A Peek Inside the Machines of Bacterial Nucleotide Excision Repair

 and
and 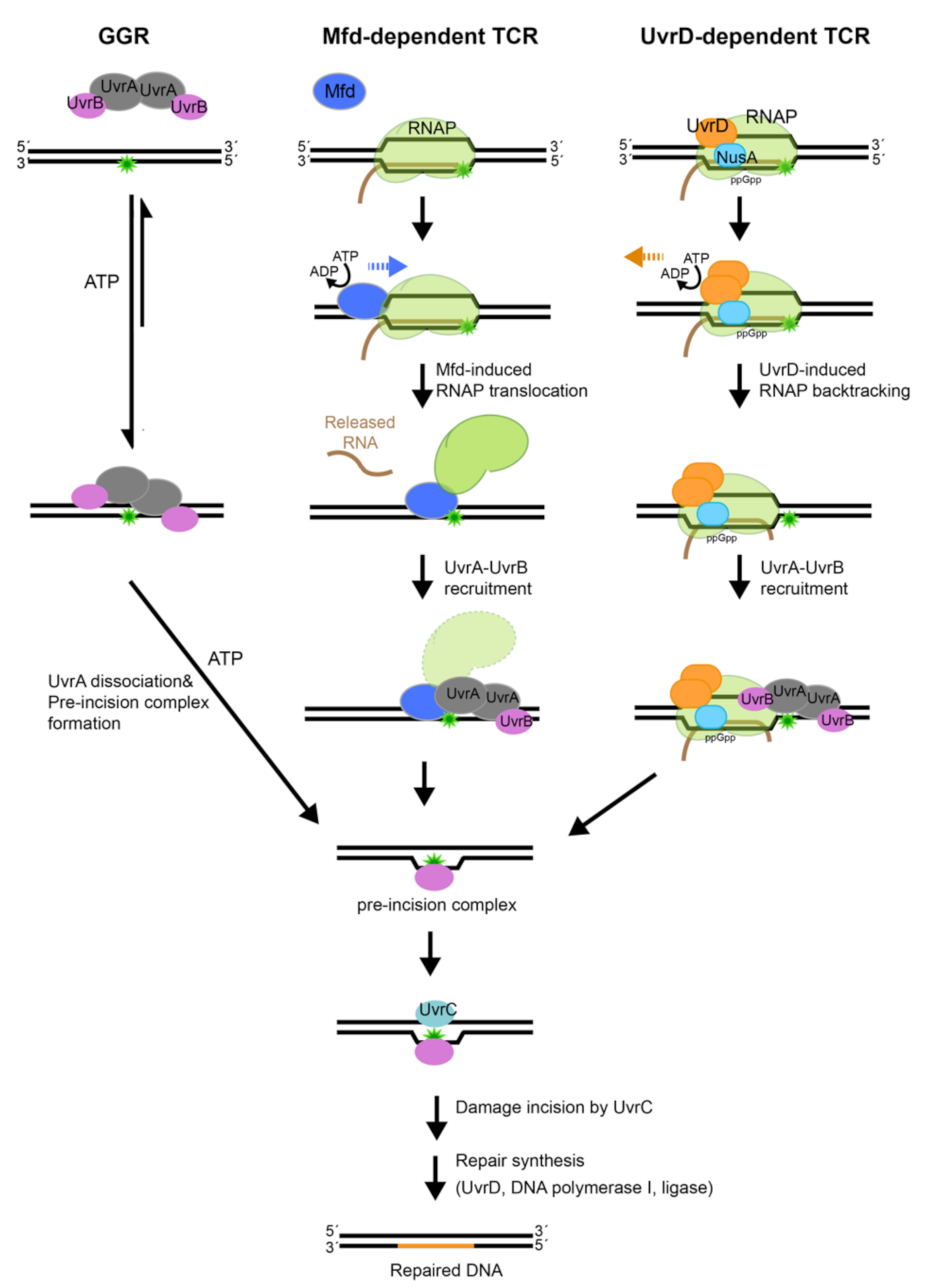{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
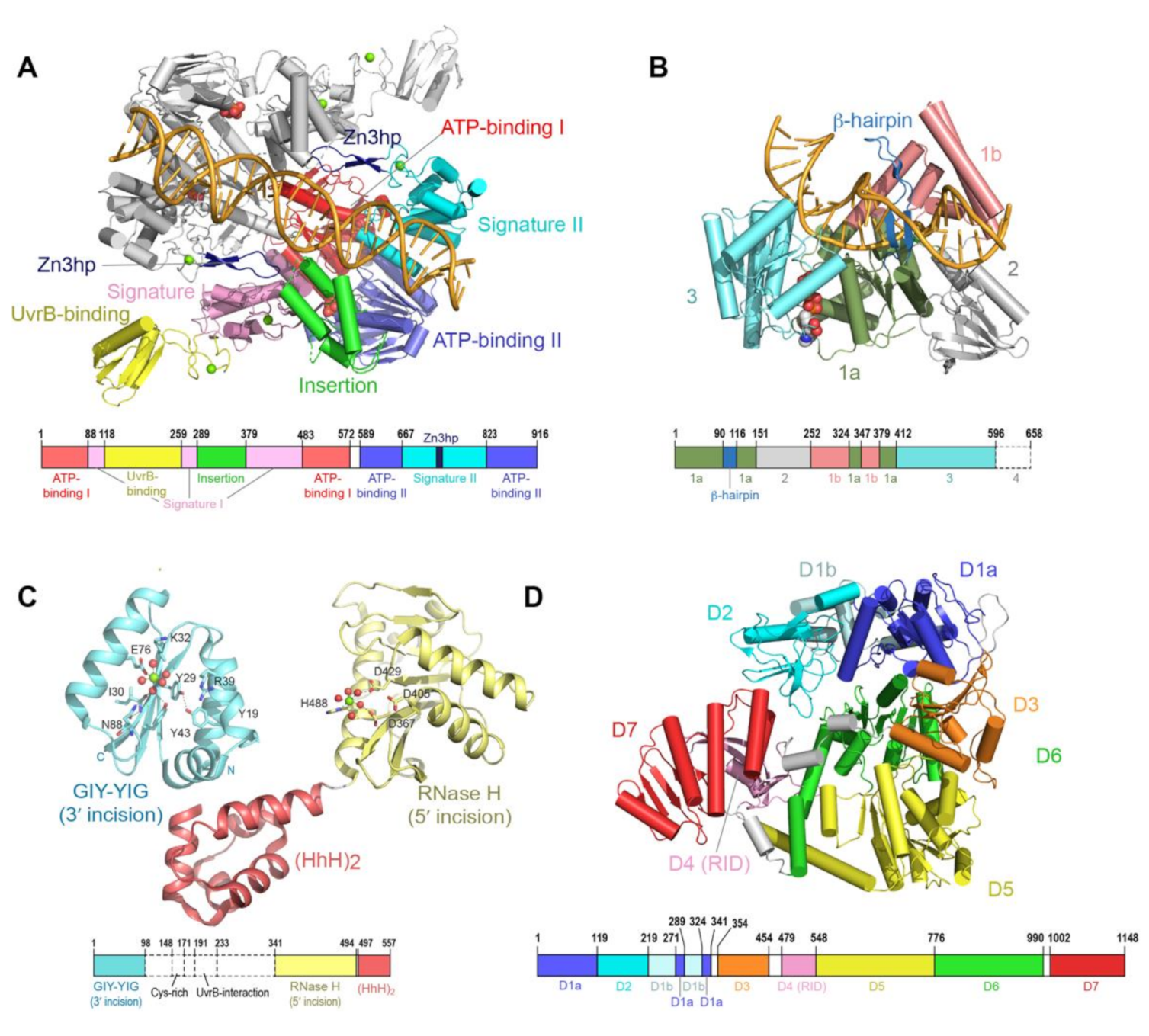
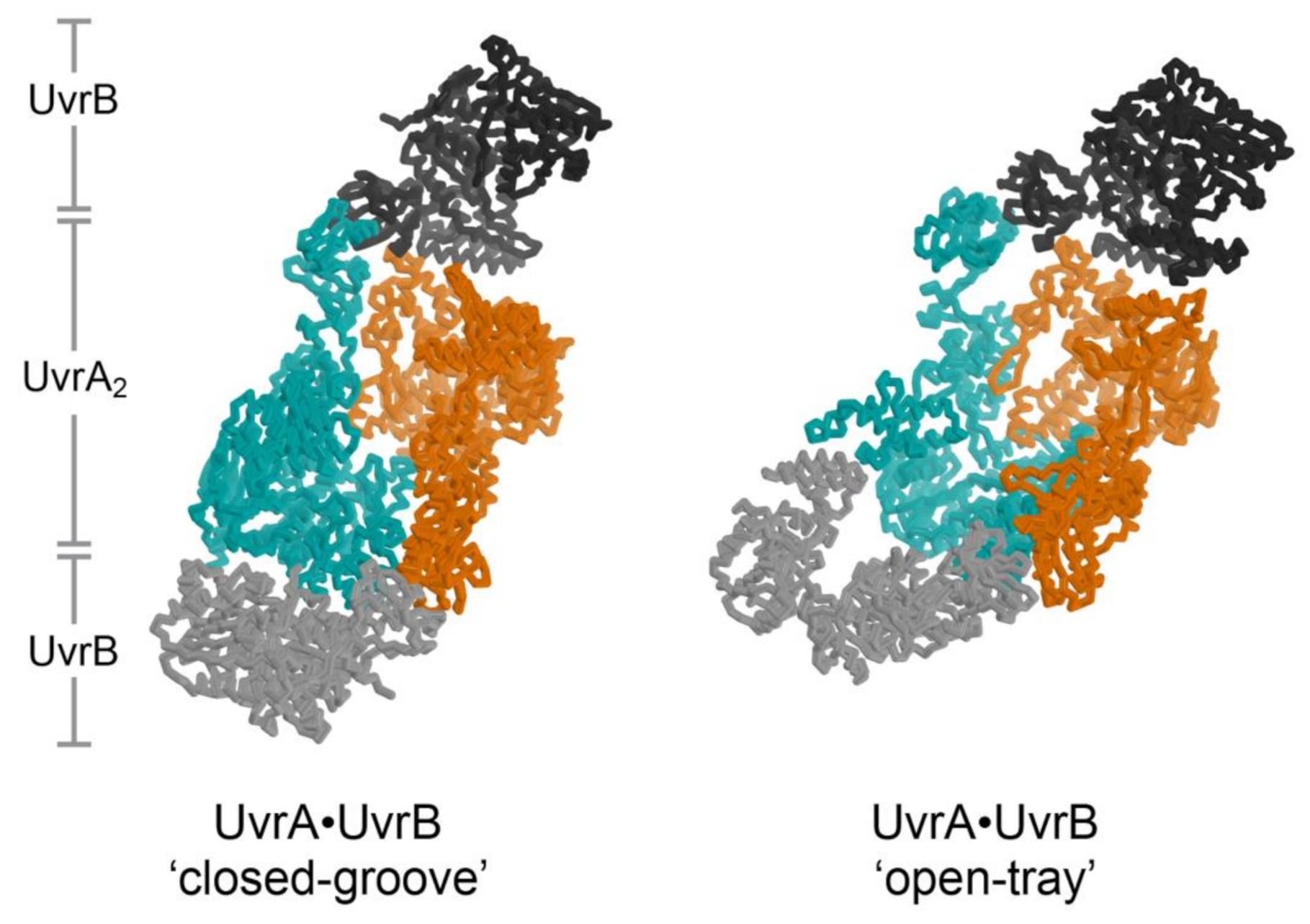
2. The UvrA DNA Damage Sensor
3. UvrB is a Damage Specific Helicase that Prepares DNA for Excision
4. UvrC is a Damage Specific Dual Nuclease
5. Mfd Structure and Function
6. Global Genome Repair (GGR)
6.1. Discrimination of Native DNA from Damaged by UvrA
6.2. UvrA Mediates a Match between Damaged DNA and UvrB
6.3. Dissociation of UvrA from DNA and Formation of the Pre-Incision Complex
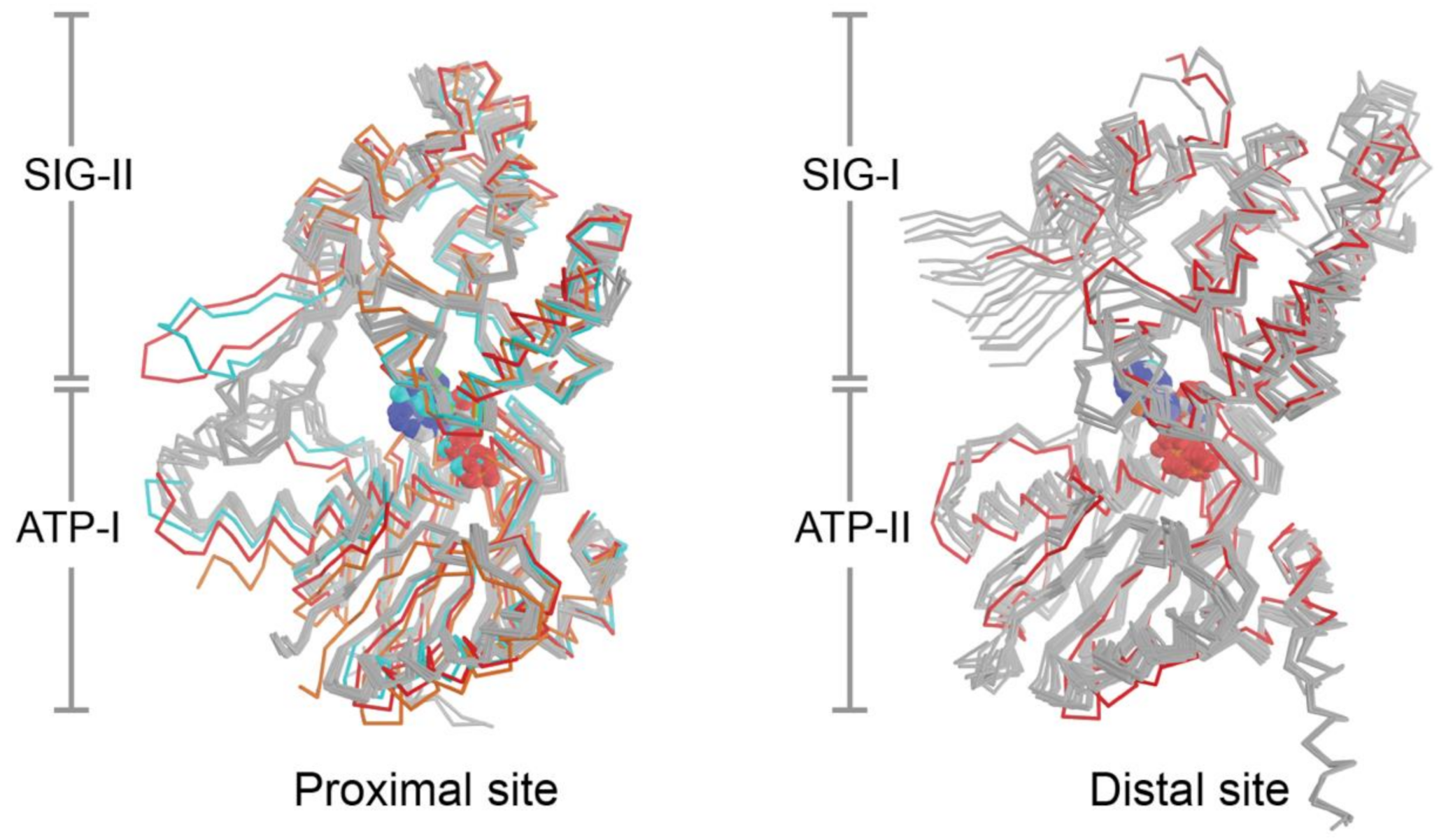
6.4. Two Types of ABC ATPase Sites on UvrA Power Damage Detection and UvrB Binding
7. Transcription-Coupled Repair (TCR): RNA Polymerase as the DNA Damage Sensor
7.1. Mfd-Dependent TCR
7.2. Does UvrD Mediate an Alternative TCR Pathway?
8. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Lindahl, T. The Intrinsic Fragility of DNA (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2016, 55, 8528–8534. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and Decay of the Primary Structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Andersson, A. Rate of Chain Breakage at Apurinic Sites in Double-Stranded Deoxyribonucleic Acid. Biochemistry 1972, 11, 3618–3623. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Nyberg, B. Rate of Depurination of Native Deoxyribonucleic Acid. Biochemistry 1972, 11, 3610–3618. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ. Mol. Mutagenesis 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis, 2nd ed.; ASM Press: Washington, DC, USA, 2005. [Google Scholar]
- Yi, C.; He, C. DNA Repair by Reversal of DNA Damage. Cold Spring Harb. Perspect. Biol. 2013, 5, a012575. [Google Scholar] [CrossRef]
- Kowalczykowski, S.C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016410. [Google Scholar]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Yasui, A. Alternative Excision Repair Pathways. Cold Spring Harb. Perspect. Biol. 2013, 5, a012617. [Google Scholar] [CrossRef] [Green Version]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Clauson, C.; Schärer, O.D.; Niedernhofer, L. Advances in Understanding the Complex Mechanisms of DNA Interstrand Cross-Link Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012732. [Google Scholar] [CrossRef] [PubMed]
- Stingele, J.; Bellelli, R.; Boulton, S.J. Mechanisms of DNA—Protein Crosslink Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Goosen, N.; Moolenaar, G.F. Role of ATP Hydrolysis by UvrA and UvrB during Nucleotide Excision Repair. Res. Microbiol. 2001, 152, 401–409. [Google Scholar] [CrossRef]
- Kisker, C.; Kuper, J.; Van Houten, B. Prokaryotic Nucleotide Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012591. [Google Scholar] [CrossRef]
- DellaVecchia, M.J.; Croteau, D.L.; Skorvaga, M.; Dezhurov, S.V.; Lavrik, O.I.; Van Houten, B. Analyzing the Handoff of DNA from UvrA to UvrB Utilizing DNA-Protein Photoaffinity Labeling. J. Biol. Chem. 2004, 279, 45245–45256. [Google Scholar] [CrossRef] [Green Version]
- Jaciuk, M.; Nowak, E.; Skowronek, K.; Tańska, A.; Nowotny, M. Structure of UvrA nucleotide excision repair protein in complex with modified DNA. Nat. Struct. Mol. Biol. 2011, 18, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Jaciuk, M.; Swuec, P.; Gaur, V.; Kasprzak, J.M.; Renault, L.; Dobrychłop, M.; Nirwal, S.; Bujnicki, J.M.; Costa, A.; Nowotny, M. A combined structural and biochemical approach reveals translocation and stalling of UvrB on the DNA lesion as a mechanism of damage verification in bacterial nucleotide excision repair. DNA Repair 2020, 85, 102746. [Google Scholar] [CrossRef]
- Pakotiprapha, D.; Samuels, M.; Shen, K.; Hu, J.H.; Jeruzalmi, D. Structure and mechanism of the UvrA-UvrB DNA damage sensor. Nat. Struct. Mol. Biol. 2012, 19, 291–298. [Google Scholar] [CrossRef]
- Yang, W. Surviving the Sun: Repair and Bypass of DNA UV Lesions. Protein Sci. 2011, 20, 1781–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhoeven, E.E.; van Kesteren, M.; Moolenaar, G.F.; Visse, R.; Goosen, N. Catalytic Sites for 3′ and 5′ Incision of Escherichia coli Nucleotide Excision Repair Are Both Located in UvrC. J. Biol. Chem. 2000, 275, 5120–5123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deaconescu, A.M.; Suhanovsky, M.M. From Mfd to TRCF and Back Again—A Perspective on Bacterial Transcription-Coupled Nucleotide Excision Repair. Photochem. Photobiol. 2017, 93, 268–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Marr, M.T.; Roberts, J.W. E. coli Transcription Repair Coupling Factor (Mfd Protein) Rescues Arrested Complexes by Promoting Forward Translocation. Cell 2002, 109, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Deaconescu, A.M.; Sevostyanova, A.; Artsimovitch, I.; Grigorieff, N. Nucleotide excision repair (NER) machinery recruitment by the transcription-repair coupling factor involves unmasking of a conserved intramolecular interface. Proc. Natl. Acad. Sci. USA 2012, 109, 3353–3358. [Google Scholar] [CrossRef] [Green Version]
- Ghodke, H.; Ho, H.N.; Van Oijen, A.M. Single-Molecule Live-Cell Imaging Visualizes Parallel Pathways of Prokaryotic Nucleotide Excision Repair. Nat. Commun. 2020, 11, 1477. [Google Scholar] [CrossRef] [Green Version]
- Epshtein, V.; Kamarthapu, V.; McGary, K.; Svetlov, V.; Ueberheide, B.; Proshkin, S.; Mironov, A.; Nudler, E. UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nat. Cell Biol. 2014, 505, 372–377. [Google Scholar] [CrossRef] [Green Version]
- Pani, B.; Nudler, E. Mechanistic Insights Into Transcription Coupled DNA Repair. DNA Repair 2017, 56, 42–50. [Google Scholar] [CrossRef]
- Sancar, A.; Reardon, J.T. Nucleotide Excision Repair in E. coli and Man. Adv. Protein Chem. 2004, 69, 43–71. [Google Scholar]
- Doolittle, R.F.; Johnson, M.S.; Husain, I.; Van Houten, B.; Thomas, D.C.; Sancar, A. Domainal evolution of a prokaryotic DNA repair protein and its relationship to active-transport proteins. Nat. Cell Biol. 1986, 323, 451–453. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Koonin, E.V. Superfamily of UvrA-Related NTP-Binding Proteins. Implications for Rational Classification of Recombination/Repair Systems. J. Mol. Biol. 1990, 213, 583–591. [Google Scholar] [CrossRef]
- Krishnan, A.; Burroughs, A.M.; Iyer, L.M.; Aravind, L. Comprehensive classification of ABC ATPases and their functional radiation in nucleoprotein dynamics and biological conflict systems. Nucleic Acids Res. 2020, 48, 10045–10075. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.-P. Invited Review: Architectures and Mechanisms of ATP Binding Cassette Proteins. Biopolymers 2016, 105, 492–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linton, K.J. Structure and Function of ABC Transporters. Physiology 2007, 22, 122–130. [Google Scholar] [CrossRef]
- Locher, K. Mechanistic Diversity in ATP-Binding Cassette (ABC) Transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Pakotiprapha, D.; Inuzuka, Y.; Bowman, B.R.; Moolenaar, G.F.; Goosen, N.; Jeruzalmi, D.; Verdine, G.L. Crystal Structure of Bacillus stearothermophilus UvrA Provides Insight into ATP-Modulated Dimerization, UvrB Interaction, and DNA Binding. Mol. Cell 2008, 29, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Stracy, M.; Jaciuk, M.; Uphoff, S.; Kapanidis, A.N.; Nowotny, M.; Sherratt, D.J.; Zawadzki, P. Single-molecule imaging of UvrA and UvrB recruitment to DNA lesions in living Escherichia coli. Nat. Commun. 2016, 7, 12568. [Google Scholar] [CrossRef] [Green Version]
- Thiagalingam, S.; Grossman, L. The Multiple Roles for ATP in the Escherichia coli UvrABC Endonuclease-Catalyzed Incision Reaction. J. Biol. Chem. 1993, 268, 18382–18389. [Google Scholar]
- Thiagalingam, S.; Grossman, L. Both ATPase Sites of Escherichia coli UvrA Have Functional Roles in Nucleotide Excision Repair. J. Biol. Chem. 1991, 266, 11395–11403. [Google Scholar]
- Kraithong, T.; Channgam, K.; Itsathitphaisarn, O.; Tiensuwan, M.; Jeruzalmi, D.; Pakotiprapha, D. Movement of the beta-hairpin in the third zinc-binding module of UvrA is required for DNA damage recognition. DNA Repair 2017, 51, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Croteau, D.L.; DellaVecchia, M.J.; Wang, H.; Bienstock, R.J.; Melton, M.A.; Van Houten, B. The C-terminal Zinc Finger of UvrA Does Not Bind DNA Directly but Regulates Damage-specific DNA Binding. J. Biol. Chem. 2006, 281, 26370–26381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, K.; Moolenaar, G.F.; Goosen, N. Role of the Insertion Domain and the Zinc-Finger Motif of Escherichia coli UvrA in Damage Recognition and ATP Hydrolysis. DNA Repair 2011, 10, 483–496. [Google Scholar] [CrossRef]
- Timmins, J.; Gordon, E.; Caria, S.; Leonard, G.; Acajjaoui, S.; Kuo, M.-S.; Monchois, V.; McSweeney, S. Structural and Mutational Analyses of Deinococcus radiodurans UvrA2 Provide Insight into DNA Binding and Damage Recognition by UvrAs. Structure 2009, 17, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Rossi, F.; Khanduja, J.S.; Bortoluzzi, A.; Houghton, J.; Sander, P.; Güthlein, C.; Davis, E.O.; Springer, B.; Böttger, E.C.; Relini, A.; et al. The biological and structural characterization of Mycobacterium tuberculosis UvrA provides novel insights into its mechanism of action. Nucleic Acids Res. 2011, 39, 7316–7328. [Google Scholar] [CrossRef] [PubMed]
- Case, B.C.; Hartley, S.; Osuga, M.; Jeruzalmi, D.; Hingorani, M.M. The ATPase mechanism of UvrA2 reveals the distinct roles of proximal and distal ATPase sites in nucleotide excision repair. Nucleic Acids Res. 2019, 47, 4136–4152. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Sung, R.J.; Verdine, G.L. Mechanism of DNA Lesion Homing and Recognition by the Uvr Nucleotide Excision Repair System. Research 2019, 2019, 5641746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truglio, J.J.; Rhau, B.; Croteau, D.L.; Wang, L.; Skorvaga, M.; Karakas, E.; DellaVecchia, M.J.; Wang, H.; Van Houten, B.; Kisker, C. Structural Insights Into the First Incision Reaction during Nucleotide Excision Repair. EMBO J. 2005, 24, 885–894. [Google Scholar] [CrossRef] [Green Version]
- Karakas, E.; Truglio, J.J.; Croteau, D.; Rhau, B.; Wang, L.; Van Houten, B.; Kisker, C. Structure of the C-terminal half of UvrC reveals an RNase H endonuclease domain with an Argonaute-like catalytic triad. EMBO J. 2007, 26, 613–622. [Google Scholar] [CrossRef] [Green Version]
- Deaconescu, A.M.; Chambers, A.L.; Smith, A.J.; Nickels, B.E.; Hochschild, A.; Savery, N.J.; Darst, S.A. Structural Basis for Bacterial Transcription-Coupled DNA Repair. Cell 2006, 124, 507–520. [Google Scholar] [CrossRef] [Green Version]
- Theis, K.; Chen, P.J.; Skorvaga, M.; Van Houten, B.; Kisker, C. Crystal structure of UvrB, a DNA helicase adapted for nucleotide excision repair. EMBO J. 1999, 18, 6899–6907. [Google Scholar] [CrossRef]
- Truglio, J.J.; Croteau, D.L.; Van Houten, B.; Kisker, C. Prokaryotic Nucleotide Excision Repair: The UvrABC System. Chem. Rev. 2006, 106, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.Y.; Grossman, L. Helicase Properties of the Escherichia coli UvrAB Protein Complex. Proc. Natl. Acad. Sci. USA 1987, 84, 3638–3642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, E.Y.; Grossman, L. Characterization of the helicase activity of the Escherichia coli UvrAB protein complex. J. Biol. Chem. 1989, 264, 1336–1343. [Google Scholar] [PubMed]
- Seeley, T.W.; Grossman, L. The Role of Escherichia coli UvrB in Nucleotide Excision Repair. J. Biol. Chem. 1990, 265, 7158–7165. [Google Scholar] [PubMed]
- Seeley, T.W.; Grossman, L. Mutations in the Escherichia coli UvrB ATPase Motif Compromise Excision Repair Capacity. Proc. Natl. Acad. Sci. USA 1989, 86, 6577–6581. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; DellaVecchia, M.J.; Skorvaga, M.; Croteau, D.L.; Erie, D.A.; Van Houten, B. UvrB Domain 4, an Autoinhibitory Gate for Regulation of DNA Binding and ATPase Activity. J. Biol. Chem. 2006, 281, 15227–15237. [Google Scholar] [CrossRef] [Green Version]
- Caron, R.; Grossman, L. Involvement of a Cryptic ATPase Activity of UvrB and Its Proteolysis Product, UvrB* in DNA Repair. Nucleic Acids Res. 1988, 16, 10891–10902. [Google Scholar] [CrossRef]
- Gordienko, I.; Rupp, W.D. The Limited Strand-Separating Activity of the UvrAB Protein Complex and Its Role in the Recognition of DNA Damage. EMBO J. 1997, 16, 889–895. [Google Scholar] [CrossRef] [Green Version]
- Moolenaar, G.F.; Höglund, L.; Goosen, N. Clue to Damage Recognition by UvrB: Residues in the Beta-Hairpin Structure Prevent Binding to Non-Damaged DNA. EMBO J. 2001, 20, 6140–6149. [Google Scholar] [CrossRef] [Green Version]
- Skorvaga, M.; Theis, K.; Mandavilli, B.S.; Kisker, C.; Van Houten, B. The beta-hairpin motif of UvrB is essential for DNA binding, damage processing, and UvrC-mediated incisions. J. Biol. Chem. 2002, 277, 1553–1559. [Google Scholar] [CrossRef] [Green Version]
- Waters, T.R.; Eryilmaz, J.; Geddes, S.; Barrett, T.E. Damage detection by the UvrABC pathway: Crystal structure of UvrB bound to fluorescein-adducted DNA. FEBS Lett. 2006, 580, 6423–6427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truglio, J.J.; Karakas, E.; Rhau, B.; Wang, H.; DellaVecchia, M.J.; Van Houten, B.; Kisker, C. Structural basis for DNA recognition and processing by UvrB. Nat. Struct. Mol. Biol. 2006, 13, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Moolenaar, G.F.; Bazuine, M.; Van Knippenberg, I.C.; Visse, R.; Goosen, N. Characterization of the Escherichia coli damage-independent UvrBC endonuclease activity. J. Biol. Chem. 1998, 273, 34896–34903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moolenaar, G.F.; Herron, M.F.P.; Monaco, V.; Van Der Marel, G.A.; Van Boom, J.H.; Visse, R.; Goosen, N. The Role of ATP Binding and Hydrolysis by UvrB during Nucleotide Excision Repair. J. Biol. Chem. 2000, 275, 8044–8050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravind, L.; Walker, D.R.; Koonin, E.V. Conserved Domains in DNA Repair Proteins and Evolution of Repair Systems. Nucleic Acids Res. 1999, 27, 1223–1242. [Google Scholar] [CrossRef]
- Lin, J.J.; Sancar, A. Active Site of (A)BC Excinuclease. I. Evidence for 5′ Incision by UvrC Through a Catalytic Site Involving Asp399, Asp438, Asp466, and His538 Residues. J. Biol. Chem. 1992, 267, 17688–17692. [Google Scholar]
- Alexandrovich, A.; Czisch, M.; Frenkiel, T.A.; Kelly, G.P.; Goosen, N.; Moolenaar, G.F.; Chowdhry, B.Z.; Sanderson, M.R.; Lane, A.N. Solution Structure, Hydrodynamics and Thermodynamics of the UvrB C-terminal Domain. J. Biomol. Struct. Dyn. 2001, 19, 219–236. [Google Scholar] [CrossRef]
- Sohi, M.; Alexandrovich, A.; Moolenaar, G.; Visse, R.; Goosen, N.; Vernede, X.; Fontecilla-Camps, J.C.; Champness, J.; Sanderson, M.R. Crystal Structure of Escherichia coli UvrB C-Terminal Domain, and a Model for UvrB-uvrC interaction. FEBS Lett. 2000, 465, 161–164. [Google Scholar] [CrossRef] [Green Version]
- Moolenaar, G.F.; Franken, K.L.M.C.; Dijkstra, D.M.; Thomas-Oates, J.E.; Visse, R.; Van De Putte, P.; Goosen, N. The C-terminal region of the UvrB protein of Escherichia coli contains an important determinant for UvrC binding to the preincision complex but not the catalytic site for 3’-incision. J. Biol. Chem. 1995, 270, 30508–30515. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Folkers, G.E.; Bonvin, A.M.J.J.; Boelens, R.; Wechselberger, R.; Niztayev, A.; Kaptein, R. Solution structure and DNA-binding properties of the C-terminal domain of UvrC from E.coli. EMBO J. 2002, 21, 6257–6266. [Google Scholar] [CrossRef] [Green Version]
- Moolenaar, G.F.; Uiterkamp, R.S.; Zwijnenburg, D.A.; Goosen, N. The C-terminal region of the Escherichia coli UvrC protein, which is homologous to the C-terminal region of the human ERCC1 protein, is involved in DNA binding and 5’-incision. Nucleic Acids Res. 1998, 26, 462–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhoeven, E.E.A.; Van Kesteren, M.; Turner, J.J.; Van Der Marel, G.A.; Van Boom, J.H.; Moolenaar, G.F.; Goosen, N. The C-terminal region of Escherichia coli UvrC contributes to the flexibility of the UvrABC nucleotide excision repair system. Nucleic Acids Res. 2002, 30, 2492–2500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargreaves, D.; Rice, D.W.; Sedelnikova, S.E.; Artymiuk, P.J.; Lloyd, R.G.; Rafferty, J.B. Crystal structure of E.coli RuvA with bound DNA Holliday junction at 6 Å resolution. Nat. Genet. 1998, 5, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Brugger, C.; Zhang, C.; Suhanovsky, M.M.; Kim, D.D.; Sinclair, A.N.; Lyumkis, D.; Deaconescu, A.M. Molecular determinants for dsDNA translocation by the transcription-repair coupling and evolvability factor Mfd. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Assenmacher, N.; Wenig, K.; Lammens, A.; Hopfner, K.-P. Structural Basis for Transcription-coupled Repair: The N Terminus of Mfd Resembles UvrB with Degenerate ATPase Motifs. J. Mol. Biol. 2006, 355, 675–683. [Google Scholar] [CrossRef]
- Selby, C.; Sancar, A. Structure and Function of Transcription-Repair Coupling Factor. II. Catalytic Properties. J. Biol. Chem. 1995, 270, 4890–4895. [Google Scholar] [CrossRef] [Green Version]
- Mahdi, A.A.; Briggs, G.S.; Sharples, G.J.; Wen, Q.; Lloyd, R.G. A model for dsDNA translocation revealed by a structural motif common to RecG and Mfd proteins. EMBO J. 2003, 22, 724–734. [Google Scholar] [CrossRef] [Green Version]
- Warren, G.M.; Stein, R.A.; Mchaourab, H.S.; Eichman, B.F. Movement of the RecG Motor Domain upon DNA Binding Is Required for Efficient Fork Reversal. Int. J. Mol. Sci. 2018, 19, 3049. [Google Scholar] [CrossRef] [Green Version]
- Chambers, A.L.; Smith, A.J.; Savery, N.J. A DNA Translocation Motif in the Bacterial Transcription-Repair Coupling Factor, Mfd. Nucleic Acids Res. 2003, 31, 6409–6418. [Google Scholar] [CrossRef] [Green Version]
- Gunz, D.; Hess, M.T.; Naegeli, H. Recognition of DNA Adducts by Human Nucleotide Excision Repair. Evidence for a Thermodynamic Probing Mechanism. J. Biol. Chem. 1996, 271, 25089–25098. [Google Scholar] [CrossRef] [Green Version]
- Hess, M.T.; Gunz, D.; Luneva, N.; Geacintov, N.E.; Naegeli, H. Base pair conformation-dependent excision of benzo[a]pyrene diol epoxide-guanine adducts by human nucleotide excision repair enzymes. Mol. Cell. Biol. 1997, 17, 7069–7076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camenisch, U.; Dip, R.; Schumacher, S.B.; Schuler, B.; Naegeli, H. Recognition of helical kinks by xeroderma pigmentosum group A protein triggers DNA excision repair. Nat. Struct. Mol. Biol. 2006, 13, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Camenisch, U.; Dip, R.; Vitanescu, M.; Naegeli, H. Xeroderma pigmentosum complementation group A protein is driven to nucleotide excision repair sites by the electrostatic potential of distorted DNA. DNA Repair 2007, 6, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Yang, W. Poor Base Stacking at DNA Lesions May Initiate Recognition by Many Repair Proteins. DNA Repair 2006, 5, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, R.J.; Spielmanna, H.P. A Model for Initial DNA Lesion Recognition by NER and MMR Based on Local Conformational Flexibility. DNA Repair 2004, 3, 455–464. [Google Scholar]
- Maillard, O.; Camenisch, U.; Clement, F.C.; Blagoev, K.B.; Naegeli, H. DNA repair triggered by sensors of helical dynamics. Trends Biochem. Sci. 2007, 32, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.; Grossman, L. The Binding of UvrAB Proteins to Bubble and Loop Regions in Duplex DNA. J. Biol. Chem. 1996, 271, 21462–21470. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Walker, R.; Bassett, H.; Geacintov, N.E.; Van Houten, B. Formation of DNA repair intermediates and incision by the ATP-dependent UvrB-UvrC endonuclease. J. Biol. Chem. 1997, 272, 4820–4827. [Google Scholar] [CrossRef] [Green Version]
- Bertrand-Burggraf, E.; Selby, C.P.; Hearst, J.E.; Sancar, A. Identification of the different intermediates in the interaction of (A)BC excinuclease with its substrates by DNase I footprinting on two uniquely modified oligonucleotides. J. Mol. Biol. 1991, 219, 27–36. [Google Scholar] [CrossRef]
- Branum, M.E.; Reardon, J.T.; Sancar, A. DNA Repair Excision Nuclease Attacks Undamaged DNA. A Potential Source of Spontaneous Mutations. J. Biol. Chem. 2001, 276, 25421–25426. [Google Scholar] [CrossRef] [Green Version]
- Lindsey-Boltz, L.A.; Sancar, A. RNA Polymerase: The Most Specific Damage Recognition Protein in Cellular Responses to DNA Damage? Proc. Natl. Acad. Sci. USA 2007, 104, 13213–13214. [Google Scholar] [CrossRef] [Green Version]
- Reardon, J.T.; Sancar, A. Thermodynamic Cooperativity and Kinetic Proofreading in DNA Damage Recognition and Repair. Cell Cycle 2004, 3, 139–142. [Google Scholar] [CrossRef]
- Kad, N.M.; Van Houten, B. Dynamics of Lesion Processing by Bacterial Nucleotide Excision Repair Proteins. Prog. Mol. Biol. Transl. Sci. 2012, 110, 1–24. [Google Scholar]
- Hu, J.; Selby, C.P.; Adar, S.; Adebali, O.; Sancar, A. Molecular mechanisms and genomic maps of DNA excision repair in Escherichia coli and humans. J. Biol. Chem. 2017, 292, 15588–15597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, S.M.; Guthrie, C. Beat the Clock: Paradigms for NTPases in the Maintenance of Biological Fidelity. Trends Biochem. Sci. 1993, 18, 381–384. [Google Scholar] [CrossRef]
- Hopfield, J.J. Kinetic Proofreading: A New Mechanism for Reducing Errors in Biosynthetic Processes Requiring High Specificity. Proc. Natl. Acad. Sci. USA 1974, 71, 4135–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kad, N.M.; Wang, H.; Kennedy, G.G.; Warshaw, D.M.; Van Houten, B. Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single- molecule imaging of quantum-dot-labeled proteins. Mol. Cell 2010, 37, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Malta, E.; Moolenaar, G.F.; Goosen, N. Dynamics of the UvrABC Nucleotide Excision Repair Proteins Analyzed by Fluorescence Resonance Energy Transfer. Biochemistry 2007, 46, 9080–9088. [Google Scholar] [CrossRef]
- Orren, D.K.; Sancar, A. The (A)BC Excinuclease of Escherichia coli Has Only the UvrB and UvrC Subunits in the Incision Complex. Proc. Natl. Acad. Sci. USA 1989, 86, 5237–5241. [Google Scholar] [CrossRef] [Green Version]
- Verhoeven, E.E.; Wyman, C.; Moolenaar, G.F.; Goosen, N. The presence of two UvrB subunits in the UvrAB complex ensures damage detection in both DNA strands. EMBO J. 2002, 21, 4196–4205. [Google Scholar] [CrossRef] [Green Version]
- Moolenaar, G.F.; Schut, M.; Goosen, N. Binding of the UvrB Dimer to Non-Damaged and Damaged DNA: Residues Y92 and Y93 Influence the Stability of Both Subunits. DNA Repair 2005, 4, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Demple, B.; Robins, P. Suicide Inactivation of the E. coli O6-methylguanine-DNA Methyltransferase. EMBO J. 1982, 1, 1359–1363. [Google Scholar] [CrossRef] [PubMed]
- Trewick, S.C.; Henshaw, T.F.; Hausinger, R.P.; Lindahl, T.; Sedgwick, B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nat. Cell Biol. 2002, 419, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Mielecki, D.; Grzesiuk, E. Ada Response—A Strategy for Repair of Alkylated DNA in Bacteria. FEMS Microbiol. Lett. 2014, 355, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mazón, G.; Philippin, G.; Cadet, J.; Gasparutto, D.; Fuchs, R.P. The alkyltransferase-like ybaZ gene product enhances nucleotide excision repair of O6-alkylguanine adducts in E. coli. DNA Repair 2009, 8, 697–703. [Google Scholar] [CrossRef]
- Tubbs, J.L.; Latypov, V.; Kanugula, S.; Butt, A.; Melikishvili, M.; Kraehenbuehl, R.; Fleck, O.; Marriott, A.; Watson, A.J.; Verbeek, B.; et al. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nat. Cell Biol. 2009, 459, 808–813. [Google Scholar] [CrossRef] [Green Version]
- Rill, N.; Mukhortava, A.; Lorenz, S.; Tessmer, I. Alkyltransferase-like protein clusters scan DNA rapidly over long distances and recruit NER to alkyl-DNA lesions. Proc. Natl. Acad. Sci. USA 2020, 117, 9318–9328. [Google Scholar] [CrossRef]
- Springall, L.; Hughes, C.D.; Simons, M.; Azinas, S.; Van Houten, B.; Kad, N.M. Recruitment of UvrBC complexes to UV-induced damage in the absence of UvrA increases cell survival. Nucleic Acids Res. 2018, 46, 1256–1265. [Google Scholar] [CrossRef] [Green Version]
- Thakur, M.; Badugu, S.; Muniyappa, K. UvrA and UvrC Subunits of the Mycobacterium tuberculosis UvrABC Excinuclease Interact Independently of UvrB and DNA. FEBS Lett. 2020, 594, 851–863. [Google Scholar] [CrossRef]
- Caron, R.; Grossman, L. Incision of Damaged Versus Nondamaged DNA by the Escherichia coli UvrABC Proteins. Nucleic Acids Res. 1988, 16, 7855–7865. [Google Scholar] [CrossRef] [Green Version]
- Sancar, A.; Hearst, J. Molecular Matchmakers. Science 1993, 259, 1415–1420. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, E.E.; Wyman, C.; Moolenaar, G.F.; Hoeijmakers, J.H.J.; Goosen, N. Architecture of nucleotide excision repair complexes: DNA is wrapped by UvrB before and after damage recognition. EMBO J. 2001, 20, 601–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Lu, M.; Tang, M.-S.; Van Houten, B.; Ross, J.B.A.; Weinfeld, M.; Le, X.C. DNA wrapping is required for DNA damage recognition in the Escherichia coli DNA nucleotide excision repair pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 12849–12854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeberg, E.; Steinum, A.L. Purification and Properties of the uvrA Protein from Escherichia coli. Proc. Natl. Acad. Sci. USA 1982, 79, 988–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moolenaar, G.F.; Monaco, V.; van der Marel, G.A.; van Boom, J.H.; Visse, R.; Goosen, N. The effect of the DNA flanking the lesion on formation of the UvrB-DNA preincision complex. Mechanism for the UvrA-mediated loading of UvrB onto a DNA damaged site. J. Biol. Chem. 2000, 275, 8038–8043. [Google Scholar] [CrossRef] [Green Version]
- Hu, J. The Role of ATP Binding and Hydrolysis by UvrA in Dimerization and DNA Binding and Repair, in Bachelor of Arts with Honors in the Field of Chemical and Physical Biology; Harvard University: Cambridge, MA, USA, 2011. [Google Scholar]
- Myles, G.M.; Hearst, J.E.; Sancar, A. Site-Specific Mutagenesis of Conserved Residues within Walker A and B Sequences of Escherichia coli UvrA Protein. Biochemistry 1991, 30, 3824–3834. [Google Scholar] [CrossRef]
- Oh, E.Y.; Claassen, L.; Thiagalingam, S.; Mazur, S.; Grossman, L. ATPase activity of the UvrA and UvrAB protein complexes of the Escherichia coli UvrABC endonuclease. Nucleic Acids Res. 1989, 17, 4145–4159. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.; Moolenaar, G.F.; Goosen, N. Role of the Two ATPase Domains of Escherichia coli UvrA in Binding Non-Bulky DNA Lesions and Interaction with UvrB. DNA Repair 2010, 9, 1176–1186. [Google Scholar] [CrossRef]
- Mazur, S.J.; Grossman, L. Dimerization of Escherichia coli UvrA and Its Binding to Undamaged and Ultraviolet Light Damaged DNA. Biochemistry 1991, 30, 4432–4443. [Google Scholar] [CrossRef]
- Wagner, K.; Moolenaar, G.; Van Noort, J.; Goosen, N. Single-molecule analysis reveals two separate DNA-binding domains in the Escherichia coli UvrA dimer. Nucleic Acids Res. 2009, 37, 1962–1972. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Shell, S.M.; Utzat, C.D.; Luo, C.; Yang, Z.; Geacintov, N.E.; Basu, A.K. Effects of DNA Adduct Structure and Sequence Context on Strand Opening of Repair Intermediates and Incision by UvrABC Nuclease. Biochemistry 2003, 42, 12654–12661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekhovich, O.; Tang, M.-S.; Romano, L.J. Rate of Incision ofN-Acetyl-2-aminofluorene andN-2-Aminofluorene Adducts by UvrABC Nuclease Is Adduct- and Sequence-Specific: Comparison of the Rates of UvrABC Nuclease Incision and Protein−DNA Complex Formation. Biochemistry 1998, 37, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Januliene, D.; Mehdipour, A.R.; Thomas, C.; Stefan, E.; Brüchert, S.; Kuhn, B.T.; Geertsma, E.R.; Hummer, G.; Tampé, R.; et al. Conformation space of a heterodimeric ABC exporter under turnover conditions. Nat. Cell Biol. 2019, 571, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Witkin, E.M. Radiation-Induced Mutations and Their Repair. Science 1966, 152, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Mellon, I.; Hanawalt, P.C. Induction of the Escherichia coli Lactose Operon Selectively Increases Repair of Its Transcribed DNA Strand. Nat. Cell Biol. 1989, 342, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Mellon, I.; Spivak, G.; Hanawalt, P.C. Selective Removal of Transcription-Blocking DNA Damage from the Transcribed Strand of the Mammalian DHFR Gene. Cell 1987, 51, 241–249. [Google Scholar] [CrossRef]
- Bohr, V.A.; Smith, C.A.; Okumoto, D.S.; Hanawalt, P.C. DNA repair in an active gene: Removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell 1985, 40, 359–369. [Google Scholar] [CrossRef]
- Selby, C.P.; Sancar, A. Gene- and Strand-Specific Repair In Vitro: Partial Purification of a Transcription-Repair Coupling Factor. Proc. Natl. Acad. Sci. USA 1991, 88, 8232–8236. [Google Scholar] [CrossRef] [Green Version]
- Selby, C.; Witkin, E.M.; Sancar, A. Escherichia coli mfd Mutant Deficient in “Mutation Frequency Decline” Lacks Strand-Specific Repair: In Vitro Complementation with Purified Coupling Factor. Proc. Natl. Acad. Sci. USA 1991, 88, 11574–11578. [Google Scholar] [CrossRef] [Green Version]
- Selby, C.; Sancar, A. Transcription-Repair Coupling and Mutation Frequency Decline. J. Bacteriol. 1993, 175, 7509–7514. [Google Scholar] [CrossRef] [Green Version]
- Selby, C.; Sancar, A. Molecular Mechanism of Transcription-Repair Coupling. Science 1993, 260, 53–58. [Google Scholar] [CrossRef]
- Ganesan, A.K.; Hanawalt, C. Transcription-Coupled Nucleotide Excision Repair of a Gene Transcribed by Bacteriophage T7 RNA Polymerase in Escherichia coli. DNA Repair 2010, 9, 958–963. [Google Scholar] [CrossRef] [Green Version]
- Selby, C.P. Mfd Protein and Transcription-Repair Coupling in Escherichia coli. Photochem. Photobiol. 2017, 93, 280–295. [Google Scholar] [CrossRef] [Green Version]
- Wade, J.T.; Grainger, D.C. Pervasive Transcription: Illuminating the Dark Matter of Bacterial Transcriptomes. Nat. Rev. Genet. 2014, 12, 647–653. [Google Scholar] [CrossRef]
- Selby, C.; Sancar, A. Structure and Function of Transcription-Repair Coupling Factor. I. Structural Domains and Binding Properties. J. Biol. Chem. 1995, 270, 4882–4889. [Google Scholar] [CrossRef] [Green Version]
- Kamarthapu, V.; Nudler, E. Rethinking Transcription Coupled DNA Repair. Curr. Opin. Microbiol. 2015, 24, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Graves, E.T.; Duboc, C.; Fan, J.; Stransky, F.; Leroux-Coyau, M.; Strick, T.R. A dynamic DNA-repair complex observed by correlative single-molecule nanomanipulation and fluorescence. Nat. Struct. Mol. Biol. 2015, 22, 452–457. [Google Scholar] [CrossRef]
- Ho, H.N.; Van Oijen, A.M.; Ghodke, H. Single-Molecule Imaging Reveals Molecular Coupling between Transcription and DNA Repair Machinery in Live Cells. Nat. Commun. 2020, 11, 1478. [Google Scholar] [CrossRef] [Green Version]
- Haines, N.M.; Kim, Y.-I.T.; Smith, A.J.; Savery, N.J. Stalled transcription complexes promote DNA repair at a distance. Proc. Natl. Acad. Sci. USA 2014, 111, 4037–4042. [Google Scholar] [CrossRef] [Green Version]
- Le, T.T.; Yang, Y.; Tan, C.; Suhanovsky, M.M.; Fulbright, R.M.; Inman, J.T.; Li, M.; Lee, J.; Perelman, S.; Roberts, J.W.; et al. Mfd Dynamically Regulates Transcription via a Release and Catch-Up Mechanism. Cell 2018, 172, 344–357.e15. [Google Scholar] [CrossRef] [Green Version]
- Washburn, R.S.; Wang, Y.; Gottesman, M.E. Role of E. Coli Transcription-Repair Coupling Factor Mfd in Nun-Mediated Transcription Termination. J. Mol. Biol. 2003, 329, 655–662. [Google Scholar] [CrossRef]
- Belitsky, B.R.; Sonenshein, A.L. Roadblock Repression of Transcription by Bacillus subtilis CodY. J. Mol. Biol. 2011, 411, 729–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.J.; Szczelkun, M.D.; Savery, N. Controlling the Motor Activity of a Transcription-Repair Coupling Factor: Autoinhibition and the Role of RNA Polymerase. Nucleic Acids Res. 2007, 35, 1802–1811. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.N.; Gong, P.; Ralto, K.; Manelyte, L.; Savery, N.; Theis, K. An N-terminal clamp restrains the motor domains of the bacterial transcription-repair coupling factor Mfd. Nucleic Acids Res. 2009, 37, 6042–6053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Leroux-Coyau, M.; Savery, N.J.; Strick, T.R. Reconstruction of bacterial transcription-coupled repair at single-molecule resolution. Nat. Cell Biol. 2016, 536, 234–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, D.J.; Hanawalt, P.C. The SOS-Dependent Upregulation of uvrD Is Not Required for Efficient Nucleotide Excision Repair of Ultraviolet Light Induced DNA Photoproducts in Escherichia coli. Mutat. Res. Repair 2001, 485, 319–329. [Google Scholar] [CrossRef]
- Ahn, B. A Physical Interaction of UvrD with Nucleotide Excision Repair Protein UvrB. Mol. Cells 2000, 10, 592–597. [Google Scholar] [CrossRef]
- Manelyte, L.; Guy, C.P.; Smith, R.M.; Dillingham, M.S.; McGlynn, P.; Savery, N. The unstructured C-terminal extension of UvrD interacts with UvrB, but is dispensable for nucleotide excision repair. DNA Repair 2009, 8, 1300–1310. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.E.; Lewis, C.A.; Mooney, R.A.; Kohanski, M.A.; Collins, J.J.; Landick, R.; Walker, G.C. Roles for the transcription elongation factor NusA in both DNA repair and damage tolerance pathways in Escherichia coli. Proc. Natl. Acad. Sci. USA 2010, 107, 15517–15522. [Google Scholar] [CrossRef] [Green Version]
- Matson, S.W.; George, J.W. DNA Helicase II of Escherichia coli. Characterization of the Single-Stranded DNA-Dependent NTPase and Helicase Activities. J. Biol. Chem. 1987, 262, 2066–2076. [Google Scholar]
- Lee, J.Y.; Yang, W. UvrD Helicase Unwinds DNA One Base Pair at a Time by a Two-Part Power Stroke. Cell 2006, 127, 1349–1360. [Google Scholar] [CrossRef] [Green Version]
- Maluf, N.K.; Fischer, C.J.; Lohman, T.M. A Dimer of Escherichia coli UvrD is the Active Form of the Helicase In Vitro. J. Mol. Biol. 2003, 325, 913–935. [Google Scholar] [CrossRef]
- Malta, E.; Verhagen, C.P.; Moolenaar, G.F.; Filippov, D.V.; Van Der Marel, G.A.; Goosen, N. Functions of base flipping in E. coli nucleotide excision repair. DNA Repair 2008, 7, 1647–1658. [Google Scholar] [CrossRef]
- Easton, A.M.; Kushner, S.R. Transcription of the uvrD Gene of Escherichia coli is Controlled by the lexA Repressor and by Attenuation. Nucleic Acids Res. 1983, 11, 8625–8640. [Google Scholar] [CrossRef] [Green Version]
- Siegel, E.C. The Escherichia coli uvrD Gene is Inducible by DNA Damage. Mol. Genet. Genom. 1983, 191, 397–400. [Google Scholar] [CrossRef]
- Drögemüller, J.; Strauß, M.; Schweimer, K.; Jurk, M.; Rösch, P.; Knauer, S.H. Determination of RNA polymerase binding surfaces of transcription factors by NMR spectroscopy. Sci. Rep. 2015, 5, 16428. [Google Scholar] [CrossRef]
- Yang, X.; Molimau, S.; Doherty, G.P.; Johnston, E.B.; Marles-Wright, J.; Rothnagel, R.; Hankamer, B.; Lewis, R.J.; Lewis, P.J. The structure of bacterial RNA polymerase in complex with the essential transcription elongation factor NusA. EMBO Rep. 2009, 10, 997–1002. [Google Scholar] [CrossRef] [Green Version]
- Ha, K.S.; Toulokhonov, I.; Vassylyev, D.G.; Landick, R. The NusA N-Terminal Domain Is Necessary and Sufficient for Enhancement of Transcriptional Pausing via Interaction with the RNA Exit Channel of RNA Polymerase. J. Mol. Biol. 2010, 401, 708–725. [Google Scholar] [CrossRef] [Green Version]
- Borukhov, S.; Lee, J.; Laptenko, O. Bacterial Transcription Elongation Factors: New Insights Into Molecular Mechanism of Action. Mol. Microbiol. 2005, 55, 1315–1324. [Google Scholar] [CrossRef]
- Potrykus, K.; Murphy, H.; Philippe, N.; Cashel, M. ppGpp is the major source of growth rate control in E. coli. Environ. Microbiol. 2011, 13, 563–575. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.; Wang, Y.; Steitz, T.A. The Mechanism of E. coli RNA Polymerase Regulation by ppGpp Is Suggested by the Structure of Their Complex. Mol. Cell 2013, 50, 430–436. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraithong, T.; Hartley, S.; Jeruzalmi, D.; Pakotiprapha, D. A Peek Inside the Machines of Bacterial Nucleotide Excision Repair. Int. J. Mol. Sci. 2021, 22, 952. https://doi.org/10.3390/ijms22020952
Kraithong T, Hartley S, Jeruzalmi D, Pakotiprapha D. A Peek Inside the Machines of Bacterial Nucleotide Excision Repair. International Journal of Molecular Sciences. 2021; 22(2):952. https://doi.org/10.3390/ijms22020952
Chicago/Turabian StyleKraithong, Thanyalak, Silas Hartley, David Jeruzalmi, and Danaya Pakotiprapha. 2021. "A Peek Inside the Machines of Bacterial Nucleotide Excision Repair" International Journal of Molecular Sciences 22, no. 2: 952. https://doi.org/10.3390/ijms22020952
APA StyleKraithong, T., Hartley, S., Jeruzalmi, D., & Pakotiprapha, D. (2021). A Peek Inside the Machines of Bacterial Nucleotide Excision Repair. International Journal of Molecular Sciences, 22(2), 952. https://doi.org/10.3390/ijms22020952