Emerging Roles of Protease-Activated Receptors (PARs) in the Modulation of Synaptic Transmission and Plasticity


Abstract
:1. Protease-Activated Receptors (PARs) and Their Ligands in the Nervous System
1.1. PARs Activation and Signaling
1.2. PARs Activation in the Brain
2. PARs’ Roles in the Regulation of Neurotransmission and Synaptic Plasticity
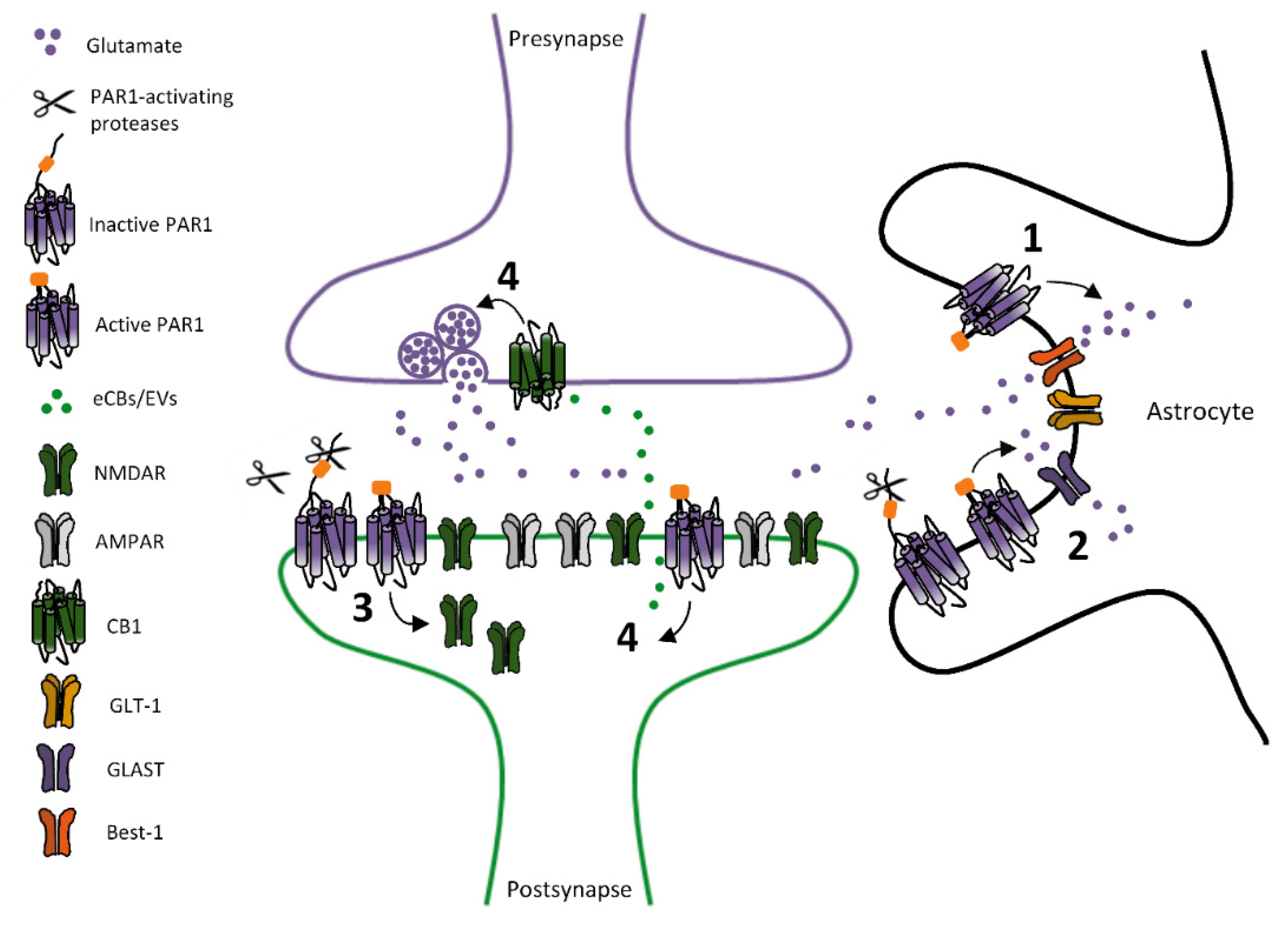
2.1. PARs-Dependent Modulation of Glutamatergic Transmission
2.2. PARs-Dependent Modulation of GABAergic Transmission
3. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Ossovskaya, V.S.; Bunnett, N.W. Protease-activated receptors: Contribution to physiology and disease. Physiol. Rev. 2004, 84, 579–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieseler, F.; Ungefroren, H.; Settmacher, U.; Hollenberg, M.D.; Kaufmann, R. Proteinase-activated receptors (PARs)—Focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun. Signal. 2013, 11, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuberger, D.M.; Schuepbach, R.A. Protease-activated receptors (PARs): Mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. J. Thromb. 2019, 17, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Fox, O.W.; Preston, R.J.S. Molecular basis of protease-activated receptor 1 signaling diversity. J. Thromb. Haemost. 2020, 18, 6–164. [Google Scholar] [CrossRef]
- Ramachandran, R.; Noorbakhsh, F.; DeFea, K.; Hollenberg, M.D. Targeting proteinase-activated receptors: Therapeutic potential and challenges. Nat. Rev. Drug Discov. 2012, 11, 69–86. [Google Scholar] [CrossRef]
- Blackhart, B.D.; Emilsson, K.; Nguyen, D.; Teng, W.; Martelli, A.J.; Nystedt, S.; Sundelin, J.; Scarborough, R.M. Ligand cross-reactivity within the protease-activated receptor family. J. Biol. Chem. 1996, 271, 16466–16471. [Google Scholar] [CrossRef] [Green Version]
- Soh, U.J.; Dores, M.R.; Chen, B.; Trejo, J. Signal transduction by protease-activated receptors. Br. J. Pharm. 2010, 160, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi-Matsui, M.; Zheng, Y.W.; Sulciner, D.J.; Weiss, E.J.; Ludeman, M.J.; Coughlin, S.R. PAR3 is a cofactor for PAR4 activation by thrombin. Nature 2000, 404, 609–613. [Google Scholar] [CrossRef]
- Seminario-Vidal, L.; Kreda, S.; Jones, L.; O’Neal, W.; Trejo, J.; Boucher, R.C.; Lazarowski, E.R. Thrombin promotes release of ATP from lung epithelial cells through coordinated activation of rho- and Ca2+-dependent signaling pathways. J. Biol Chem. 2009, 284, 20638–20648. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Metcalf, M.; Bunnett, N.W. Biased signaling of protease-activated receptors. Front. Endocrinol. 2014, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.R.; Gold, S.J.; Cunningham, D.D.; Gall, C.M. Cellular localization of thrombin receptor mRNA in rat brain: Expression by mesencephalic dopaminergic neurons and codistribution with prothrombin mRNA. J. Neurosci. 1995, 15, 2906–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, M.R.; Derian, C.K.; Leturcq, D.; Baker, S.M.; Brunmark, A.; Ling, P.; Darrow, A.L.; Santulli, R.J.; Brass, L.F.; Andrade-Gordon, P. Characterization of protease-activated receptor-2 immunoreactivity in normal human tissues. J. Histochem. Cytochem. 1998, 46, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niclou, S.P.; Suidan, H.S.; Pavlik, A.; Vejsada, R.; Monard, D. Changes in the expression of protease-activated receptor 1 and protease nexin-1 mRNA during rat nervous system development and after nerve lesion. Eur. J. Neurosci. 1998, 10, 1590–1607. [Google Scholar] [CrossRef] [PubMed]
- Striggow, F.; Riek-Burchardt, M.; Kiesel, A.; Schmidt, W.; Henrich-Noack, P.; Breder, J.; Krug, M.; Reymann, K.G.; Reiser, G. Four different types of protease-activated receptors are widely expressed in the brain and are up-regulated in hippocampus by severe ischemia. Eur. J. Neurosci. 2001, 14, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ubl, J.J.; Reiser, G. Four subtypes of protease-activated receptors, co-expressed in rat astrocytes, evoke different physiological signaling. Glia 2002, 37, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, E.; Reiser, G. Prothrombin/thrombin and the thrombin receptors PAR-1 and PAR-4 in the brain: Localization, expression and participation in neurodegenerative diseases. Thromb. Haemost. 2008, 100, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.K.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068. [Google Scholar] [CrossRef]
- Dihanich, M.; Kaser, M.; Reinhard, E.; Cunningham, D.; Monard, D. Prothrombin mRNA is expressed by cells of the nervous system. Neuron 1991, 6, 575–581. [Google Scholar] [CrossRef]
- Balcaitis, S.; Xie, Y.; Weinstein, J.R.; Andersen, H.; Hanisch, U.; Ransom, B.R.; Möller, T. Expression of proteinase-activated receptors in mouse microglial cells. Neuroreport 2003, 14, 2373–2377. [Google Scholar] [CrossRef]
- Junge, C.E.; Lee, C.J.; Hubbard, K.B.; Zhang, Z.; Olson, J.J.; Hepler, J.R.; Brat, D.J.; Traynelis, S.F. Protease-activated receptor-1 in human brain: Localization and functional expression in astrocytes. Exp. Neurol. 2004, 188, 94–103. [Google Scholar] [CrossRef]
- Nystedt, S.; Emilsson, K.; Wahlestedt, C.; Sundelin, J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 9208–9212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushell, T.J.; Plevin, R.; Cobb, S.; Irving, A.J. Characterization of proteinase-activated receptor 2 signalling and expression in rat hippocampal neurons and astrocytes. Neuropharmacology 2006, 50, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Lohman, R.J.; O’Brien, T.J.; Cocks, T.M. Protease-activated receptor-2 regulates trypsin expression in the brain and protects against seizures and epileptogenesis. Neurobiol. Dis. 2008, 30, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, H.; Connolly, A.J.; Zeng, D.; Kahn, M.L.; Zheng, Y.W.; Timmons, C.; Tram, T.; Coughlin, S.R. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature 1997, 386, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Turgeon, V.L.; Houenou, L.J. The role of thrombin-like (serine) proteases in the development, plasticity and pathology of the nervous system. Brain Res. Brain Res. Rev. 1997, 25, 85–95. [Google Scholar] [CrossRef]
- Turgeon, V.L.; Salman, N.; Houenou, L.J. Thrombin: A neuronal cell modulator. Thromb Res. 2000, 99, 417–427. [Google Scholar] [CrossRef]
- Almonte, A.G.; Sweatt, J.D. Serine proteases, serine protease inhibitors, and protease-activated receptors: Roles in synaptic function and behavior. Brain Res. 2011, 1407, 107–122. [Google Scholar] [CrossRef] [Green Version]
- Qian, Z.; Gilbert, M.E.; Colicos, M.A.; Kandel, E.R.; Kuhl, D. Tissue plasminogen activator is induced as an immediate-early gene during seizure, kindling, and long-term potentiation. Nature 1993, 361, 453–457. [Google Scholar] [CrossRef]
- Sallés, F.J.; Strickland, S. Localization and regulation of the tissue plasminogen activator-plasmin system in the hippocampus. J. Neurosci. 2002, 22, 2125–2134. [Google Scholar] [CrossRef] [Green Version]
- Shin, C.Y.; Kundel, M.; Wells, D.G. Rapid, activity-induced increase in tissue plasminogen activator is mediated by metabotropic glutamate receptor-dependent mRNA translation. J. Neurosci. 2004, 24, 9425–9433. [Google Scholar] [CrossRef]
- Yepes, M.; Lawrence, D.A. New functions for an old enzyme: Nonhemostatic roles for tissue-type plasminogen activator in the central nervous system. Exp. Biol. Med. 2004, 226, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Lochner, J.E.; Honigman, L.S.; Grant, W.F.; Gessford, S.K.; Hansen, A.B.; Silverman, M.A.; Scalettar, B.A. Activity-dependent release of tissue plasminogen activator from the dendritic spines of hippocampal neurons revealed by live-cell imaging. J. Neurobiol. 2006, 66, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gschwend, T.P.; Krueger, S.R.; Kozlov, S.V.; Wolfer, D.P.; Sonderegger, P. Neurotrypsin, a novel multidomain serine protease expressed in the nervous system. Mol. Cell Neurosci. 1997, 9, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Scarisbrick, I.A.; Isackson, P.J.; Ciric, B.; Windebank, A.J.; Rodriguez, M. MSP, a trypsin-like serine protease, is abundantly expressed in the human nervous system. J. Comp. Neurol. 2001, 431, 347–361. [Google Scholar] [CrossRef]
- Bernett, M.J.; Blaber, S.I.; Scarisbrick, I.A.; Dhanarajan, P.; Thompson, S.M.; Blaber, M. Crystal structure and biochemical characterization of human kallikrein 6 reveals that a trypsin-like kallikrein is expressed in the central nervous system. J. Biol. Chem. 2002, 277, 24562–24570. [Google Scholar] [CrossRef] [Green Version]
- Brkic, M.; Balusu, S.; Libert, C.; Vandenbroucke, R.E. MMPs in brain disease friends or foes: Matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediat. Inflam. 2015, 2015, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.L.; Van Nostrand, W.E.; Lau, A.L.; Farrow, J.S.; Suzuki, M.; Bartus, R.T.; Schuppek, R.; Nguyen, A.; Cotman, C.W.; Cunningham, D.D. Co-distribution of protease nexin-1 and protease nexin-2 in brains of non-human primates. Brain Res. 1993, 626, 90–98. [Google Scholar] [CrossRef]
- Reinhard, E.; Suidan, H.S.; Pavlik, A.; Monard, D. Glia-derived nexin/protease nexin-1 is expressed by a subset of neurons in the rat brain. J. Neurosci Res. 1994, 37, 256–270. [Google Scholar] [CrossRef]
- Miranda, E.; Lomas, D.A. Neuroserpin: A serpin to think about. Cell Mol. Life Sci. 2006, 63, 709–722. [Google Scholar] [CrossRef]
- Junge, C.E.; Sugawara, T.; Mannaioni, G.; Alagarsamy, S.; Conn, P.J.; Brat, D.J.; Chan, P.H.; Traynelis, S.F. The contribution of protease-activated receptor 1 to neuronal damage caused by transient focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2003, 100, 13019–13024. [Google Scholar] [CrossRef] [Green Version]
- Nicole, O.; Goldshmidt, A.; Hamill, C.E.; Sorensen, S.D.; Sastre, A.; Lyuboslavsky, P.; Helper, J.R.; McKeon, R.J.; Traynelis, S.F. Activation of protease-activated receptor-1 triggers astrogliosis after brain injury. J. Neurosci. 2005, 25, 4319–4329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Wang, Y.; Reiser, G. Protease-activated receptors in the brain: Receptor expression, activation, and functions in neurodegeneration and neuroprotection. Brain Res. Rev. 2007, 56, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Shrimpton, A.E.; Holohan, P.D.; Bradshaw, C.; Feiglink, D.; Collins, G.H.; Sonderegger, P.; Kinter, J.; Becker, L.M.; Lacbawan, F.; et al. Familial dementia caused by polymerization of mutant Neuroserpin. Nature 1999, 401, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Molinari, F.; Meskanaite, V.; Munnich, A.; Sonderegger, P.; Colleaux, L. Extracellular proteases and their inhibitors in genetic diseases of the central nervous system. Hum. Mol. Genet. 2003, 12, R195–R200. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, S.; Seeds, N.W. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J. Neurochem. 2009, 109, 303–315. [Google Scholar] [CrossRef]
- Frey, U.; Müller, M.; Kuhl, D. A different form of long-lasting potentiation revealed in tissue plasminogen activator mutant mice. J. Neurosci. 1996, 16, 2057–2063. [Google Scholar] [CrossRef] [Green Version]
- Lüthi, A.; Van Der Putten, H.; Botteri, F.M.; Mansuy, I.M.; Meins, M.; Frey, U.; Sansig, G.; Portet, C.; Schmutz, M.; Schröder, M.; et al. Endogenous serine protease inhibitor modulates epileptic activity and hippocampal long-term potentiation. J. Neurosci. 1997, 17, 4688–4699. [Google Scholar] [CrossRef]
- Baranes, D.; Lederfein, D.; Huang, Y.Y.; Chen, M.; Bailey, C.H.; Kandel, E.R. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron 1998, 21, 813–825. [Google Scholar] [CrossRef] [Green Version]
- Madani, R.; Hulo, S.; Toni, N.; Madani, H.; Steimer, T.; Muller, D.; Vassalli, J. Enhanced hippocampal long-term potentiation and learning by increased neuronal expression of tissue-type plasminogen activator in transgenic mice. EMBO J. 1999, 18, 3007–3012. [Google Scholar] [CrossRef] [Green Version]
- Calabresi, P.; Napolitano, M.; Centonze, D.; Marfia, G.A.; Gubellini, P.; Teule, M.A.; Berretta, N.; Bernardi, G.; Frati, L.; Tolu, M.; et al. Tissue plasminogen activator controls multiple forms of synaptic plasticity and memory. Eur. J. Neurosci. 2000, 12, 1002–1012. [Google Scholar] [CrossRef]
- Maggio, N.; Itsekson, Z.; Dominissini, D.; Blatt, I.; Amariglio, N.; Rechavi, G.; Tanne, D.; Chapman, J. Thrombin regulation of synaptic plasticity: Implications for physiology and pathology. Exp. Neurol. 2013, 247, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Ben Shimon, M.; Lenz, M.; Ikenberg, B.; Becker, D.; Shavit Stein, E.; Chapman, J.; Tanne, D.; Pick, C.G.; Blatt, I.; Neufeld, M.; et al. Thrombin regulation of synaptic transmission and plasticity: Implications for health and disease. Front. Cell Neurosci. 2015, 9, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlak, R.; Magarinos, A.M.; Melchor, J.; McEwen, B.; Strickland, S. Tissue plasminogen activator in the amygdala is critical for stress-induced anxiety-like behavior. Nat. Neurosci. 2003, 6, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, R.; Rao, B.S.; Melchor, J.P.; Chattarji, S.; McEwen, B.; Strickland, S. Tissue plasminogen activator and plasminogen mediate stress-induced decline of neuronal and cognitive functions in the mouse hippocampus. Proc. Natl. Acad. Sci. USA 2005, 102, 18201–18206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matys, T.; Pawlak, R.; Matys, E.; Pavlides, C.; McEwen, B.S.; Strickland, S. Tissue plasminogen activator promotes the effects of corticotropin-releasing factor on the amygdala and anxiety-like behavior. Proc. Natl. Acad. Sci. USA 2004, 101, 16345–16350. [Google Scholar] [CrossRef] [Green Version]
- Gingrich, M.B.; Junge, C.E.; Lyuboslavsky, P.; Traynelis, S.F. Potentiation of NMDA receptor function by the serine protease thrombin. J. Neurosci. 2000, 20, 4582–4595. [Google Scholar] [CrossRef]
- Lee, C.J.; Mannaioni, G.; Yuan, H.; Woo, D.H.; Gingrich, M.B.; Traynelis, S.F. Astrocytic control of synaptic NMDA receptors. J. Physiol. 2007, 581, 1057–1081. [Google Scholar] [CrossRef]
- Mannaioni, G.; Orr, A.G.; Hamill, C.E.; Yuan, H.; Pedone, K.H.; McCoy, K.L.; Palmini, R.B.; Junge, C.E.; Lee, C.J.; Yepes, M.; et al. Plasmin potentiates synaptic N-methyl-D-aspartate receptor function in hippocampal neurons through activation of protease-activated receptor-1. J. Biol. Chem. 2008, 283, 20600–20611. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Han, K.; Seo, J.; Lee, J.; Dravid, S.M.; Woo, J.; Chun, H.; Cho, S.; Bae, J.Y.; An, H.; et al. Channel-mediated astrocytic glutamate modulates hippocampal synaptic plasticity by activating postsynaptic NMDA receptors. Mol. Brain 2015, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Maggio, N.; Shavit, E.; Chapman, J.; Segal, M. Thrombin induces long-term potentiation of reactivity to afferent stimulation and facilitates epileptic seizures in rat hippocampal slices: Toward understanding the functional consequences of cerebrovascular insults. J. Neurosci. 2008, 28, 732–736. [Google Scholar] [CrossRef]
- Almonte, A.G.; Qadri, L.H.; Sultan, F.A.; Watson, J.A.; Mount, D.J.; Rumbaugh, G.; Sweatt, J.D. Protease-activated receptor-1 modulates hippocampal memory formation and synaptic plasticity. J. Neurochem. 2013, 124, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, A.M.; Fleming, K.E.; McCauley, J.P.; Rodriguez, M.F.; Martin, E.T.; Sousa, A.A.; Leapman, R.D.; Scimemi, A. PAR1 activation induces rapid changes in glutamate uptake and astrocyte morphology. Sci. Rep. 2017, 7, 43606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, K.M.; Rogers, R.C.; Hermann, G.E. PAR1-activated astrocytes in the nucleus of the solitary tract stimulate adjacent neurons via NMDA receptors. J. Neurosci. 2015, 35, 776–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huda, R.; Chang, Z.; Do, J.; McCrimmon, D.R.; Martina, M. Activation of astrocytic PAR1 receptors in the rat nucleus of the solitary tract regulates breathing through modulation of presynaptic TRPV1. J. Physiol. 2018, 596, 497–513. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Liu, T.; Nakatsuka, T.; Kumamoto, E. Proteinase-activated receptor-1 activation presynaptically enhances spontaneous glutamatergic excitatory transmission in adult rat substantia gelatinosa neurons. J. Neurophysiol. 2009, 102, 312–319. [Google Scholar] [CrossRef] [Green Version]
- Price, R.; Ferrari, E.; Gardoni, F.; Mercuri, N.B.; Ledonne, A. Protease-activated receptor 1 (PAR1) inhibits synaptic NMDARs in mouse nigral dopaminergic neurons. Pharm. Res. 2020, 160, 105185. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Sehati, N.; Romano, C.; Cotman, C.W. Exposure of astrocytes to thrombin reduces levels of the metabotropic glutamate receptor mGluR5. J. Neurochem. 1996, 67, 1435–1447. [Google Scholar] [CrossRef]
- Gan, J.; Greenwood, S.M.; Cobb, S.R.; Bushell, T.J. Indirect modulation of neuronal excitability and synaptic transmission in the hippocampus by activation of proteinase-activated receptor-2. Br. J. Pharmacol. 2011, 163, 984–994. [Google Scholar] [CrossRef] [Green Version]
- Shavit-Stein, E.; Artan-Furman, A.; Feingold, E.; Ben Shimon, M.; Itzekson-Hayosh, Z.; Chapman, J.; Vlachos, A.; Maggio, N. Protease Activated Receptor 2 (PAR2) Induces Long-Term Depression in the Hippocampus through Transient Receptor Potential Vanilloid 4 (TRPV4). Front. Mol. Neurosci. 2017, 10, 42. [Google Scholar] [CrossRef] [Green Version]
- Hashimotodani, Y.; Ohno-Shosaku, T.; Yamazaki, M.; Sakimura, K.; Kano, M. Neuronal Protease-Activated Receptor 1 Drives Synaptic Retrograde Signaling Mediated by the Endocannabinoid 2-Arachidonoylglycerol. J. Neurosci. 2011, 31, 3104–3109. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Tao, K.; Zhu, H.; Miao, X.; Wang, Z.; Yu, W.; Lu, Z. Acute PAR2 activation reduces GABAergic inhibition in the spinal dorsal horn. Brain Res. 2011, 1425, 20–26. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Receptor | Activating Proteases | Inactivating Proteases | Activating Peptides | Signaling Pathways | Cerebral Localization |
|---|---|---|---|---|---|
| PAR1 | Thrombin Factor VIIa (FVIIa) Factor Xa (FXa) Plasmin MMP-1, -2, -3, -8, -9, -12, -13 Activated protein C (APC) Elastase Proteinase 3 Kallikrein 4, -5, -6, -14 Granzyme A, B, K Calpain-1 | Cathepsin G Proteinase 3 Elastase Plasmin Chymase | SFLLR-NH2 TFLLR-NH2 NPNDKYEPF-NH2 PRSFLLR-NH2 | Gq Gi G12/13 β-arrestins | Hippocampus Cortex Amydgala Substantia nigra Ventral tegmental Area Thalamus Hypothalamus Striatum Dorsal root ganglion |
| PAR2 | Trypsin I/II Trypsin IV Tryptase Factor VIIa (FVIIa) Factor Xa (FXa) Elastase Proteinase 3 Cathepsin G Acrosin Granzyme A Kallikrein 2, -4,-5, -6, 14 | Chymase | SLIGRL-NH2 SLIGKV-NH2 AC-98170 AC-55541 | Gq Gi G12/13 β-arrestins | Hippocampus Cortex Amydgala Substantia nigra Thalamus Hypothalamus Striatum Dorsal root ganglion |
| PAR3 | Thrombin Trypsin Activated protein C (APC) | Cathepsin G | TFRGAP-NH2 | Gq | Hippocampus Cortex Amydgala Thalamus Hypothalamus Striatum Dorsal root ganglion |
| PAR4 | Thrombin Trypsin Plasmin Cathepsin G MT-SP1 | GYPGQV-NH2 GYPGKF-NH2 AYPGKF | Gq G12/13 | Hippocampus Cortes Amydgala Thalamus Hypothalamus |
| Receptor | Neurotransmitter System | Effect | Main Response/Mechanism | Brain Area/Cellular Population | References |
|---|---|---|---|---|---|
| PAR1 | Glutamatergic Transmission | ↑ | Increased NMDAR-mediated spontaneous EPSCs | Hippocampus CA1 area, Pyramidal neurons | [56] |
| ↑ | Potentiated NMDA-activated currents and NMDARs-mediated spontaneous EPSCs, due to PAR1-induced glutamate release | Hippocampus CA1 Area, Pyramidal neurons | [57,58] | ||
| ↑ | NMDAR-dependent LTP of field EPSPs | Hippocampus, CA3-CA1 synapses | [60] | ||
| ↑ | Increased NMDAR-mediated currents and LTP of fEPSPs, due to astrocyte-released glutamate via Best-1 channels | Hippocampus, CA3-CA1 synapses | [59] | ||
| ↑ | Impaired NMDAR-dependent TBS-induced LTP of fEPSPs in PAR1 knockout mice | Hippocampus, CA3-CA1 synapses | [61] | ||
| ↑ | Potentiated NMDAR-mediated spontaneous transmission, due to astrocytic PAR1-induced glutamate release | Nucleus of solitary tract, neurons | [63] | ||
| ↑ | Increased glutamate release, elicited by astrocytic PAR1-released endovanilloids (EVs) and TRPV1 activation | Nucleus of solitary tract, neurons | [64] | ||
| ↑ | Increased spontaneous EPSCs, due to enhanced glutamate release | Spinal cord, Substantia gelatinosa neurons | [65] | ||
| ↓ | Reduced NMDAR-mediated EPSCs and decreased NMDAR-dependent LTP of field EPSPs | Hippocampal CA3-CA1 synapses | [62] | ||
| ↓ | Reduced AMPAR-mediated EPSCs | Hippocampal CA1 area, pyramidal neurons | [62] | ||
| ↓ | LTD of NMDAR-mediated EPSCs, due to PAR1-induced NMDARs endocytosis | Substantia nigra compacta, DA neurons | [66] | ||
| ↓ | Reduced NMDA-activated currents | Substantia nigra compacta, DA neurons | [66] | ||
| = | Unaffected synaptic and extrasynaptic AMPAR-mediated currents | Substantia nigra compacta, DA neurons | [66] | ||
| ↓ | Reduced mGluR5 expression | Astrocytic cultures | [67] | ||
| PAR1 | GABAergic transmission | ↓ | Reduced spontaneous and evoked IPSCs due to PAR1-dependent eCB-mediated decrease of GABA release | Hippocampal neuronal cultures | [70] |
| PAR2 | Glutamatergic transmission | ↓ | LTD of fEPSPs (NMDAR-mediated) | Hippocampus, CA3-CA1 synapses | [68] |
| ↓ | LTD of fEPSPs (TRPV4-mediated) | Hippocampus, CA3-CA1 synapses | [69] | ||
| GABAergic transmission | ↓ | Reduced spontaneous IPSCs | Spinal cord dorsal horn, neurons | [71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Price, R.; Mercuri, N.B.; Ledonne, A. Emerging Roles of Protease-Activated Receptors (PARs) in the Modulation of Synaptic Transmission and Plasticity. Int. J. Mol. Sci. 2021, 22, 869. https://doi.org/10.3390/ijms22020869
Price R, Mercuri NB, Ledonne A. Emerging Roles of Protease-Activated Receptors (PARs) in the Modulation of Synaptic Transmission and Plasticity. International Journal of Molecular Sciences. 2021; 22(2):869. https://doi.org/10.3390/ijms22020869
Chicago/Turabian StylePrice, Rachel, Nicola Biagio Mercuri, and Ada Ledonne. 2021. "Emerging Roles of Protease-Activated Receptors (PARs) in the Modulation of Synaptic Transmission and Plasticity" International Journal of Molecular Sciences 22, no. 2: 869. https://doi.org/10.3390/ijms22020869