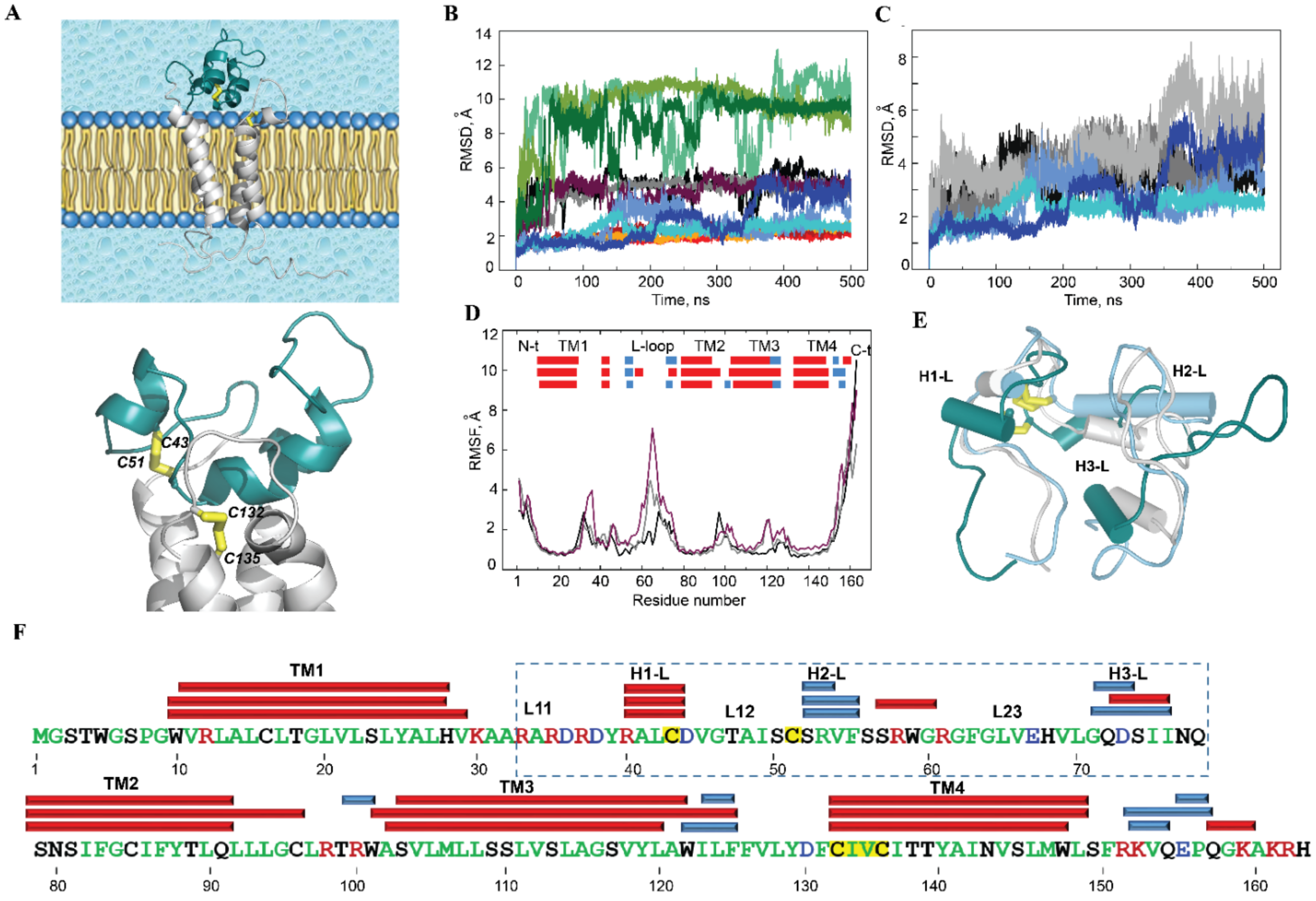
2.2. Human VKORC1 Viewed as the Target of a Trx-Fold Protein
2.2.1. General Characterisation
2.2.2. The Luminal Loop of hVKORC1: Structure and Dynamics
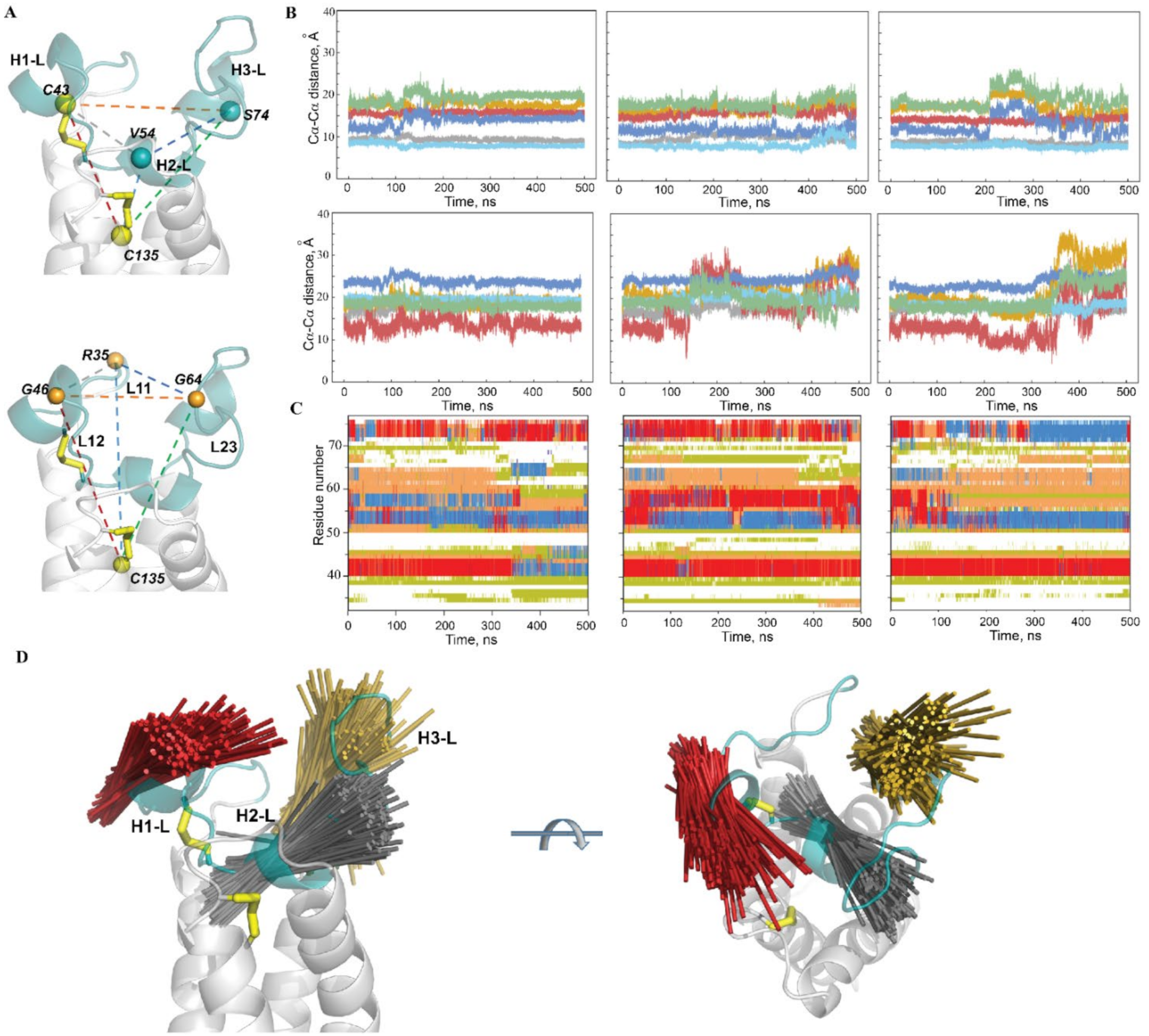
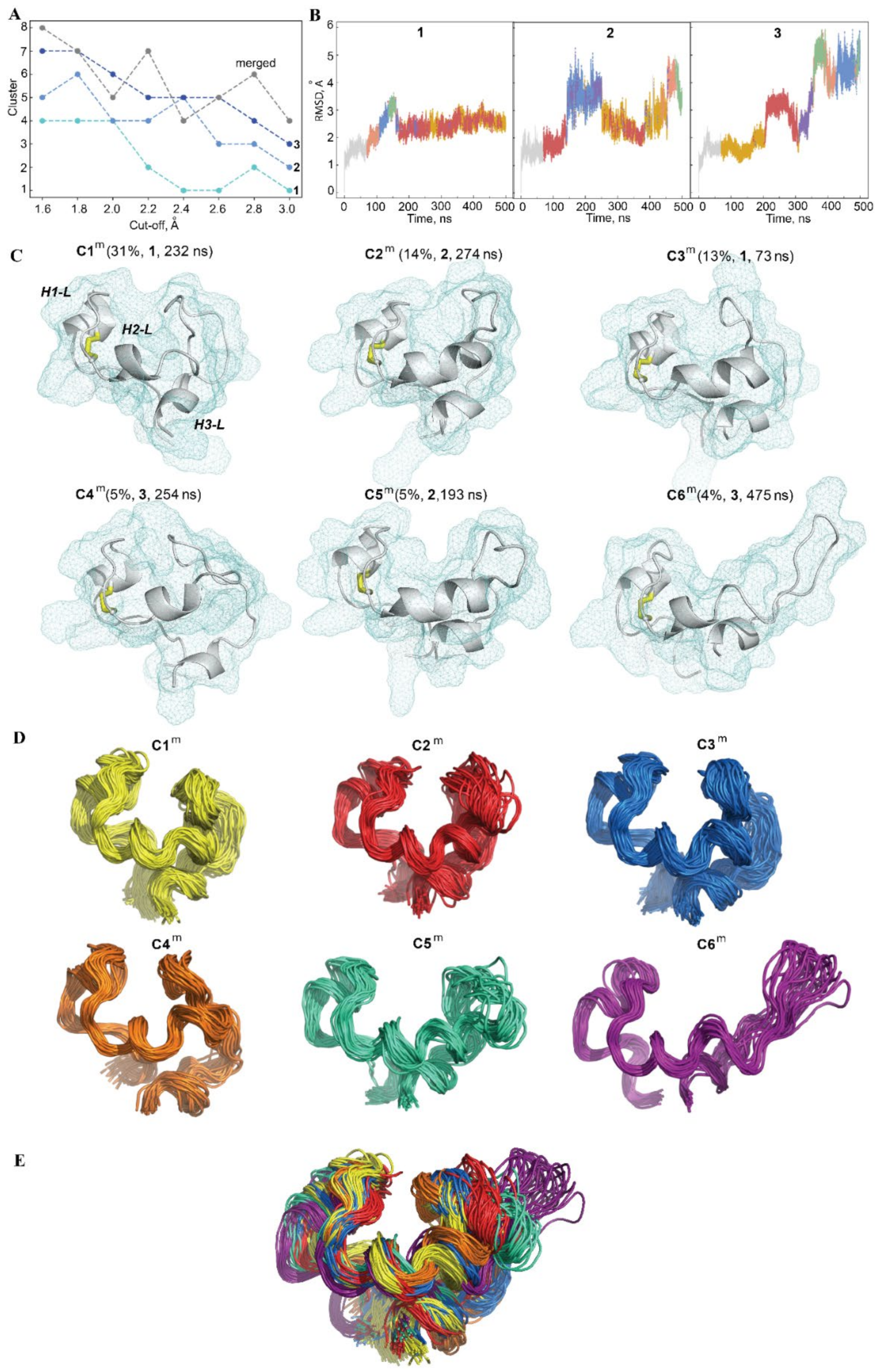
2.2.3. Conformational Variability of the hVKORC1 L-Loop
2.2.4. Intra-L-Loop Interactions
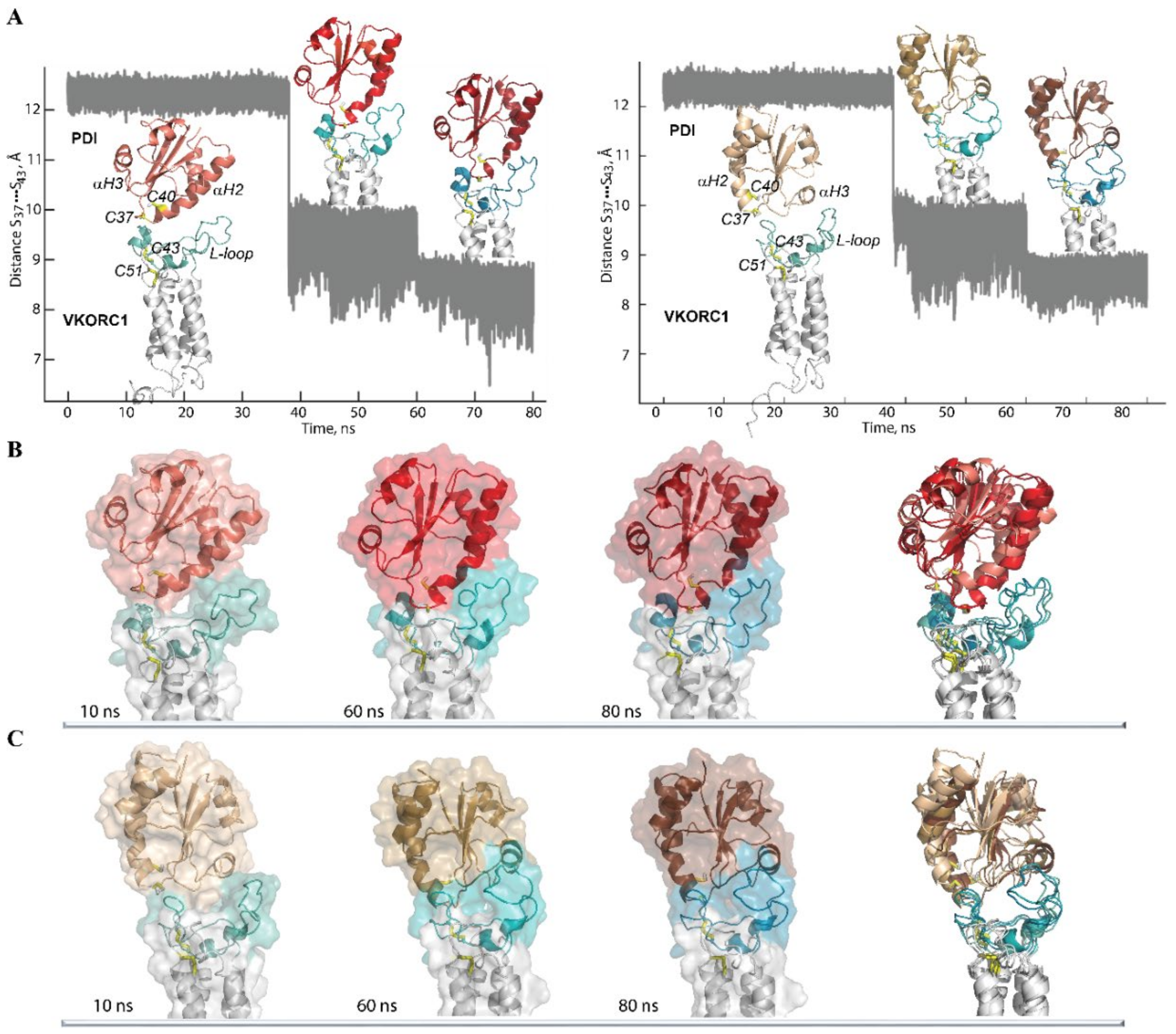
2.3. Modelling of Molecular Complex Formed by hVKORC1 and Its Redox Partners
3. Discussion
4. Materials and Methods
4.1. 3D Models
4.2. Molecular Dynamics Simulation
4.2.1. Preparation of the Systems
4.2.2. Set-Up of the Systems
4.2.3. Production of the MD Trajectories
4.2.4. The Stepped MD Simulations of the PDI–hVKORC1 Complex
4.3. Data Analysis
4.3.1. Conventional Analysis of the MD Trajectories
- (1)
- The RMSD and RMSF values were calculated for the Cα-atoms using the initial model (at t = 0 ns) as a reference. All analysis was performed on the MD conformations (every 10 ps) by considering either all simulations or the production part of the simulation, which was generated after the removal of non-well-equilibrated conformations (0–70 ns), as was shown by the RMSDs, or on residues with a fluctuation of less than 4 Å, as shown by the RMSFs. For hVKORC1, the RMSDs were individually calculated for each domain after least-square fittings of the MD conformations to the initial conformation of a domain, thus removing rigid-body motion from the analysis.
- (2)
- Secondary structural propensities for all residues were calculated using the Define Secondary Structure of Proteins (DSSP) method [71]. The secondary structure types were assigned for residues based on backbone -NH and -CO atom positions. Secondary structures were assigned every 10 and 20 ps for the individual and concatenated trajectories, respectively.
- (3)
- The dynamic cross-correlation (DCC) between all atoms within a molecule quantifies the correlation coefficients of motions between atoms, i.e., the degree to which the atoms move together [72]. Calculations were performed on backbone Cα-atoms on the productive simulation time of each MD trajectory using an ensemble-based approach [28]. The correlation values vary between −1 and 1, where 1 illustrates a complete correlation, −1 a complete anti-correlation and 0 no correlation.
- (4)
- The collective motions of proteins were investigated by principal component analysis (PCA). For an N-atom system, a trajectory matrix contains, in each column, Cartesian coordinates for a given atom at each time step . Fitting the coordinate data to a reference structure results in the proper trajectory matrix . The trajectory data is then used to generate a covariance matrix , elements of which are defined as in Equation (1)where denotes an average performed over all the time steps of the trajectory.The principal components (PCs) are obtained by a diagonalisation of the covariance matrix (see Equation (2)).This results in a diagonal matrix containing eigenvalues as diagonal entries and a matrix containing the corresponding eigenvectors. If the eigenvectors are sorted such that their eigenvalues are in decreasing order, the eigenvector with the largest eigenvalues (i.e., the first PCs) accounts for the highest proportion of variance within the data. The second component is orthogonal to the first one and accounts for the second-highest proportion of variance, and so on.
- (5)
- The extent to which the fluctuations of a system are correlated depends on the magnitude of the cross-correlation coefficient (CCij). The CCij of the atomic fluctuations obtained from the MD simulations (CCPCA) were computed using Equation (3):where i and j are two atoms of Cα; Δri and Δrj are displacement vectors of i and j; denotes the transpose of a column vector. If CC(ij) = 1, the fluctuations of i and j are completely correlated.If CC(ij) = −1, the fluctuations of i and j are completely anticorrelated. If CC(ij) = 0, the fluctuations of i and j are not correlated. All snapshots were fitted using the transmembrane domain Cα as a reference before performing cross-correlation analysis.
- (6)
- Clustering analysis was performed on the productive simulation time of each MD trajectory using an ensemble-based approach [28]. The first 70 ns were omitted from the analysis of Trx-fold proteins. The analysis was performed every 100 ps.The algorithm extracts representative MD conformations from a trajectory by clustering the recorded snapshots according to their Cα-atom RMSDs. The procedure for each trajectory can be described as follows: (i) a reference structure is randomly chosen in the MD conformational ensemble, and all conformations within an arbitrary cutoff r are removed from the ensemble; this step is repeated until no conformation remains in the ensemble, providing a set of reference structures at a distance of at least r; (ii) the MD conformations are grouped into n reference clusters based on their RMSDs from each reference structure. The cut-off was set to 2 Å for both clustered proteins or domains (Trx-fold and L-loop) to allow the comparison.
- (7)
- Drift analysis of helices was performed on the L-loop from h-VKORC1 using the centroids (Ci) defined for the main-chain atoms for amino acids (aas) at the top and bottom of each helix. Positions of these centroids were monitored over the MD simulations, and their coordinates were projected on the x–z and y–z planes. The geometry of the CX1X2C motif from the Trx-fold proteins was described by the distance S⋯S between two sulphur atoms from cysteine residues C37 and C40 and the dihedral angle determined as an absolute value of pseudotorsion angle S− Cα37− C40α−S.
- (8)
- H-bonds between heavy atoms (N, O, and S) as potential donors/acceptors were calculated with the following geometric criteria: donor/acceptor distance cut-off was set to 3.6 Å, and the bond angle cut-off was set to 120°. Hydrophobic contacts were considered for all hydrophobic residues with side chains within a distance of 4 Å of each other.Visual inspection of the conformations and figure preparation was made with PyMOL (https://pymol.org/2/). The VMD 1.9.3 program [74] was used to prepare the protein MD animations. To visualise the motions along the principal components, the Normal Mode Wizard (NMWiz) plugin [73], which is distributed with the VMD program, was utilised.
4.3.2. Advanced Methods of Analysis
- (1)
- Metric multidimensional scaling (MDS) is an algorithm for dimension reduction and visualization; it computes an embedding of a set of points (a shape trajectory in our case) in a lower dimension space with respect to the pairwise distances (Kendall’s ones in our case) in the original set [19].The algorithm consists of a minimisation of the cost (see Equation (4)):where is the pairwise distance matrix, and are the embedded points. It can be implemented using the manifold. MDS class in Python’s scikit learn library.
- (2)
- The Fréchet mean of a set is a point minimising the sum of squared distances to each point of the set. As an example, the Fréchet mean of one set of tetrahedrons is defined as in Equation (5):When the distance is the Euclidean distance, the Fréchet mean is no other than the classical mean we know.
- (3)
- Kendall’s shape space of 3D triangles is isometric to the northern hemisphere of a 3D sphere of radius where the equilateral triangle is at the north pole [20]. We use a planar representation of the half-sphere as a disk with the equilateral triangle at the centre by the transformation from the spherical coordinates to the polar coordinates. Each 3D triangle, up to translation rotation and scaling, is represented by a unique point of the disk.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| bVKOR | Vitamin K epoxide reductase from bacteria |
| ERp18 | Endoplasmic reticulum oxidoreductase |
| hVKORC1 | Human vitamin K epoxide reductase |
| ID | Identification code |
| L-loop | Luminal loop |
| MD | Molecular dynamics |
| PCA | Principal component analysis |
| PDB | Protein Database |
| PDI | Protein disulphide isomerase |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| Tmx1 | Thioredoxin-related transmembrane protein 1 |
| Tmx4 | Thioredoxin-related transmembrane protein 4 |
| Trx | Thioredoxin |
References
- Hatahet, F.; Ruddock, L.W. Protein Disulfide Isomerase: A Critical Evaluation of Its Function in Disulfide Bond Formation. Antioxidants Redox Signal. 2009, 11, 2807–2850. [Google Scholar] [CrossRef]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and Thioredoxin Target Proteins: From Molecular Mechanisms to Functional Significance. Antioxidants Redox Signal. 2013, 18, 1165–1207. [Google Scholar] [CrossRef]
- Hudson, D.A.; Gannon, S.A.; Thorpe, C. Oxidative protein folding: From thiol–disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radic. Biol. Med. 2015, 80, 171–182. [Google Scholar] [CrossRef]
- Winther, J.R.; Thorpe, C. Quantification of thiols and disulfides. Biochim. Biophys. Acta 2014, 1840, 838–846. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Chatron, N.; Chalmond, B.; Trouvé, A.; Benoît, E.; Caruel, H.; Lattard, V.; Tchertanov, L. Identification of the functional states of human vitamin K epoxide reductase from molecular dynamics simulations. RSC Adv. 2017, 7, 52071–52090. [Google Scholar] [CrossRef]
- Martin, J.L. Thioredoxin—A fold for all reasons. Structure 1995, 3, 245–250. [Google Scholar] [CrossRef]
- Dobson, C.M.; Karplus, M. The fundamentals of protein folding: Bringing together theory and experiment. Curr. Opin. Struct. Biol. 1999, 9, 92–101. [Google Scholar] [CrossRef]
- Wang, C.L.; Li, W.; Ren, J.; Fang, J.; Ke, H.; Gong, W.; Feng, W.; Wang, C.-C. Structural Insights into the Redox-Regulated Dynamic Conformations of Human Protein Disulfide Isomerase. Antioxidants Redox Signal. 2013, 19, 36–45. [Google Scholar] [CrossRef]
- Guddat, L.W.; Bardwell, J.C.; Martin, J.L. Crystal structures of reduced and oxidized DsbA: Investigation of domain motion and thiolate stabilization. Structure 1998, 6, 757–767. [Google Scholar] [CrossRef]
- Goodstadt, L.; Ponting, C.P. Vitamin K epoxide reductase: Homology, active site and catalytic mechanism. Trends Biochem. Sci. 2004, 29, 289–292. [Google Scholar] [CrossRef]
- Schulman, S.; Wang, B.; Li, W.; Rapoport, T.A. Vitamin K epoxide reductase prefers ER membrane-anchored thioredoxin-like redox partners. Proc. Natl. Acad. Sci. USA 2010, 107, 15027–15032. [Google Scholar] [CrossRef]
- Wajih, N.; Hutson, S.M.; Wallin, R. Disulfide-dependent Protein Folding Is Linked to Operation of the Vitamin K Cycle in the Endoplasmic Reticulum. J. Biol. Chem. 2007, 282, 2626–2635. [Google Scholar] [CrossRef]
- Tie, J.-K.; Jin, D.-Y.; Stafford, D.W. Mycobacterium tuberculosis Vitamin K Epoxide Reductase Homologue Supports Vitamin K–Dependent Carboxylation in Mammalian Cells. Antioxid. Redox Signal. 2012, 16, 329–338. [Google Scholar] [CrossRef]
- Rowe, M.L.; Ruddock, L.W.; Kelly, G.; Schmidt, J.M.; Williamson, R.A.; Howard, M.J. Solution Structure and Dynamics of ERp18, a Small Endoplasmic Reticulum Resident Oxidoreductase. Biochemistry 2009, 48, 4596–4606. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Imai, K.; Mitaku, S. Mechanisms of secondary structure breakers in soluble proteins. Biophysics 2005, 1, 55–65. [Google Scholar] [CrossRef]
- Schwartz, R.; King, J. Frequencies of hydrophobic and hydrophilic runs and alternations in proteins of known structure. Protein Sci. 2006, 15, 102–112. [Google Scholar] [CrossRef]
- Kendall, D.G. Shape Manifolds, Procrustean Metrics, and Complex Projective Spaces. Bull. Lond. Math. Soc. 1984, 16, 81–121. [Google Scholar] [CrossRef]
- Dryden, I.L.; Mardia, K.V. Statistical Shape Analysis: With Applications in R, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016; 496p. [Google Scholar]
- Kortemmea, T.; Creighton, T.E. Ionisation of Cysteine Residues at the Termini of Model α-Helical Peptides. Relevance to Unusual Thiol pKaValues in Proteins of the Thioredoxin Family. J. Mol. Biol. 1995, 253, 799–812. [Google Scholar] [CrossRef]
- Xu, S.; Sankar, S.; Neamati, N. Protein disulfide isomerase: A promising target for cancer therapy. Drug Discov. Today 2014, 19, 222–240. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Jeng, M.-F.; Tennant, L.L.; Slaby, I.; Lindell, M.; Cui, D.-S.; Kuprin, A.S.; Holmgren, A. Effects of Buried Charged Groups on Cysteine Thiol Ionization and Reactivity in Escherichia coli Thioredoxin: Structural and Functional Characterization of Mutants of Asp 26 and Lys 57. Biochemistry 1997, 36, 2622–2636. [Google Scholar] [CrossRef] [PubMed]
- Pinitglang, S.; Noble, M.; Verma, C.; Thomas, E.W.; Brocklehurst, K. Studies on the enhancement of the reactivity of the (Cys-25)-S-/(His159)-Im+H ion-pair of papain by deprotonation across pKa 4. Biochem. Soc. Trans. 1996, 24, 468s. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.-K.; Stafford, D.W. Structural and functional insights into enzymes of the vitamin K cycle. J. Thromb. Haemost. 2016, 14, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.-K.; Stafford, D.W. Functional Study of the Vitamin K Cycle Enzymes in Live Cells. Methods Enzymol. 2017, 584, 349–394. [Google Scholar] [CrossRef]
- Hatahet, F.; Boyd, D.; Beckwith, J. Disulfide bond formation in prokaryotes: History, diversity and design. Biochim. Biophys. Acta 2014, 1844, 1402–1414. [Google Scholar] [CrossRef]
- Lyman, E.; Zuckerman, D.M. Ensemble-Based Convergence Analysis of Biomolecular Trajectories. Biophys. J. 2006, 91, 164–172. [Google Scholar] [CrossRef]
- Pylaeva, S.; Brehm, M.; Sebastiani, D. Salt Bridge in Aqueous Solution: Strong Structural Motifs but Weak Enthalpic Effect. Sci. Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Pace, C.N.; Fu, H.; Fryar, K.L.; Landua, J.; Trevino, S.R.; Shirley, B.A.; Hendricks, M.M.; Iimura, S.; Gajiwala, K.; Scholtz, J.M.; et al. Contribution of Hydrophobic Interactions to Protein Stability. J. Mol. Biol. 2011, 408, 514–528. [Google Scholar] [CrossRef]
- Rishavy, M.A.; Usubalieva, A.; Hallgren, K.W.; Berkner, K.L. Novel Insight into the Mechanism of the Vitamin K Oxidoreductase (VKOR). J. Biol. Chem. 2011, 286, 7267–7278. [Google Scholar] [CrossRef]
- Bevans, C.G.; Krettler, C.; Reinhart, C.; Watzka, M.; Oldenburg, J. Phylogeny of the Vitamin K 2,3-Epoxide Reductase (VKOR) Family and Evolutionary Relationship to the Disulfide Bond Formation Protein B (DsbB) Family. Nutrients 2015, 7, 6224–6249. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.J.; Davies, E.A.; Tucknott, M.G.; Jones, E.Y.; Van Der Merwe, P.A. The role of charged residues mediating low affinity protein-protein recognition at the cell surface by CD2. Proc. Natl. Acad. Sci. USA 1998, 95, 5490–5494. [Google Scholar] [CrossRef] [PubMed]
- Slagle, S.P.; Kozack, R.E.; Subramaniam, S. Role of Electrostatics in Antibody-Antigen Association: Anti-Hen Egg Lysozyme/Lysozyme Complex (HyHEL-5/HEL). J. Biomol. Struct. Dyn. 1994, 12, 439–456. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.A.; Viner, N.J.; Young, S.P.; Petzold, S.J.; Unanue, E.R. A negatively charged anchor residue promotes high affinity binding to the MHC class II molecule I-Ak. J. Immunol. 1996, 157, 755–762. [Google Scholar]
- Stenlund, P.; Lindberg, M.J.; Tibell, L.A.E. Structural Requirements for High-Affinity Heparin Binding: Alanine Scanning Analysis of Charged Residues in the C-Terminal Domain of Human Extracellular Superoxide Dismutase. Biochemistry 2002, 41, 3168–3175. [Google Scholar] [CrossRef]
- Schreiber, G. Kinetic studies of protein–protein interactions. Curr. Opin. Struct. Biol. 2002, 12, 41–47. [Google Scholar] [CrossRef]
- Wade, R.C.; Gabdoulline, R.R.; Lüdemann, S.K.; Lounnas, V. Electrostatic steering and ionic tethering in enzyme-ligand binding: Insights from simulations. Proc. Natl. Acad. Sci. USA 1998, 95, 5942–5949. [Google Scholar] [CrossRef]
- Haberland, J.; Gerke, V. Conserved charged residues in the leucine-rich repeat domain of the Ran GTPase activating protein are required for Ran binding and GTPase activation. Biochem. J. 1999, 343, 653–662. [Google Scholar] [CrossRef]
- Unkles, S.E.; Rouch, D.A.; Wang, Y.; Siddiqi, M.Y.; Glass, A.D.M.; Kinghorn, J.R. Two perfectly conserved arginine residues are required for substrate binding in a high-affinity nitrate transporter. Proc. Natl. Acad. Sci. USA 2004, 101, 17549–17554. [Google Scholar] [CrossRef]
- Zhao, N.; Pang, B.; Shyu, C.-R.; Korkin, D. Charged residues at protein interaction interfaces: Unexpected conservation and orchestrated divergence. Protein Sci. 2011, 20, 1275–1284. [Google Scholar] [CrossRef]
- De Planque, M.R.R.; Bonev, B.B.; Demmers, J.A.A.; Greathouse, D.V.; Koeppe, R.E.; Separovic, F.; Watts, A.; Killian, J.A. Interfacial Anchor Properties of Tryptophan Residues in Transmembrane Peptides Can Dominate over Hydrophobic Matching Effects in Peptide–Lipid Interactions. Biochemistry 2003, 42, 5341–5348. [Google Scholar] [CrossRef]
- Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nat. Cell Biol. 2005, 437, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of Molecular Recognition Features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Davey, N.E.; Gibson, T.J.; Babu, M.M. A Million Peptide Motifs for the Molecular Biologist. Mol. Cell 2014, 55, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Van Der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.W.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Wong, E.T.C.; So, V.; Guron, M.; Kuechler, E.R.; Malhis, N.; Bui, J.M.; Gsponer, J. Protein–Protein Interactions Mediated by Intrinsically Disordered Protein Regions Are Enriched in Missense Mutations. Biomolecules 2020, 10, 1097. [Google Scholar] [CrossRef]
- Dosztányi, Z.; Chen, J.; Dunker, A.K.; Simon, I.; Tompa, P. Disorder and Sequence Repeats in Hub Proteins and Their Implications for Network Evolution. J. Proteome Res. 2006, 5, 2985–2995. [Google Scholar] [CrossRef]
- Vangone, A.; Bonvin, A.M.J.J. Contacts-based prediction of binding affinity in protein–protein complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of Protein–Protein Interactions: What Are the Preferred Ways for Proteins to Interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef]
- Keskin, O.; Ma, B.; Nussinov, R. Hot Regions in Protein–Protein Interactions: The Organization and Contribution of Structurally Conserved Hot Spot Residues. J. Mol. Biol. 2005, 345, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; Bromberg, S.; Yue, K.; Chan, H.S.; Ftebig, K.M.; Yee, D.P.; Thomas, P.D. Principles of protein folding—A perspective from simple exact models. Protein Sci. 2008, 4, 561–602. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-J.; Lin, S.L.; Wolfson, H.J.; Nussinov, R. Studies of protein-protein interfaces: A statistical analysis of the hydrophobic effect. Protein Sci. 1997, 6, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-J.; Ma, B.; Sham, Y.Y.; Kumar, S.; Nussinov, R. Structured disorder and conformational selection. Proteins Struct. Funct. Bioinform. 2001, 44, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-J.; Nussinov, R. Hydrophobic folding units at protein-protein interfaces: Implications to protein folding and to protein-protein association. Protein Sci. 1997, 6, 1426–1437. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Laskowski, R.A. PDBsum: Summaries and analyses of PDB structures. Nucleic Acids Res. 2001, 29, 221–222. [Google Scholar] [CrossRef]
- Case, D.A.; Iii, T.E.C.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Peters, E.; Goga, N.; Berendsen, H.J.C. Stochastic Dynamics with Correct Sampling for Constrained Systems. J. Chem. Theory Comput. 2014, 10, 4208–4220. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The Nose–Hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F.; Berendsen, H.J.C. A Leap-frog Algorithm for Stochastic Dynamics. Mol. Simul. 1988, 1, 173–185. [Google Scholar] [CrossRef]
- Grubmüller, H.; Heller, H.; Windemuth, A.; Schulten, K. Generalized Verlet Algorithm for Efficient Molecular Dynamics Simulations with Long-range Interactions. Mol. Simul. 1991, 6, 121–142. [Google Scholar] [CrossRef]
- Hockney, R.W.; Eastwood, J.W. Computer Simulation Using Particles; McGraw-Hill: New York, NY, USA, 1981. [Google Scholar]
- Roe, D.R.; Cheatham, I.T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Hünenberger, P.; Mark, A.; Van Gunsteren, W. Fluctuation and Cross-correlation Analysis of Protein Motions Observed in Nanosecond Molecular Dynamics Simulations. J. Mol. Biol. 1995, 252, 492–503. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stolyarchuk, M.; Ledoux, J.; Maignant, E.; Trouvé, A.; Tchertanov, L. Identification of the Primary Factors Determining the Specificity of Human VKORC1 Recognition by Thioredoxin-Fold Proteins. Int. J. Mol. Sci. 2021, 22, 802. https://doi.org/10.3390/ijms22020802
Stolyarchuk M, Ledoux J, Maignant E, Trouvé A, Tchertanov L. Identification of the Primary Factors Determining the Specificity of Human VKORC1 Recognition by Thioredoxin-Fold Proteins. International Journal of Molecular Sciences. 2021; 22(2):802. https://doi.org/10.3390/ijms22020802
Chicago/Turabian StyleStolyarchuk, Maxim, Julie Ledoux, Elodie Maignant, Alain Trouvé, and Luba Tchertanov. 2021. "Identification of the Primary Factors Determining the Specificity of Human VKORC1 Recognition by Thioredoxin-Fold Proteins" International Journal of Molecular Sciences 22, no. 2: 802. https://doi.org/10.3390/ijms22020802
APA StyleStolyarchuk, M., Ledoux, J., Maignant, E., Trouvé, A., & Tchertanov, L. (2021). Identification of the Primary Factors Determining the Specificity of Human VKORC1 Recognition by Thioredoxin-Fold Proteins. International Journal of Molecular Sciences, 22(2), 802. https://doi.org/10.3390/ijms22020802