The NFκB Antagonist CDGSH Iron-Sulfur Domain 2 Is a Promising Target for the Treatment of Neurodegenerative Diseases



{kind=link}
Abstract
:1. Preface
2. Inflammation and Mitochondrial Dysfunction Implicated in NDs
3. NFκB-Driven Inflammation and Mitochondrial Dysfunction in NDs
4. The Evolutionary Conserved NEET Family Member CISD2 Regulates Important Homeostatic CNS Functions such as pH and Oxidation State
5. Physiological Function of CISD2
- (1)
- Calcium excitotoxicity. CISD2 has been shown to depress excitotoxic Ca2+ increase at the ER through the binding of CISD2 to BCL2 along with the inositol 1,4,5-triphosphate (IP3) receptor (a kind of calcium channel in the ER membrane) [72,73]. CISD2 deficiency can trigger an increase in ER and cytoplasmic Ca2+ levels in CISD2 knockout mice compared with wild-type mice [72]. Furthermore, results obtained from CISD2 knockout mice showed that CISD2 along with GTPase of immune-associated protein 5 (Gimap5) in MAMs decreased cytosolic Ca2+ surge and enhanced mitochondrial Ca2+ uptake [63].
- (2)
- Apoptosis. CISD2 along with the IP3 receptor has been shown to connect with a combination of Bcl-2-Beclin-1 complex in the ER membrane. Bcl-2 exerts its antiapoptotic effect via antagonism of Beclin-1. As a Bcl-2-interacting protein, CISD2 functions as an autophagy regulator. The binding of CISD2 to Bcl-2 regulates Bcl-2 to antagonize autophagy/apoptosis in response to stress [71,74]. CISD2 enhances Bcl-2-Beclin-1 interaction and prevents the potential apoptotic cellular damage. When CISD2 is attenuated, the interaction between Bcl-2 and Beclin-1 is greatly reduced, and autophagy is triggered. As mentioned earlier, the effect generated by the combination of CISD2 and Bcl-2 is invalid when CISD2 loses the [2Fe-2S] cluster [71].
- (3)
- OMM breakdown and subsequent mitochondrial abnormality. The most prominent study on mitochondrial CISD2 function from CISD2 knockout mice was published by a group of Taiwanese neuroscientists headed by Ting-Fen Tsai in 2009 [75]. As a part of OMMs, CISD2 proteins play a vital role in the maintenance of mitochondrial function. Studies on CISD2-/- mice have demonstrated that CISD2 knockout induces mitochondrial degeneration, autophagy, and consequent enhancement of the aging process. As a kind of ER/mitochondria-related disease, WFS2, a subtype among WFS (featured in diabetes insipidus, diabetes, optic atrophy, and deafness [DIDMOAD]), has been linked to the recessive mutation of CISD2. WFS2 may clinically manifest as diabetes mellitus, optic atrophy, and a bleeding tendency [76].
6. CISD2 as a NFκB Antagonist against Inflammation and Mitochondrial Dysfunction: A Promising Target for NDs
7. CISD2 Attenuation on Neural Pathology
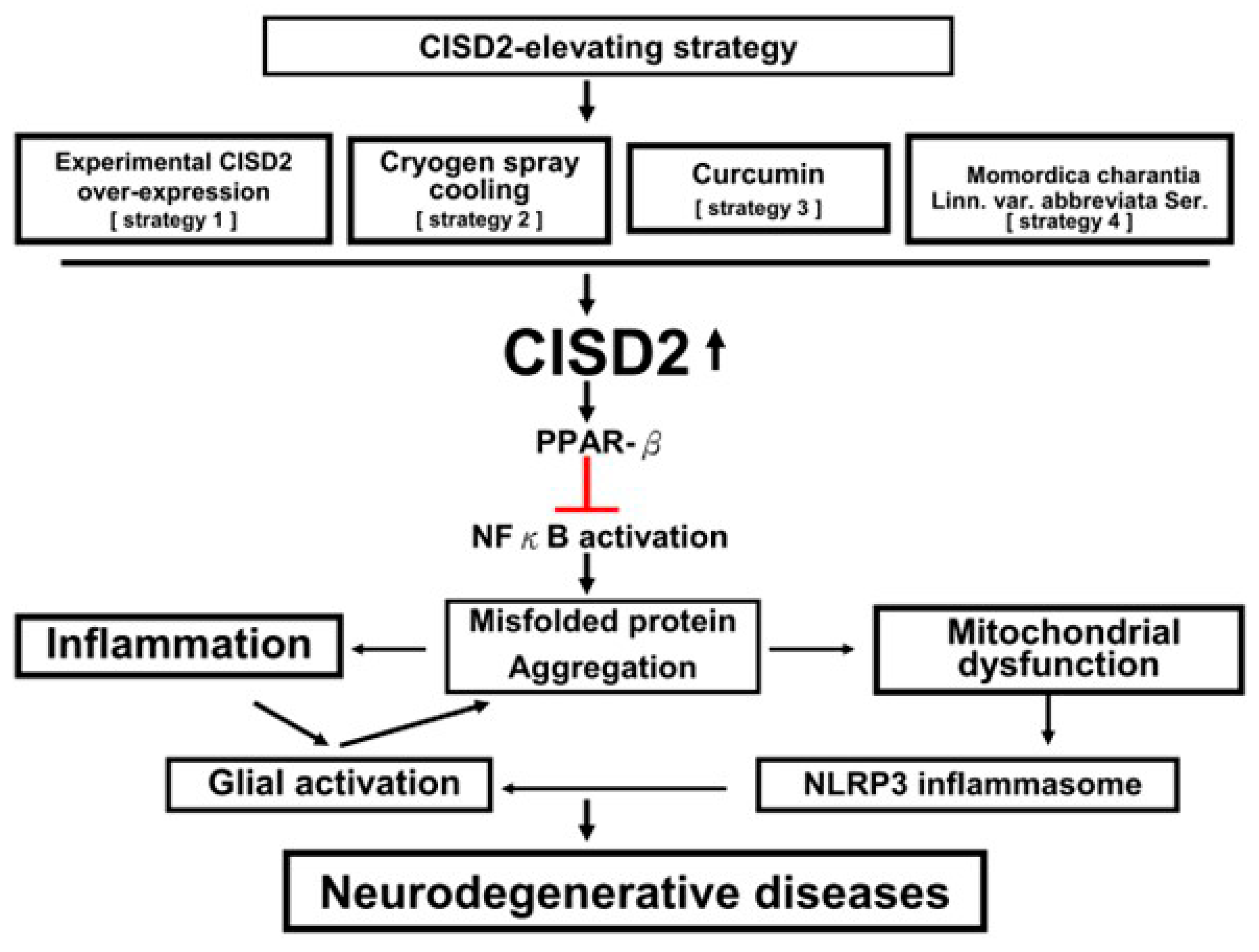
8. CISD2-Elevating Strategy as the Potential Future Therapy in NDs
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sanada, F.; Taniyama, Y.; Muratsu, J.; Otsu, R.; Shimizu, H.; Rakugi, H.; Morishita, R. Source of Chronic Inflammation in Aging. Front. Cardiovasc. Med. 2018, 5, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. J. Clin. Med. 2020, 9, 740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laforge, M.; Rodrigues, V.; Silvestre, R.; Gautier, C.; Weil, R.; Corti, O.; Estaquier, J. NF-kB pathway controls mitochondrial dynamics. Cell Death Differ. 2016, 23, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Albensi, B.C. What Is Nuclear Factor Kappa B (NF-kB) Doing in and to the Mitochondrion? Front. Cell Dev. Biol. 2019, 7, 154. [Google Scholar] [CrossRef]
- Datta, G.; Colasanti, A.; Rabiner, E.A.; Gunn, R.N.; Malik, O.; Ciccarelli, O.; Nicholas, R.; Van, V.E.; Van, H.W.; Searle, G.; et al. Neuroinflammation and its relationship to changes in brain volume and white matter lesions in multiple sclerosis. Brain 2017, 140, 2927–2938. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [Green Version]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Orr, H.T. Spinocerebellar Ataxia Type 1: Molecular Mechanisms of Neurodegeneration and Preclinical Studies. Adv. Exp. Med. Biol. 2018, 1049, 135–145. [Google Scholar]
- Jia, L.; Fu, Y.; Shen, L.; Zhang, H.; Zhu, M.; Qiu, Q.; Wang, Q.; Yan, X.; Kong, C.; Hao, J.; et al. PSEN1, PSEN2, and APP mutations in 404 Chinese pedigrees with familial Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 178–191. [Google Scholar] [CrossRef]
- Olgiati, S.; Thomas, A.; Quadri, M.; Breedveld, G.J.; Graafland, J.; Eussen, H.; Douben, H.; de Klein, A.; Onofrj, M.; Bonifati, V. Early-onset parkinsonism caused by alpha-synuclein gene triplication: Clinical and genetic findings in a novel family. Parkinsonism Relat. Disord. 2015, 21, 981–986. [Google Scholar] [CrossRef] [Green Version]
- Hauser, D.N.; Mamais, A.; Conti, M.M.; Primiani, C.T.; Kumaran, R.; Dillman, A.A.; Langston, R.G.; Beilina, A.; Garcia, J.H.; Diaz-Ruiz, A.; et al. Hexokinases link DJ-1 to the PINK1/parkin pathway. Mol. Neurodegener. 2017, 12, 70. [Google Scholar] [CrossRef] [PubMed]
- Abati, E.; Bresolin, N.; Comi, G.; Corti, S. Silence superoxide dismutase 1 (SOD1): A promising therapeutic target for amyotrophic lateral sclerosis (ALS). Expert Opin. Ther. Targets. 2020, 24, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Greaves, C.V.; Rohrer, J.D. An update on genetic frontotemporal dementia. J. Neurol. 2019, 266, 2075–2086. [Google Scholar] [CrossRef] [Green Version]
- Olmedillas Del, M.M.; Asavapanumas, N.; Uzcátegui, N.L.; Garaschuk, O. Healthy Brain Aging Modifies Microglial Calcium Signaling In Vivo. Int. J. Mol. Sci. 2019, 20, 589. [Google Scholar] [CrossRef] [Green Version]
- Matt, S.M.; Johnson, R.W. Neuro-immune dysfunction during brain aging: New insights in microglial cell regulation. Curr. Opin. Pharmacol. 2016, 26, 96–101. [Google Scholar] [CrossRef] [Green Version]
- Tcw, J.; Goate, A.M. Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024539. [Google Scholar] [CrossRef]
- Ma, B.; Zhao, J.; Nussinov, R. Conformational selection in amyloid-based immunotherapy: Survey of crystal structures of antibody-amyloid complexes. Biochim. Biophys. Acta (BBA) Gen. Subj. 2016, 1860, 2672–2681. [Google Scholar] [CrossRef] [Green Version]
- Venegas, C.; Heneka, M.T. Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol. 2017, 101, 87–98. [Google Scholar] [CrossRef]
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElroy, P.B.; Liang, L.P.; Day, B.J.; Patel, M. Scavenging reactive oxygen species inhibits status epilepticus-induced neuroinflammation. Exp. Neurol. 2017, 298, 13–22. [Google Scholar] [CrossRef]
- Norden, D.M.; Muccigrosso, M.M.; Godbout, J.P. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 2015, 96, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.S.; Liang, P.Z.; Lu, S.Z.; Chen, R.; Yin, Y.Q.; Zhou, J.W. Extracellular ab-crystallin modulates the inflammatory responses. Biochem. Biophys. Res. Commun. 2019, 508, 282–288. [Google Scholar] [CrossRef]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Zhou, D.; Jiang, Y. Sirtuin 3 attenuates neuroinflammation-induced apoptosis in BV-2 microglia. Aging 2019, 11, 9075–9089. [Google Scholar] [CrossRef]
- Yoo, S.M.; Park, J.; Kim, S.H.; Jung, Y.K. Emerging perspectives on mitochondrial dysfunction and inflammation in Alzheimer’s disease. BMB Rep. 2020, 53, 35–46. [Google Scholar] [CrossRef]
- Santoni, G.; Cardinali, C.; Morelli, M.B.; Santoni, M.; Nabissi, M.; Amantini, C. Danger- and pathogen-associated molecular patterns recognition by pattern-recognition receptors and ion channels of the transient receptor potential family triggers the inflammasome activation in immune cells and sensory neurons. J. Neuroinflamm. 2015, 12, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El, K.J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Ravari, A.; Mirzaei, T.; Kennedy, D.; Kazemi, A.M. Chronoinflammaging in Alzheimer; A systematic review on the roles of toll like receptor 2. Life Sci. 2017, 171, 16–20. [Google Scholar] [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.C.; Chang, J.H.; Jin, J. Regulation of nuclear factor-kB in autoimmunity. Trends Immunol. 2013, 34, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Latimer, M.; Ernst, M.K.; Dunn, L.L.; Drutskaya, M.; Rice, N.R. The N-terminal domain of IkappaB alpha masks the nuclear localization signal(s) of p50 and c-Rel homodimers. Mol. Cell Biol. 1998, 18, 2640–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene 2006, 25, 6706–6716. [Google Scholar] [CrossRef] [Green Version]
- Shifera, A.S. The zinc finger domain of IKK gamma (NEMO) protein in health and disease. J. Cell. Mol. Med. 2010, 14, 2404–2414. [Google Scholar] [CrossRef] [Green Version]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Tong, Z.; Jiang, S.; Zheng, W.; Zhao, J.; Zhou, X. The Roles of Endoplasmic Reticulum in NLRP3 Inflammasome Activation. Cells 2020, 9, 1219. [Google Scholar] [CrossRef]
- Amr, S.; Heisey, C.; Zhang, M.; Xia, X.J.; Shows, K.H.; Ajlouni, K.; Pandya, A.; Satin, L.S.; El-Shanti, H.; Shiang, R. A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am. J. Hum. Genet. 2007, 81, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, M.; La, S.L.; Rondinelli, M.; Errichiello, E.; Zuffardi, O.; Puca, A.A.; Genovese, S.; Ceriello, A. A donor splice site mutation in CISD2 generates multiple truncated, non-functional isoforms in Wolfram syndrome type 2 patients. BMC Med. Genet. 2017, 18, 147. [Google Scholar] [CrossRef]
- Moreno-Navarrete, J.M.; Moreno, M.; Ortega, F.; Sabater, M.; Xifra, G.; Ricart, W.; Fernández-Real, J.M. CISD1 in association with obesity-associated dysfunctional adipogenesis in human visceral adipose tissue. Obesity 2016, 24, 139–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Zhang, L.; Lai, S.; Ye, K. Structure and molecular evolution of CDGSH iron-sulfur domains. PLoS ONE 2011, 6, e24790. [Google Scholar] [CrossRef] [PubMed]
- Pesce, L.; Calandrini, V.; Marjault, H.B.; Lipper, C.H.; Rossetti, G.; Mittler, R.; Jennings, P.A.; Bauer, A.; Nechushtai, R.; Carloni, P. Molecular Dynamics Simulations of the [2Fe-2S] Cluster-Binding Domain of NEET Proteins Reveal Key Molecular Determinants That Induce Their Cluster Transfer/Release. J. Phys. Chem. B 2017, 121, 10648–10656. [Google Scholar] [CrossRef]
- Wiley, S.E.; Murphy, A.N.; Ross, S.A.; van der Geer, P.; Dixon, J.E. MitoNEET is an iron-containing outer mitochondrial membrane protein that regulates oxidative capacity. Proc. Natl. Acad. Sci. USA 2007, 104, 5318–5323. [Google Scholar] [CrossRef] [Green Version]
- Inupakutika, M.A.; Sengupta, S.; Nechushtai, R.; Jennings, P.A.; Onuchic, J.N.; Azad, R.K.; Padilla, P.; Mittler, R. Phylogenetic analysis of eukaryotic NEET proteins uncovers a link between a key gene duplication event and the evolution of vertebrates. Sci. Rep. 2017, 7, 42571. [Google Scholar] [CrossRef]
- Karmi, O.; Marjault, H.B.; Pesce, L.; Carloni, P.; Onuchic, J.N.; Jennings, P.A.; Mittler, R.; Nechushtai, R. The unique fold and lability of the [2Fe-2S] clusters of NEET proteins mediate their key functions in health and disease. J. Biol. Inorg. Chem. 2018, 23, 599–612. [Google Scholar] [CrossRef] [Green Version]
- Kusminski, C.M.; Holland, W.L.; Sun, K.; Park, J.; Spurgin, S.B.; Lin, Y.; Askew, G.R.; Simcox, J.A.; McClain, D.A.; Li, C.; et al. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat. Med. 2012, 18, 1539–1549. [Google Scholar] [CrossRef] [Green Version]
- Nechushtai, R.; Conlan, A.R.; Harir, Y.; Song, L.; Yogev, O.; Eisenberg-Domovich, Y.; Livnah, O.; Michaeli, D.; Rosen, R.; Ma, V.; et al. Characterization of Arabidopsis NEET reveals an ancient role for NEET proteins in iron metabolism. Plant Cell 2012, 24, 2139–2154. [Google Scholar] [CrossRef] [Green Version]
- Wiley, S.E.; Andreyev, A.Y.; Divakaruni, A.S.; Karisch, R.; Perkins, G.; Wall, E.A.; van der Geer, P.; Chen, Y.F.; Tsai, T.F.; Simon, M.I.; et al. Wolfram Syndrome protein, Miner1, regulates sulphydryl redox status, the unfolded protein response, and Ca2+ homeostasis. EMBO Mol. Med. 2013, 5, 904–918. [Google Scholar] [CrossRef]
- Dicus, M.M.; Conlan, A.; Nechushtai, R.; Jennings, P.A.; Paddock, M.L.; Britt, R.D.; Stoll, S. Binding of histidine in the (Cys)3(His)1-coordinated [2Fe-2S] cluster of human mitoNEET. J. Am. Chem. Soc. 2010, 132, 2037–2049. [Google Scholar] [CrossRef] [Green Version]
- Shimomura, Y.; Wada, K.; Fukuyama, K.; Takahashi, Y. The asymmetric trimeric architecture of [2Fe-2S] IscU: Implications for its scaffolding during iron-sulfur cluster biosynthesis. J. Mol. Biol. 2008, 383, 133–143. [Google Scholar] [CrossRef]
- Tamir, S.; Paddock, M.L.; Darash-Yahana-Baram, M.; Holt, S.H.; Sohn, Y.S.; Agranat, L.; Michaeli, D.; Stofleth, J.T.; Lipper, C.H.; Morcos, F.; et al. Structure-function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochim. Biophys. Acta 2015, 1853, 1294–1315. [Google Scholar] [CrossRef] [Green Version]
- Wiley, S.E.; Paddock, M.L.; Abresch, E.C.; Gross, L.; van der Geer, P.; Nechushtai, R.; Murphy, A.N.; Jennings, P.A.; Dixon, J.E. The outer mitochondrial membrane protein mitoNEET contains a novel redox-active 2Fe-2S cluster. J. Biol. Chem. 2007, 282, 23745–23749. [Google Scholar] [CrossRef] [Green Version]
- Bak, D.W.; Elliott, S.J. Conserved hydrogen bonding networks of MitoNEET tune Fe-S cluster binding and structural stability. Biochemistry 2013, 52, 4687–4696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferecatu, I.; Gonçalves, S.; Golinelli-Cohen, M.P.; Clémancey, M.; Martelli, A.; Riquier, S.; Guittet, E.; Latour, J.M.; Puccio, H.; Drapier, J.C.; et al. The diabetes drug target MitoNEET governs a novel trafficking pathway to rebuild an Fe-S cluster into cytosolic aconitase/iron regulatory protein 1. J. Biol. Chem. 2014, 289, 28070–28086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lill, R. Function and biogenesis of iron-sulphur proteins. Nature 2009, 460, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Golinelli-Cohen, M.P.; Lescop, E.; Mons, C.; Gonçalves, S.; Clémancey, M.; Santolini, J.; Guittet, E.; Blondin, G.; Latour, J.M.; Bouton, C. Redox Control of the Human Iron-Sulfur Repair Protein MitoNEET Activity via Its Iron-Sulfur Cluster. J. Biol. Chem. 2016, 291, 7583–7593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, G.; Liu, D.; Pan, F.; Zhao, J.; Li, T.; Ma, Y.; Shen, B.; Lyu, J. His-87 ligand in mitoNEET is crucial for the transfer of iron sulfur clusters from mitochondria to cytosolic aconitase. Biochem. Biophys. Res. Commun. 2016, 470, 226–232. [Google Scholar] [CrossRef]
- Zuris, J.A.; Harir, Y.; Conlan, A.R.; Shvartsman, M.; Michaeli, D.; Tamir, S.; Paddock, M.L.; Onuchic, J.N.; Mittler, R.; Cabantchik, Z.I.; et al. Facile transfer of [2Fe-2S] clusters from the diabetes drug target mitoNEET to an apo-acceptor protein. Proc. Natl. Acad. Sci. USA 2011, 108, 13047–13052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eom, K.S.; Cheong, J.S.; Lee, S.J. Structural Analyses of Zinc Finger Domains for Specific Interactions with DNA. J. Microbiol. Biotechnol. 2016, 26, 2019–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobili, A.; Krashia, P.; D’Amelio, M. Cisd2: A promising new target in Alzheimer’s disease. J. Pathol. 2020, 251, 113–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.H.; Chen, Y.F.; Wu, C.Y.; Wu, P.C.; Huang, Y.L.; Kao, C.H.; Lin, C.H.; Kao, L.S.; Tsai, T.F.; Wei, Y.H. Cisd2 modulates the differentiation and functioning of adipocytes by regulating intracellular Ca2+ homeostasis. Hum. Mol. Genet. 2014, 23, 4770–4785. [Google Scholar] [CrossRef] [Green Version]
- Conlan, A.R.; Axelrod, H.L.; Cohen, A.E.; Abresch, E.C.; Zuris, J.; Yee, D.; Nechushtai, R.; Jennings, P.A.; Paddock, M.L. Crystal structure of Miner1: The redox-active 2Fe-2S protein causative in Wolfram Syndrome 2. J. Mol. Biol. 2009, 392, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimberg, G.D.; Michalek, J.L.; Oluyadi, A.A.; Rodrigues, A.V.; Zucconi, B.E.; Neu, H.M.; Ghosh, S.; Sureschandra, K.; Wilson, G.M.; Stemmler, T.L.; et al. Cleavage and polyadenylation specificity factor 30: An RNA-binding zinc-finger protein with an unexpected 2Fe-2S cluster. Proc. Natl. Acad. Sci. USA 2016, 113, 4700–4705. [Google Scholar] [CrossRef] [Green Version]
- Han, G.; Lu, C.; Guo, J.; Qiao, Z.; Sui, N.; Qiu, N.; Wang, B. C2H2 Zinc Finger Proteins: Master Regulators of Abiotic Stress Responses in Plants. Front. Plant Sci. 2020, 11, 115. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Cooper, K.L.; Sun, X.; Liu, K.J.; Hudson, L.G. Selective Sensitization of Zinc Finger Protein Oxidation by Reactive Oxygen Species through Arsenic Binding. J. Biol. Chem. 2015, 290, 18361–18369. [Google Scholar] [CrossRef] [Green Version]
- Mozzillo, E.; Delvecchio, M.; Carella, M.; Grandone, E.; Palumbo, P.; Salina, A.; Aloi, C.; Buono, P.; Izzo, A.; D’Annunzio, G.; et al. A novel CISD2 intragenic deletion, optic neuropathy and platelet aggregation defect in Wolfram syndrome type 2. BMC Med. Genet. 2014, 15, 88. [Google Scholar] [CrossRef] [Green Version]
- Karmi, O.; Holt, S.H.; Song, L.; Tamir, S.; Luo, Y.; Bai, F.; Adenwalla, A.; Darash-Yahana, M.; Sohn, Y.S.; Jennings, P.A.; et al. Interactions between mitoNEET and NAF-1 in cells. PLoS ONE 2017, 12, e0175796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rash-Yahana, M.; Pozniak, Y.; Lu, M.; Sohn, Y.S.; Karmi, O.; Tamir, S.; Bai, F.; Song, L.; Jennings, P.A.; Pikarsky, E.; et al. Breast cancer tumorigenicity is dependent on high expression levels of NAF-1 and the lability of its Fe-S clusters. Proc. Natl. Acad. Sci. USA 2016, 113, 10890–10895. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.C.; Nguyen, M.; Germain, M.; Shore, G.C. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2010, 29, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C.; Nguyen, M.; Shore, G.C. BCL2-CISD2: An ER complex at the nexus of autophagy and calcium homeostasis? Autophagy 2012, 8, 856–857. [Google Scholar] [CrossRef]
- Chang, N.C.; Nguyen, M.; Bourdon, J.; Risse, P.A.; Martin, J.; Danialou, G.; Rizzuto, R.; Petrof, B.J.; Shore, G.C. Bcl-2-associated autophagy regulator Naf-1 required for maintenance of skeletal muscle. Hum. Mol. Genet. 2012, 21, 2277–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Chen, Y.F.; Kao, C.H.; Chen, Y.T.; Wang, C.H.; Wu, C.Y.; Tsai, C.Y.; Liu, F.C.; Yang, C.W.; Wei, Y.H.; Hsu, M.T.; et al. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 2009, 23, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouzier, C.; Moore, D.; Delorme, C.; Lacas-Gervais, S.; Ait-El-Mkadem, S.; Fragaki, K.; Burte, F.; Serre, V.; Bannwarth, S.; Chaussenot, A.; et al. A novel CISD2 mutation associated with a classical Wolfram syndrome phenotype alters Ca2+ homeostasis and ER-mitochondria interactions. Hum. Mol. Genet. 2017, 26, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Chiang, T.H.; Chen, W.J.; Sun, Y.Y.; Lee, Y.H.; Lin, M.S. CISD2 serves a novel role as a suppressor of nitric oxide signalling and curcumin increases CISD2 expression in spinal cord injuries. Injury 2015, 46, 2341–2350. [Google Scholar] [CrossRef]
- Lin, C.C.; Chiang, T.H.; Sun, Y.Y.; Lin, M.S. Protective Effects of CISD2 and Influence of Curcumin on CISD2 Expression in Aged Animals and Inflammatory Cell Model. Nutrients 2019, 11, 700. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.S. CISD2 Attenuates Inflammation and Regulates Microglia Polarization in EOC Microglial Cells-As a Potential Therapeutic Target for Neurodegenerative Dementia. Front. Aging Neurosci. 2020, 12, 260. [Google Scholar] [CrossRef]
- Kung, W.M.; Lin, C.C.; Kuo, C.Y.; Juin, Y.C.; Wu, P.C.; Lin, M.S. Wild Bitter Melon Exerts Anti-Inflammatory Effects by Upregulating Injury-Attenuated CISD2 Expression following Spinal Cord Injury. Behav. Neurol. 2020, 2020, 1080521. [Google Scholar] [CrossRef]
- Ding, G.; Cheng, L.; Qin, Q.; Frontin, S.; Yang, Q. PPARdelta modulates lipopolysaccharide-induced TNFalpha inflammation signaling in cultured cardiomyocytes. J. Mol. Cell Cardiol. 2006, 40, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, M.; Di, P.R.; Mazzon, E.; Esposito, E.; Paterniti, I.; Kapoor, A.; Thiemermann, C.; Cuzzocrea, S. GW0742, a high affinity PPAR-b/d agonist reduces lung inflammation induced by bleomycin instillation in mice. Int. J. Immunopathol. Pharmacol. 2010, 23, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Chen, Y.F.; Wang, C.H.; Kao, C.H.; Zhuang, H.W.; Chen, C.C.; Chen, L.K.; Kirby, R.; Wei, Y.H.; Tsai, S.F.; et al. A persistent level of Cisd2 extends healthy lifespan and delays aging in mice. Hum. Mol. Genet. 2012, 21, 3956–3968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.F.; Chou, T.Y.; Lin, I.H.; Chen, C.G.; Kao, C.H.; Huang, G.J.; Chen, L.K.; Wang, P.N.; Lin, C.P.; Tsai, T.F. Upregulation of Cisd2 attenuates Alzheimer’s-related neuronal loss in mice. J. Pathol. 2020, 250, 299–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, W.M.; Chang, C.J.; Chen, T.Y.; Lin, M.S. Cryogen spray cooling mitigates inflammation and injury-induced CISD2 decline in rat spinal cord hemisection model. J. Integr. Neurosci. 2020, 19, 619–628. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kung, W.-M.; Lin, M.-S. The NFκB Antagonist CDGSH Iron-Sulfur Domain 2 Is a Promising Target for the Treatment of Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 934. https://doi.org/10.3390/ijms22020934
Kung W-M, Lin M-S. The NFκB Antagonist CDGSH Iron-Sulfur Domain 2 Is a Promising Target for the Treatment of Neurodegenerative Diseases. International Journal of Molecular Sciences. 2021; 22(2):934. https://doi.org/10.3390/ijms22020934
Chicago/Turabian StyleKung, Woon-Man, and Muh-Shi Lin. 2021. "The NFκB Antagonist CDGSH Iron-Sulfur Domain 2 Is a Promising Target for the Treatment of Neurodegenerative Diseases" International Journal of Molecular Sciences 22, no. 2: 934. https://doi.org/10.3390/ijms22020934
APA StyleKung, W.-M., & Lin, M.-S. (2021). The NFκB Antagonist CDGSH Iron-Sulfur Domain 2 Is a Promising Target for the Treatment of Neurodegenerative Diseases. International Journal of Molecular Sciences, 22(2), 934. https://doi.org/10.3390/ijms22020934