
Vaccines for Non-Viral Cancer Prevention

 ,
,  , and
, and
Abstract
:1. Introduction
2. Immunology of Cancer Prevention Vaccines
3. Antigen Targets
3.1. Tumor Associated Antigens (TAAs)
3.2. Cancer-Testis Antigens (CTAs)
3.3. Tumor Specific Antigens and Neoantigens
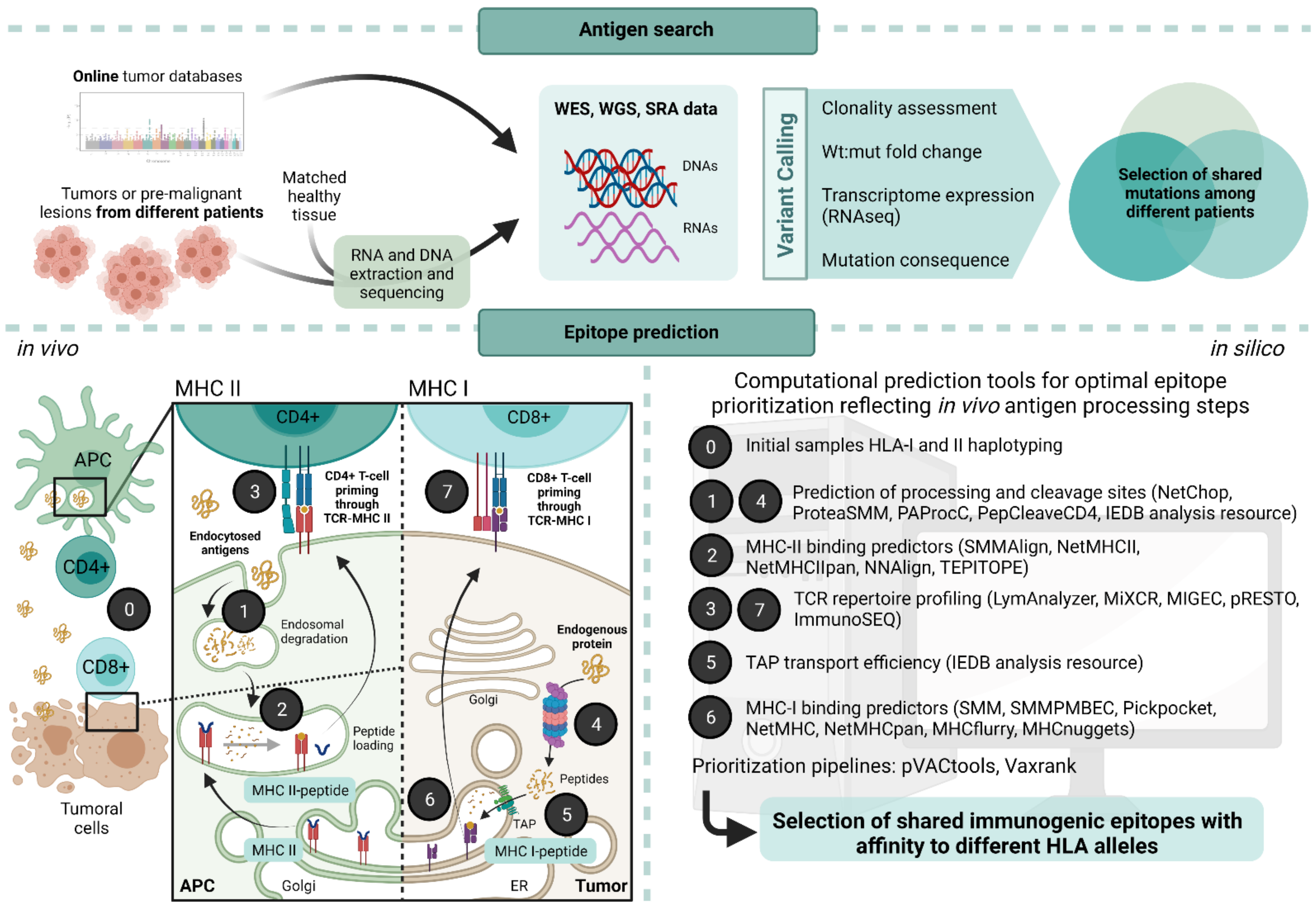
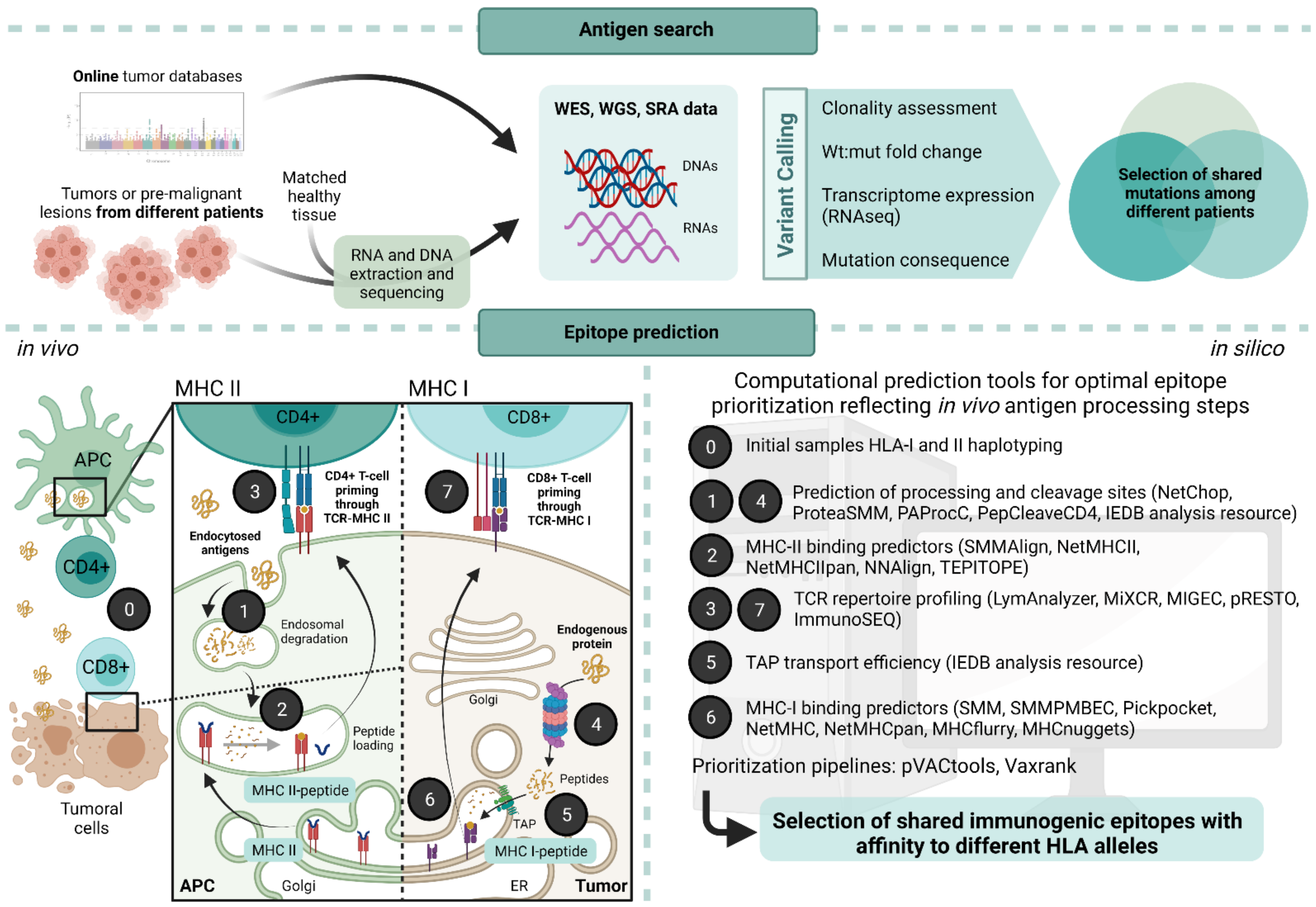
4. Challenges and Approaches for the Identification of Neoantigens
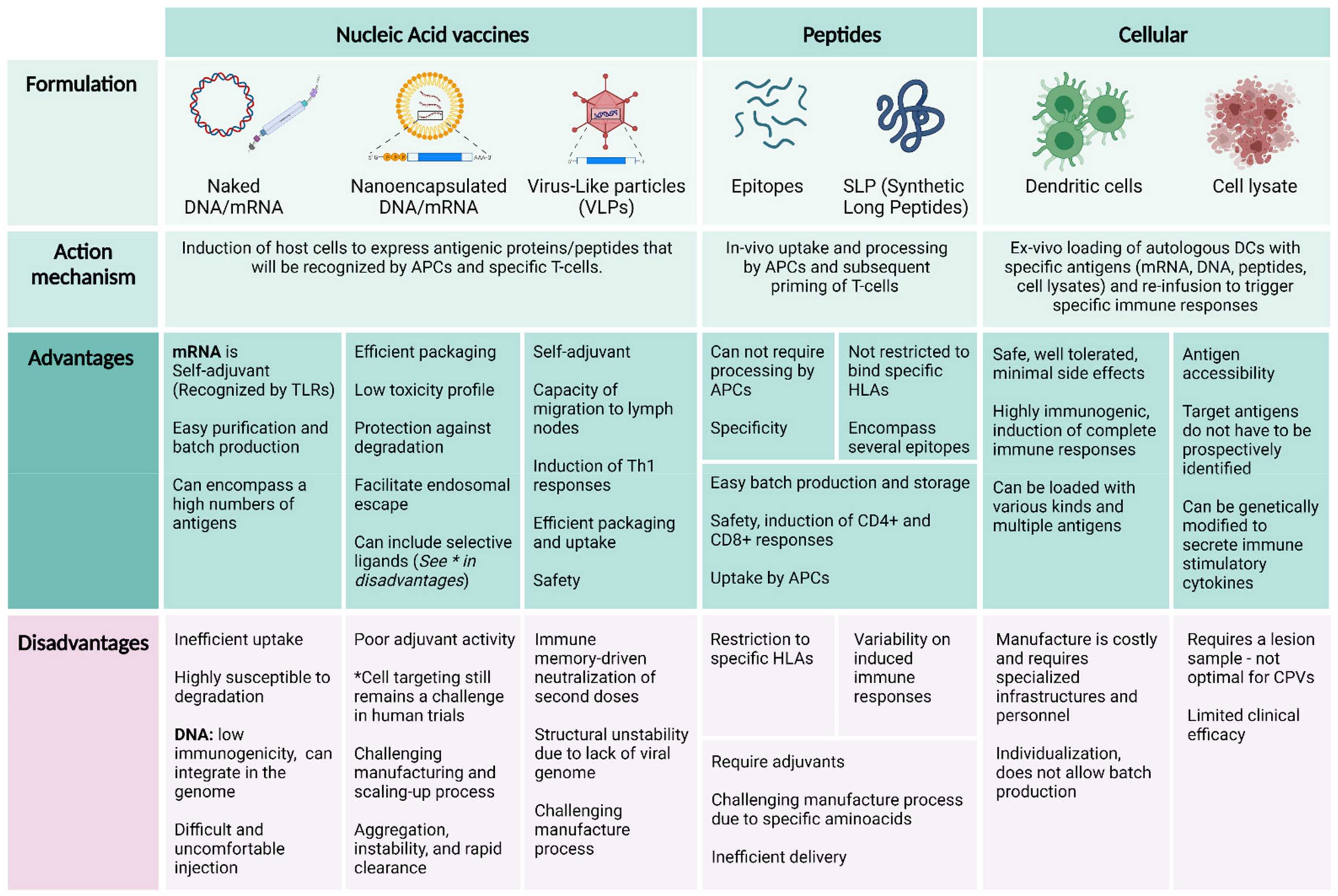
5. Vaccine Vectors
5.1. Nucleic Acid Vaccines and Peptides
Virus-like Particles
5.2. Cellular Vaccines
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goydos, J.; Elder, E.M.; Whiteside, T.L.; Finn, O.J.; Lotze, M.T. A phase I trial of a synthetic mucin peptide vaccine: Induction of specific immune reactivity in patients with adenocarcinoma. J. Surg. Res. 1996, 63, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Marchand, M.; van Baren, N.; Weynants, P.; Brichard, V.; Rankin, E.; Parmiani, G.; Arienti, F.; Humblet, Y.; Vanwijck, R.; Beauduin, M.; et al. Tumor regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by geneMAGE-3 and presented by HLA-A1. Int. J. Cancer 1999, 80, 219–230. [Google Scholar] [CrossRef]
- Disis, M.L.; Grabstein, K.H.; Sleath, P.R.; Cheever, M.A. Generation of immunity to the HER-2/neu oncogenic protein in patients with breast and ovarian cancer using a peptide-based vaccine. Clin. Cancer Res. 1999, 5, 1289–1297. [Google Scholar]
- Coughlin, C.M.; Vonderheide, R.H. Targeting adult and pediatric cancers via cell-based vaccines and the prospect of activated B lymphocytes as a novel modality. Cancer Biol. Ther. 2003, 2, 466–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, O.J. Cancer vaccines: Between the idea and the reality. Nat. Rev. Immunol. 2003, 3, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Kvistborg, P.; Yewdell, J.W. Enhancing responses to cancer immunotherapy. Science 2018, 359, 516–517. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 516–517. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Coulie, P.G.; Eynde, B.J.V.D.; Agostinis, P. Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol. 2017, 38, 577–593. [Google Scholar] [CrossRef]
- Karaki, S.; Anson, M.; Tran, T.; Giusti, D.; Blanc, C.; Oudard, S.; Tartour, E. Is there still room for cancer vaccines at the era of checkpoint inhibitors. Vaccines 2016, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Vreeland, T.J.; Clifton, G.T.; Herbert, G.S.; Hale, D.F.; Jackson, D.O.; Berry, J.S.; Peoples, G.E. Gaining ground on a cure through synergy: Combining checkpoint inhibitors with cancer vaccines. Expert Rev. Clin. Immunol. 2016, 12, 1347–1357. [Google Scholar] [CrossRef]
- Jou, J.; Harrington, K.J.; Zocca, M.-B.; Ehrnrooth, E.; Cohen, E.E. The changing landscape of therapeutic cancer vaccines—novel platforms and neoantigen identification. Clin. Cancer Res. 2021, 27, 689–703. [Google Scholar] [CrossRef]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Tan, A.C.; Goubier, A.; Kohrt, H.E. A quantitative analysis of therapeutic cancer vaccines in phase 2 or phase 3 trial. J. Immunother. Cancer 2015, 3, 48. [Google Scholar] [CrossRef] [Green Version]
- Janowitz, T.; Lu, L.; Yan, H.; Shyam-Sundar, V. Cross-sectional and longitudinal analysis of cancer vaccination trials registered on the US clinical trials database demonstrates paucity of immunological trial endpoints and decline in registration since 2008. Drug Des. Dev. Ther. 2014, 8, 1539–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shemesh, C.S.; Hsu, J.C.; Hosseini, I.; Shen, B.-Q.; Rotte, A.; Twomey, P.; Girish, S.; Wu, B. Personalized cancer vaccines: Clinical landscape, challenges, and opportunities. Mol. Ther. 2021, 29, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Umar, A.; Dunn, B.K.; Greenwald, P. Future directions in cancer prevention. Nat. Rev. Cancer 2012, 12, 835–848. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, Y.; Du, J. Human papillomavirus vaccines: An updated review. Vaccines 2020, 8, 391. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.H.; You, S.-L.; Chen, C.-J.; Liu, C.-J.; Lai, M.-W.; Wu, T.-C.; Wu, S.-F.; Lee, C.-M.; Yang, S.-S.; Chu, H.-C.; et al. Long-term effects of hepatitis B immunization of infants in preventing liver cancer. Gastroenterology 2016, 151, 472–480.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rühl, J.; Leung, C.S.; Münz, C. Vaccination against the Epstein–Barr virus. Cell. Mol. Life Sci. 2020, 77, 4315–4324. [Google Scholar] [CrossRef]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Leet, D.E.; Allesøe, R.L.; Oliveira, G.; Li, S.; Luoma, A.M.; Liu, J.; Forman, J.; Huang, T.; Iorgulescu, J.B.; et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med. 2021, 27, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Abelin, J.; Harjanto, D.; Malloy, M.; Suri, P.; Colson, T.; Goulding, S.P.; Creech, A.L.; Serrano, L.R.; Nasir, G.; Nasrullah, Y.; et al. Defining HLA-II ligand processing and binding rules with mass spectrometry enhances cancer epitope prediction. Immunity 2019, 51, 766–779.e17. [Google Scholar] [CrossRef]
- Majumder, S.; Shah, R.; Elias, J.; Manoharan, M.; Shah, P.; Kumari, A.; Chakraborty, P.; Kode, V.; Mistry, Y.; Coral, K.; et al. A cancer vaccine approach for personalized treatment of Lynch syndrome. Sci. Rep. 2018, 8, 12122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloor, M.; Reuschenbach, M.; Pauligk, C.; Karbach, J.; Rafiyan, M.-R.; Al-Batran, S.-E.; Tariverdian, M.; Jaeger, E.; Doeberitz, M.V.K. A frameshift peptide neoantigen-based vaccine for mismatch repair-deficient cancers: A phase I/IIa clinical trial. Clin. Cancer Res. 2020, 26, 4503–4510. [Google Scholar] [CrossRef]
- Kloor, M.; Reuschenbach, M.; Karbach, J.; Rafiyan, M.; Al-Batran, S.-E.; Pauligk, C.; Jaeger, E.; Doeberitz, M.V.K. Vaccination of MSI-H colorectal cancer patients with frameshift peptide antigens: A phase I/IIa clinical trial. J. Clin. Oncol. 2015, 33, 3020. [Google Scholar] [CrossRef]
- Gonzalez-Galarza, F.F.; McCabe, A.; Dos Santos, E.J.M.; Jones, J.; Takeshita, L.; Ortega-Rivera, N.D.; Del Cid-Pavon, G.M.; Ramsbottom, K.; Ghattaoraya, G.; Alfirevic, A.; et al. Allele frequency net database (AFND) 2020 update: Gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 2020, 48, D783–D788. [Google Scholar] [CrossRef]
- Willis, J.A.; Reyes-Uribe, L.; Chang, K.; Lipkin, S.M.; Vilar, E. Immune activation in mismatch repair–deficient carcinogenesis: More than just mutational rate. Clin. Cancer Res. 2020, 26, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Rossjohn, J.; Gras, S.; Miles, J.; Turner, S.J.; Godfrey, D.; McCluskey, J. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K.P.; Mills, K. Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol. 2013, 34, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef]
- Wirth, T.C.; Xue, H.-H.; Rai, D.; Sabel, J.T.; Bair, T.; Harty, J.T.; Badovinac, V.P. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8+ T cell differentiation. Immunity 2010, 33, 128–140. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van Den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Clevers, H. Modeling development and disease with organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Fujii, M.; Clevers, H.; Sato, T. Modeling human digestive diseases with CRISPR-Cas9–modified organoids. Gastroenterology 2019, 156, 562–576. [Google Scholar] [CrossRef] [Green Version]
- Wojtowicz, M.E.; Dunn, B.K.; Umar, A. Immunologic approaches to cancer prevention—current status, challenges, and future perspectives. Semin. Oncol. 2016, 43, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Coulie, P.G.; Van den Eynde, B.J.; Van Der Bruggen, P.; Boon-Falleur, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Gjerstorff, M.F.; Andersen, M.H.; Ditzel, H.J. Oncogenic cancer/testis antigens: Prime candidates for immunotherapy. Oncotarget 2015, 6, 15772–15787. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Cen, Q.; Lei, H. A review on development of MUC1-based cancer vaccine. Biomed. Pharmacother. 2020, 132, 110888. [Google Scholar] [CrossRef] [PubMed]
- Arab, A.; Yazdian-Robati, R.; Behravan, J. HER2-positive breast cancer immunotherapy: A focus on vaccine development. Arch. Immunol. Ther. Exp. 2020, 68, 2. [Google Scholar] [CrossRef]
- Shetty, K.; Ott, P.A. Personal neoantigen vaccines for the treatment of cancer. Annu. Rev. Cancer Biol. 2021, 5, 259–276. [Google Scholar] [CrossRef]
- Beatty, P.L.; Narayanan, S.; Gariépy, J.; Ranganathan, S.; Finn, O.J. Vaccine against MUC1 antigen expressed in inflammatory bowel disease and cancer lessens colonic inflammation and prevents progression to colitis-associated colon cancer. Cancer Prev. Res. 2010, 3, 438–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; McKolanis, J.R.; Dzubinski, L.A.; Islam, K.; Potter, D.M.; Salazar, A.M.; Schoen, R.E.; Finn, O.J. MUC1 Vaccine for individuals with advanced adenoma of the colon: A cancer immunoprevention feasibility study. Cancer Prev. Res. 2013, 6, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.-Y.; Bang, Y.-J. HER2-targeted therapie—a role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R.; Esteva, F.J. Trastuzumab: Triumphs and tribulations. Oncogene 2007, 26, 3637–3643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nocera, N.F.; Lee, M.C.; De La Cruz, L.M.; Rosemblit, C.; Czerniecki, B.J. Restoring lost anti-HER-2 Th1 immunity in breast cancer: A crucial role for Th1 cytokines in therapy and prevention. Front. Pharmacol. 2016, 7, 356. [Google Scholar] [CrossRef] [Green Version]
- Mittendorf, E.A.; Clifton, G.T.; Holmes, J.P.; Schneble, E.; van Echo, D.; Ponniah, S.; Peoples, G.E. Final report of the phase I/II clinical trial of the E75 (nelipepimut-S) vaccine with booster inoculations to prevent disease recurrence in high-risk breast cancer patients. Ann. Oncol. 2014, 25, 1735–1742. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Lu, B.; Melisko, M.; Hiller, J.P.; Bondarenko, I.; Brunt, A.M.; Sergii, G.; Petrakova, K.; Peoples, G.E. Efficacy and safety analysis of nelipepimut-s vaccine to prevent breast cancer recurrence: A randomized, multicenter, phase III clinical trial. Clin. Cancer Res. 2019, 25, 4248–4254. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, P.M.; Clifton, G.T.; Vreeland, T.J.; Adams, A.M.; O’Shea, A.E.; Peoples, G.E. AE37: A HER2-targeted vaccine for the prevention of breast cancer recurrence. Expert Opin. Investig. Drugs 2021, 30, 5–11. [Google Scholar] [CrossRef]
- Holmes, J.P.; Benavides, L.C.; Gates, J.D.; Carmichael, M.G.; Hueman, M.T.; Mittendorf, E.A.; Murray, J.L.; Amin, A.; Craig, D.; von Hofe, E.; et al. Results of the first phase I clinical trial of the novel Ii-key hybrid preventive HER-2/neu peptide (AE37) vaccine. J. Clin. Oncol. 2008, 26, 3426–3433. [Google Scholar] [CrossRef]
- Brown, T.A.; Mittendorf, E.A.; Hale, D.F.; Myers, J.W.; Peace, K.M.; Jackson, D.O.; Greene, J.M.; Vreeland, T.J.; Clifton, G.T.; Ardavanis, A.; et al. Prospective, randomized, single-blinded, multi-center phase II trial of two HER2 peptide vaccines, GP2 and AE37, in breast cancer patients to prevent recurrence. Breast Cancer Res. Treat. 2020, 181, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Roszik, J.; Wang, W.-L.; Livingston, J.A.; Roland, C.L.; Ravi, V.; Yee, C.; Hwu, P.; Futreal, A.; Lazar, A.J.; Patel, S.R.; et al. Overexpressed PRAME is a potential immunotherapy target in sarcoma subtypes. Clin. Sarcoma Res. 2017, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Zou, R.; Wang, J.; Wang, Z.-W.; Zhu, X. The role of the cancer testis antigen PRAME in tumorigenesis and immunotherapy in human cancer. Cell Prolif. 2020, 53, e12770. [Google Scholar] [CrossRef]
- Pujol, J.-L.; De Pas, T.; Rittmeyer, A.; Vallières, E.; Kubisa, B.; Levchenko, E.; Wiesemann, S.; Masters, G.A.; Shen, R.; Tjulandin, S.A.; et al. Safety and immunogenicity of the PRAME cancer immunotherapeutic in patients with resected non–small cell lung cancer: A phase I dose escalation study. J. Thorac. Oncol. 2016, 11, 2208–2217. [Google Scholar] [CrossRef] [Green Version]
- Gutzmer, R.; Rivoltini, L.; Levchenko, E.; Testori, A.; Utikal, J.; Ascierto, P.A.; Demidov, L.; Grob, J.-J.; Ridolfi, R.; Schadendorf, D.; et al. Safety and immunogenicity of the PRAME cancer immunotherapeutic in metastatic melanoma: Results of a phase I dose escalation study. ESMO Open 2016, 1, e000068. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 based immunotherapy of cancer: Current perspectives. Front. Immunol. 2018, 9, 947. [Google Scholar] [CrossRef] [PubMed]
- Kakimi, K.; Isobe, M.; Uenaka, A.; Wada, H.; Sato, E.; Doki, Y.; Nakajima, J.; Seto, Y.; Yamatsuji, T.; Naomoto, Y.; et al. A phase I study of vaccination with NY-ESO-1f peptide mixed with picibanil OK-432 and montanide ISA-51 in patients with cancers expressing the NY-ESO-1 antigen. Int. J. Cancer 2011, 129, 2836–2846. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K.; Matsuzaki, J.; James, S.R.; Mhawech-Fauceglia, P.; Tsuji, T.; Miller, A.; Zhang, W.; Akers, S.N.; Griffiths, E.A.; Miliotto, A.; et al. Epigenetic potentiation of NY-ESO-1 vaccine therapy in human ovarian cancer. Cancer Immunol. Res. 2014, 2, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Raza, A.; Merhi, M.; Inchakalody, V.P.; Krishnankutty, R.; Relecom, A.; Uddin, S.; Dermime, S. Unleashing the immune response to NY-ESO-1 cancer testis antigen as a potential target for cancer immunotherapy. J. Transl. Med. 2020, 18, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerkar, S.P.; Wang, Z.-F.; Lasota, J.; Park, T.; Patel, K.; Groh, E.; Rosenberg, S.A.; Miettinen, M.M. MAGE-A is more highly expressed than NY-ESO-1 in a systematic immunohistochemical analysis of 3668 cases. J. Immunother. 2016, 39, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Vansteenkiste, J.F.; Cho, B.C.; Vanakesa, T.; De Pas, T.; Zielinski, M.; Kim, M.S.; Jassem, J.; Yoshimura, M.; Dahabreh, J.; Nakayama, H.; et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016, 17, 822–835. [Google Scholar] [CrossRef]
- Dreno, B.; Thompson, J.; Smithers, B.M.; Santinami, M.; Jouary, T.; Gutzmer, R.; Levchenko, E.; Rutkowski, P.; Grob, J.-J.; Korovin, S.; et al. MAGE-A3 immunotherapeutic as adjuvant therapy for patients with resected, MAGE-A3-positive, stage III melanoma (DERMA): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 916–929. [Google Scholar] [CrossRef] [Green Version]
- Krishnadas, D.K.; Shusterman, S.; Bai, F.; Diller, L.; Sullivan, J.E.; Cheerva, A.C.; George, R.E.; Lucas, K.G. A phase I trial combining decitabine/dendritic cell vaccine targeting MAGE-A1, MAGE-A3 and NY-ESO-1 for children with relapsed or therapy-refractory neuroblastoma and sarcoma. Cancer Immunol. Immunother. 2015, 64, 1251–1260. [Google Scholar] [CrossRef]
- Chen, X.; Wang, L.; Li, P.; Song, M.; Qin, G.; Gao, Q.; Zhang, Z.; Yue, D.; Wang, D.; Nan, S.; et al. Dual TGF-β and PD-1 blockade synergistically enhances MAGE-A3-specific CD8+T cell response in esophageal squamous cell carcinoma. Int. J. Cancer 2018, 143, 2561–2574. [Google Scholar] [CrossRef] [Green Version]
- Marquez-Manriquez, J.P.; Stanton, S.E.; Disis, M.L. The antigenic repertoire of premalignant and high-risk lesions. Cancer Prev. Res. 2015, 8, 266–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Chen, Y.; Ding, Z.-Y.; Liu, J.-Y. Safety and efficacy of therapeutic cancer vaccines alone or in combination with immune checkpoint inhibitors in cancer treatment. Front. Pharmacol. 2019, 10, 1184. [Google Scholar] [CrossRef]
- Finn, O.J. The dawn of vaccines for cancer prevention. Nat. Rev. Immunol. 2018, 18, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Patel, M.R.; Cho, D.C.; Clarke, J.M.; Gutierrez, M.; Zaks, T.Z.; Frederick, J.; Hopson, K.; Mody, K.; Binanti-Berube, A.; et al. A phase I multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in patients with resected solid tumors and in combination with pembrolizumab in patients with unresectable solid tumors. J. Clin. Oncol. 2019, 37, 2523. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu-Lieskovan, S.; Chmielowski, B.; Govindan, R.; Naing, A.; Bhardwaj, N.; Margolin, K.; Awad, M.M.; Hellmann, M.D.; Lin, J.J.; et al. A phase Ib trial of personalized neoantigen therapy plus anti-PD-1 in patients with advanced melanoma, non-small cell lung cancer, or bladder cancer. Cell 2020, 183, 347–362.e24. [Google Scholar] [CrossRef]
- Roudko, V.; Bozkus, C.C.; Orfanelli, T.; McClain, C.B.; Carr, C.; O’Donnell, T.; Chakraborty, L.; Samstein, R.; Huang, K.-L.; Blank, S.V.; et al. Shared immunogenic poly-epitope frameshift mutations in microsatellite unstable tumors. Cell 2020, 183, 1634–1649.e17. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Sendabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch repair pathway, genome stability and cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Bagshaw, A.T. Functional mechanisms of microsatellite DNA in eukaryotic genomes. Genome Biol. Evol. 2017, 9, 2428–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, G.; D’Alise, A.M.; Cotugno, G.; Langone, F.; Garzia, I.; De Lucia, M.; Fichera, I.; Vitale, R.; Bignone, V.; Tucci, F.G.; et al. A genetic vaccine encoding shared cancer neoantigens to treat tumors with microsatellite instability. Cancer Res. 2020, 80, 3972–3982. [Google Scholar] [CrossRef]
- Kloor, M.; Doeberitz, M.V.K. The immune biology of microsatellite-unstable cancer. Trends Cancer 2016, 2, 121–133. [Google Scholar] [CrossRef] [Green Version]
- Buckowitz, A.; Knaebel, H.-P.; Benner, A.; Bläker, H.; Gebert, J.; Kienle, P.; Doeberitz, M.V.K.; Kloor, M. Microsatellite instability in colorectal cancer is associated with local lymphocyte infiltration and low frequency of distant metastases. Br. J. Cancer 2005, 92, 1746–1753. [Google Scholar] [CrossRef] [Green Version]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Mardis, E.R. Neoantigens and genome instability: Impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.; Balaguer, F.; Lindor, N.; De La Chapelle, A.; Hampel, H.; Aaltonen, L.A.; Hopper, J.L.; Le Marchand, L.; Gallinger, S.; Newcomb, P.A.; et al. Identification of lynch syndrome among patients with colorectal cancer. JAMA 2012, 308, 1555–1565. [Google Scholar] [CrossRef]
- Alvarez, M.D.; Quintana, I.; Terradas, M.; Mur, P.; Balaguer, F.; Valle, L. The inherited and familial component of early-onset colorectal cancer. Cells 2021, 10, 710. [Google Scholar] [CrossRef]
- Sánchez, A.; Roos, V.H.; Navarro, M.; Pineda, M.; Caballol, B.; Moreno, L.; Carballal, S.; Rodríguez-Alonso, L.; Cajal, T.R.Y.; Llort, G.; et al. Quality of colonoscopy is associated with adenoma detection and postcolonoscopy colorectal cancer prevention in Lynch syndrome. Clin. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppala, T.T.; Ten Broeke, S.W.; Plazzer, J.P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the prospective Lynch syndrome database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Van Leerdam, M.E.; Roos, V.H.; Van Hooft, J.E.; Balaguer, F.; Dekker, E.; Kaminski, M.; Latchford, A.; Neumann, H.; Ricciardiello, L.; Rupińska, M.; et al. Endoscopic management of Lynch syndrome and of familial risk of colorectal cancer: European society of gastrointestinal endoscopy (ESGE) guideline. Endoscopy 2019, 51, 1082–1093. [Google Scholar] [CrossRef] [Green Version]
- Schmeler, K.M.; Lynch, H.T.; Chen, L.-M.; Munsell, M.F.; Soliman, P.T.; Clark, M.B.; Daniels, M.; White, K.G.; Boyd-Rogers, S.G.; Conrad, P.G.; et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N. Engl. J. Med. 2006, 354, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Gebert, J.; Gelincik, O.; Oezcan-Wahlbrink, M.; Marshall, J.D.; Hernandez-Sanchez, A.; Urban, K.; Long, M.; Cortes, E.; Tosti, E.; Katzenmaier, E.-M.; et al. Recurrent frameshift neoantigen vaccine elicits protective immunity with reduced tumor burden and improved overall survival in a Lynch syndrome mouse model. Gastroenterology 2021, 161, 1288–1302.e13. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Herman, M.; Ciancanelli, M.; de Diego, R.P.; Sancho-Shimizu, V.; Abel, L.; Casanova, J.-L. TLR3 immunity to infection in mice and humans. Curr. Opin. Immunol. 2013, 25, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-κB by toll-like receptor 3. Nat. Cell Biol. 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Mints, M.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2018, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Erdmann, M.; Haendle, I.; Voland, S.; Berger, T.; Schultz, E.; Strasser, E.; Dankerl, P.; Janka, R.; Schliep, S.; et al. Twelve-year survival and immune correlates in dendritic cell–vaccinated melanoma patients. JCI Insight 2017, 2, e91438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [Green Version]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 1–21. [Google Scholar] [CrossRef]
- Litchfield, K.; Reading, J.L.; Lim, E.L.; Xu, H.; Liu, P.; Al-Bakir, M.; Wong, Y.N.S.; Rowan, A.; Funt, S.A.; Merghoub, T.; et al. Escape from nonsense-mediated decay associates with anti-tumor immunogenicity. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Leddon, S.A.; Sant, A.J. Generation of MHC class II–peptide ligands for CD4 T-cell allorecognition of MHC class II molecules. Curr. Opin. Organ Transplant. 2010, 15, 505–511. [Google Scholar] [CrossRef]
- Roudko, V.; Greenbaum, B.; Bhardwaj, N. Computational prediction and validation of tumor-associated neoantigens. Front. Immunol. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Ali, M.; Foldvari, Z.; Giannakopoulou, E.; Böschen, M.-L.; Strønen, E.; Yang, W.; Toebes, M.; Schubert, B.; Kohlbacher, O.; Schumacher, T.; et al. Induction of neoantigen-reactive T cells from healthy donors. Nat. Protoc. 2019, 14, 1926–1943. [Google Scholar] [CrossRef]
- Gee, M.H.; Han, A.; Lofgren, S.M.; Beausang, J.F.; Mendoza, J.; Birnbaum, M.; Bethune, M.T.; Fischer, S.; Yang, X.; Gomez-Eerland, R.; et al. Antigen identification for orphan T cell receptors expressed on tumor-infiltrating lymphocytes. Cell 2018, 172, 549–563.e16. [Google Scholar] [CrossRef] [Green Version]
- Lam, H.; McNeil, L.K.; Starobinets, H.; DeVault, V.L.; Cohen, R.B.; Twardowski, P.; Johnson, M.L.; Gillison, M.L.; Stein, M.N.; Vaishampayan, U.N.; et al. An empirical antigen selection method identifies neoantigens that either elicit broad antitumor T-cell responses or drive tumor growth. Cancer Discov. 2021, 11, 696–713. [Google Scholar] [CrossRef]
- Guo, Y.; Lei, K.; Tang, L. Neoantigen vaccine delivery for personalized anticancer immunotherapy. Front. Immunol. 2018, 9, 1499. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Baldin, A.; Isayev, O.; Werner, J.; Zamyatnin, A.; Bazhin, A. Cancer vaccines: Antigen selection strategy. Vaccines 2021, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Tay, B.; Wright, Q.; Ladwa, R.; Perry, C.; Leggatt, G.; Simpson, F.; Wells, J.; Panizza, B.; Frazer, I.; Cruz, J. Evolution of cancer vaccines—challenges, achievements, and future directions. Vaccines 2021, 9, 535. [Google Scholar] [CrossRef] [PubMed]
- Keikha, R.; Daliri, K.; Jebali, A. The use of nanobiotechnology in immunology and vaccination. Vaccines 2021, 9, 74. [Google Scholar] [CrossRef]
- Thi, T.; Suys, E.; Lee, J.; Nguyen, D.; Park, K.; Truong, N. Lipid-based nanoparticles in the clinic and clinical trials: From cancer nanomedicine to Covid-19 vaccines. Vaccines 2021, 9, 359. [Google Scholar] [CrossRef]
- Fifis, T.; Gamvrellis, A.; Crimeen-Irwin, B.; Pietersz, G.A.; Li, J.; Mottram, P.L.; McKenzie, I.F.C.; Plebanski, M. Size-dependent immunogenicity: Therapeutic and protective properties of nano-vaccines against tumors. J. Immunol. 2004, 173, 3148–3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.-R.; Hildebrand, W.H.; Mardis, E.R.; et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaddanapudi, K.; Meng, S.; Whitt, A.G.; Al Rayyan, N.; Richie, J.; Tu, A.; Eaton, J.W.; Li, C. Exosomes from GM-CSF expressing embryonic stem cells are an effective prophylactic vaccine for cancer prevention. OncoImmunology 2018, 8, 1561119. [Google Scholar] [CrossRef] [PubMed]
- Overwijk, W.W. Cancer vaccines in the era of checkpoint blockade: The magic is in the adjuvant. Curr. Opin. Immunol. 2017, 47, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Jorritsma, S.; Gowans, E.; Grubor-Bauk, B.; Wijesundara, D. Delivery methods to increase cellular uptake and immunogenicity of DNA vaccines. Vaccine 2016, 34, 5488–5494. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Sasso, E.; D’Alise, A.M.; Zambrano, N.; Scarselli, E.; Folgori, A.; Nicosia, A. New viral vectors for infectious diseases and cancer. Semin. Immunol. 2020, 50, 101430. [Google Scholar] [CrossRef]
- Yamasaki, S.; Miura, Y.; Davydova, J.; Vickers, S.M.; Yamamoto, M. Intravenous genetic mesothelin vaccine based on human adenovirus 40 inhibits growth and metastasis of pancreatic cancer. Int. J. Cancer 2013, 133, 88–97. [Google Scholar] [CrossRef]
- Harari, A.; Graciotti, M.; Bassani-Sternberg, M.; Kandalaft, L.E. Antitumour dendritic cell vaccination in a priming and boosting approach. Nat. Rev. Drug Discov. 2020, 19, 635–652. [Google Scholar] [CrossRef]
- Madan, R.A.; Arlen, P.M.; Mohebtash, M.; Hodge, J.W.; Gulley, J.L. Prostvac-VF: A vector-based vaccine targeting PSA in prostate cancer. Expert Opin. Investig. Drugs 2009, 18, 1001–1011. [Google Scholar] [CrossRef] [Green Version]
- Parsons, J.K.; Pinto, P.A.; Pavlovich, C.P.; Uchio, E.; Kim, H.L.; Nguyen, M.N.; Gulley, J.L.; Jamieson, C.; Hsu, P.; Wojtowicz, M.; et al. A randomized, double-blind, phase II Trial of PSA-TRICOM (PROSTVAC) in patients with localized prostate cancer: The immunotherapy to prevent progression on active surveillance study. Eur. Urol. Focus 2018, 4, 636–638. [Google Scholar] [CrossRef]
- Le Gall, C.; Weiden, J.; Eggermont, L.; Figdor, C.G. Dendritic cells in cancer immunotherapy. Nat. Mater. 2018, 17, 474–475. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Target Antigen | Examples | Tumor Specificity | Central Tolerance | Prevalence among Patients | Optimal Preventive Setting | Suitable for CPV |
|---|---|---|---|---|---|---|
| Tumor associated antigens (TAAs) | MUC1 (epithelial tumors: colon, pancreatic, cervix), HER2 (breast, bladder, gastric cancers), EGFR (NSCLC, glioma), CEA (colorectal, pancreatic, gastric, lung and breast cancers), cyclin B1 (gynecological and colorectal cancers) | Medium | Yes | Variable | Individuals with genetic predisposition, pre-malignant lesions, prevention of recurrences | Yes |
| Cancer testis antigens (CTAs) | NY-ESO (melanoma and carcinomas of lung, esophageal, liver, gastric, prostrate, ovarian, and bladder), MAGE-A 1–4 (NSCLC, bladder, esophageal and head and neck cancers, sarcomas, triple negative breast cancers, myeloma, Hodgkin’s disease), PRAME | Medium | Partial | High | Individuals with genetic predisposition or risk practices (such as smoking), pre-malignant lesions, prevention of recurrences | Yes |
| Shared or common neoantigens | Mutated oncogenes, passenger/driver mutations in Lynch syndrome (AIM2, ACVR2A, TGFBRII, CASP5, …) | High | No | High | Individuals with genetic predisposition (such as Lynch syndrome), pre-malignant lesions | Yes |
| Personalized neoantigens | Depending on each tumor and patient | High | No | Very low | None | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayó, C.; Jung, G.; Español-Rego, M.; Balaguer, F.; Benitez-Ribas, D. Vaccines for Non-Viral Cancer Prevention. Int. J. Mol. Sci. 2021, 22, 10900. https://doi.org/10.3390/ijms222010900
Bayó C, Jung G, Español-Rego M, Balaguer F, Benitez-Ribas D. Vaccines for Non-Viral Cancer Prevention. International Journal of Molecular Sciences. 2021; 22(20):10900. https://doi.org/10.3390/ijms222010900
Chicago/Turabian StyleBayó, Cristina, Gerhard Jung, Marta Español-Rego, Francesc Balaguer, and Daniel Benitez-Ribas. 2021. "Vaccines for Non-Viral Cancer Prevention" International Journal of Molecular Sciences 22, no. 20: 10900. https://doi.org/10.3390/ijms222010900
APA StyleBayó, C., Jung, G., Español-Rego, M., Balaguer, F., & Benitez-Ribas, D. (2021). Vaccines for Non-Viral Cancer Prevention. International Journal of Molecular Sciences, 22(20), 10900. https://doi.org/10.3390/ijms222010900