
Deletion of the Gamma Subunit of ENaC in Endothelial Cells Does Not Protect against Renal Ischemia Reperfusion Injury

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
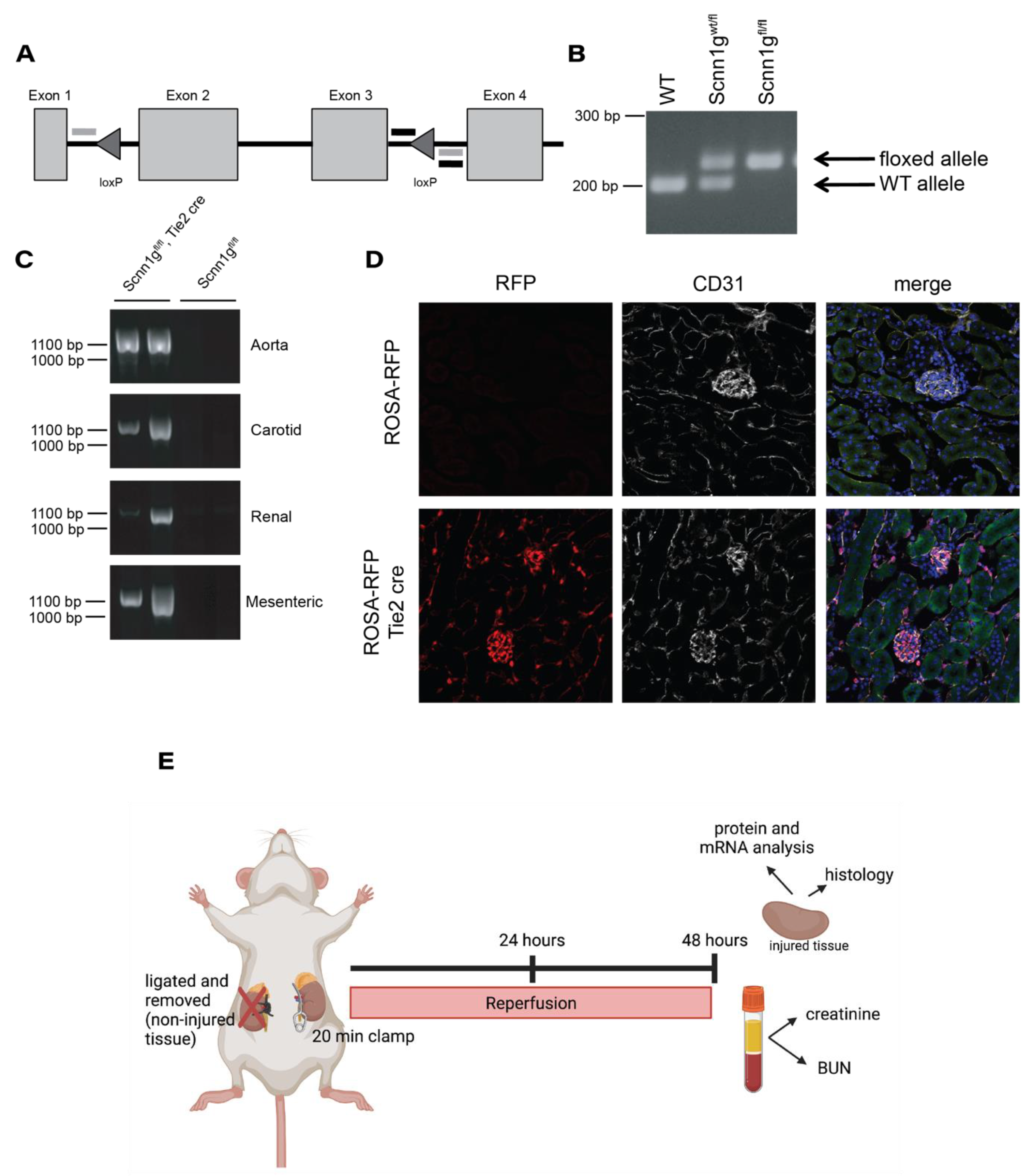
2.1. Generating an Endothelial ENaC γ Subunit Knockout
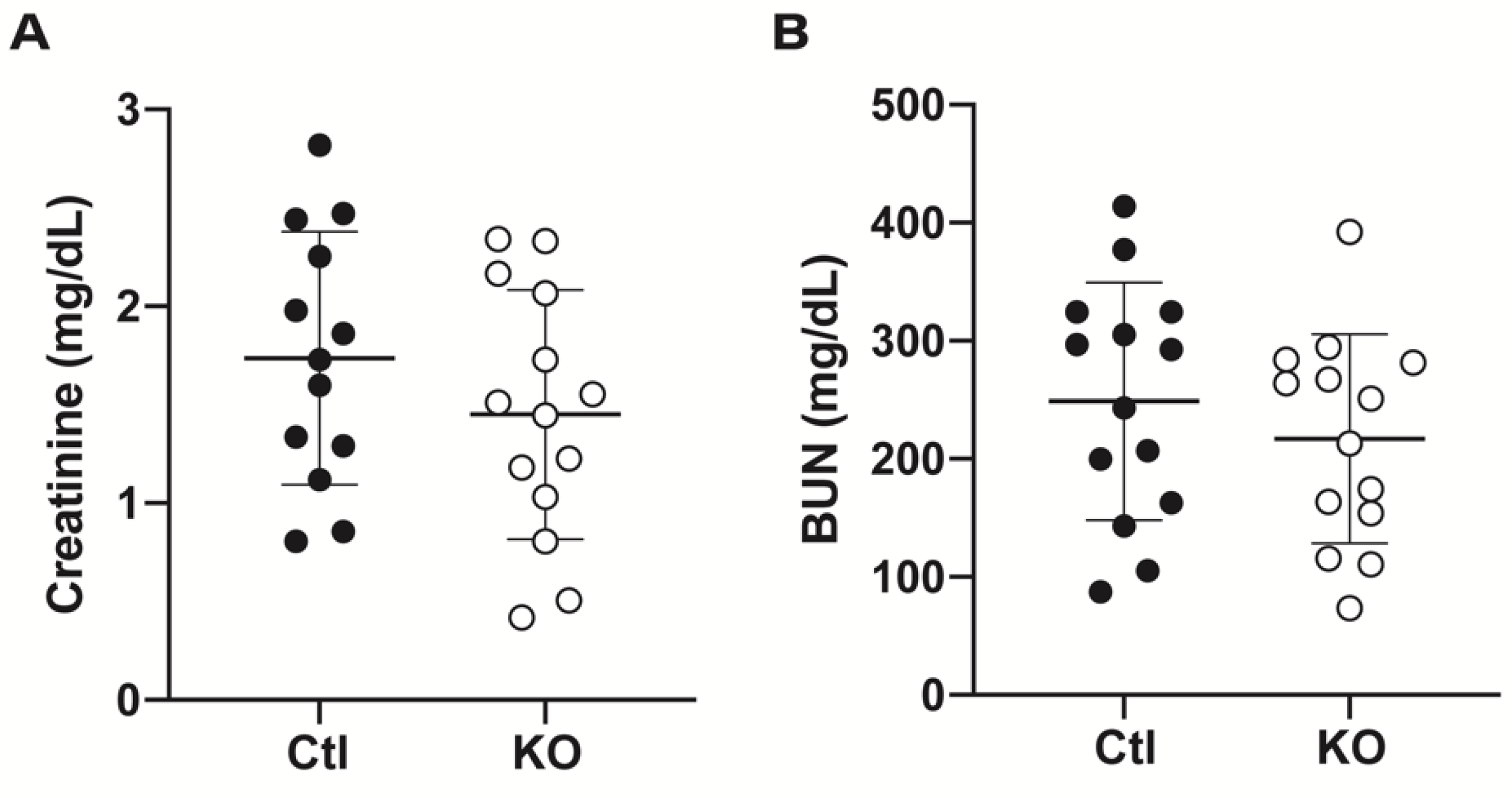
2.2. Serum and Tissue Indicators of Renal Damage Do Not Differ between Groups after IRI
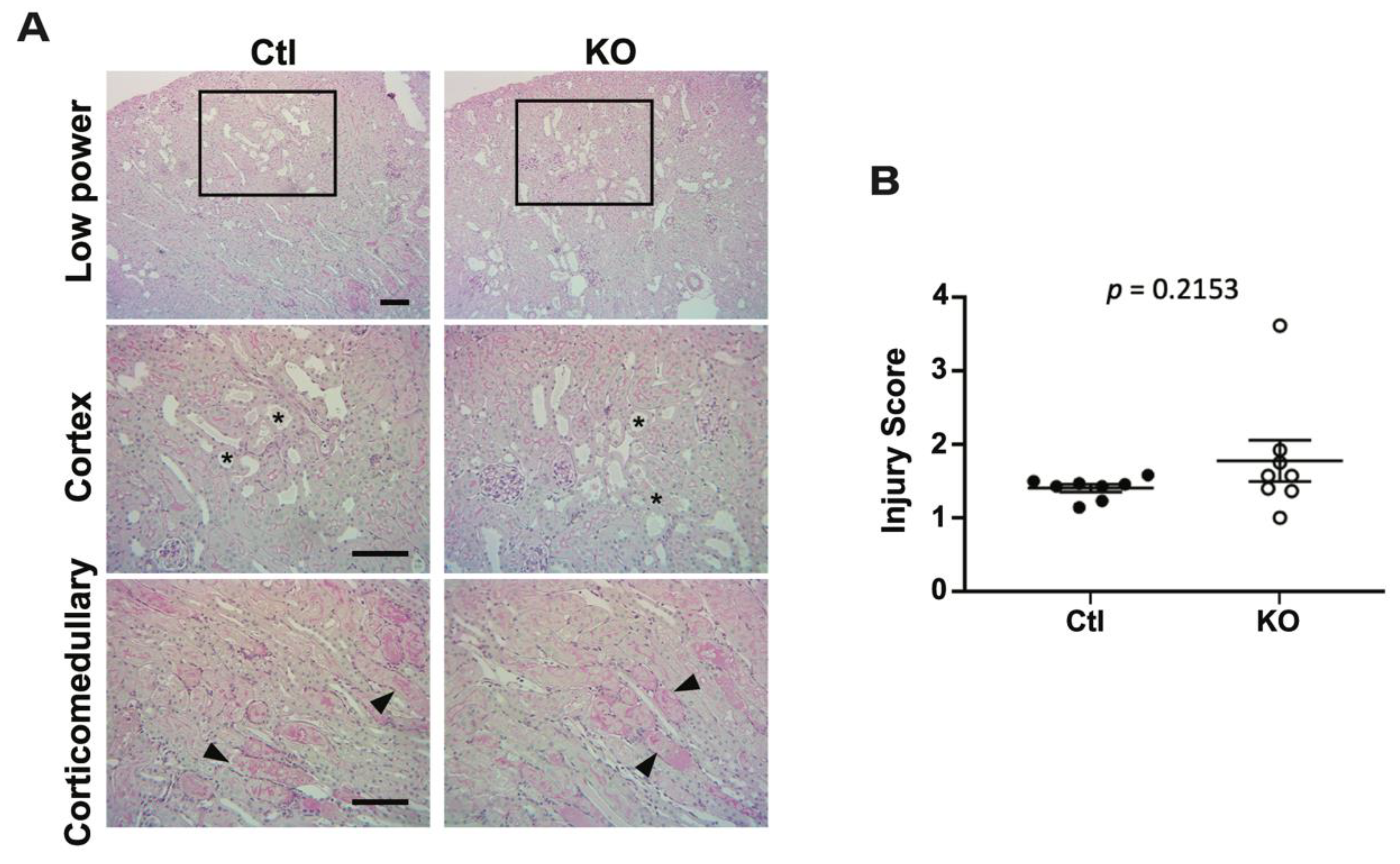
2.3. Histologic Analysis Reveals No Differences after IRI between Control and Knockout Animals
2.4. eNOS Phosphorylation Is Increased in Injured Kidneys of Knockout Animals
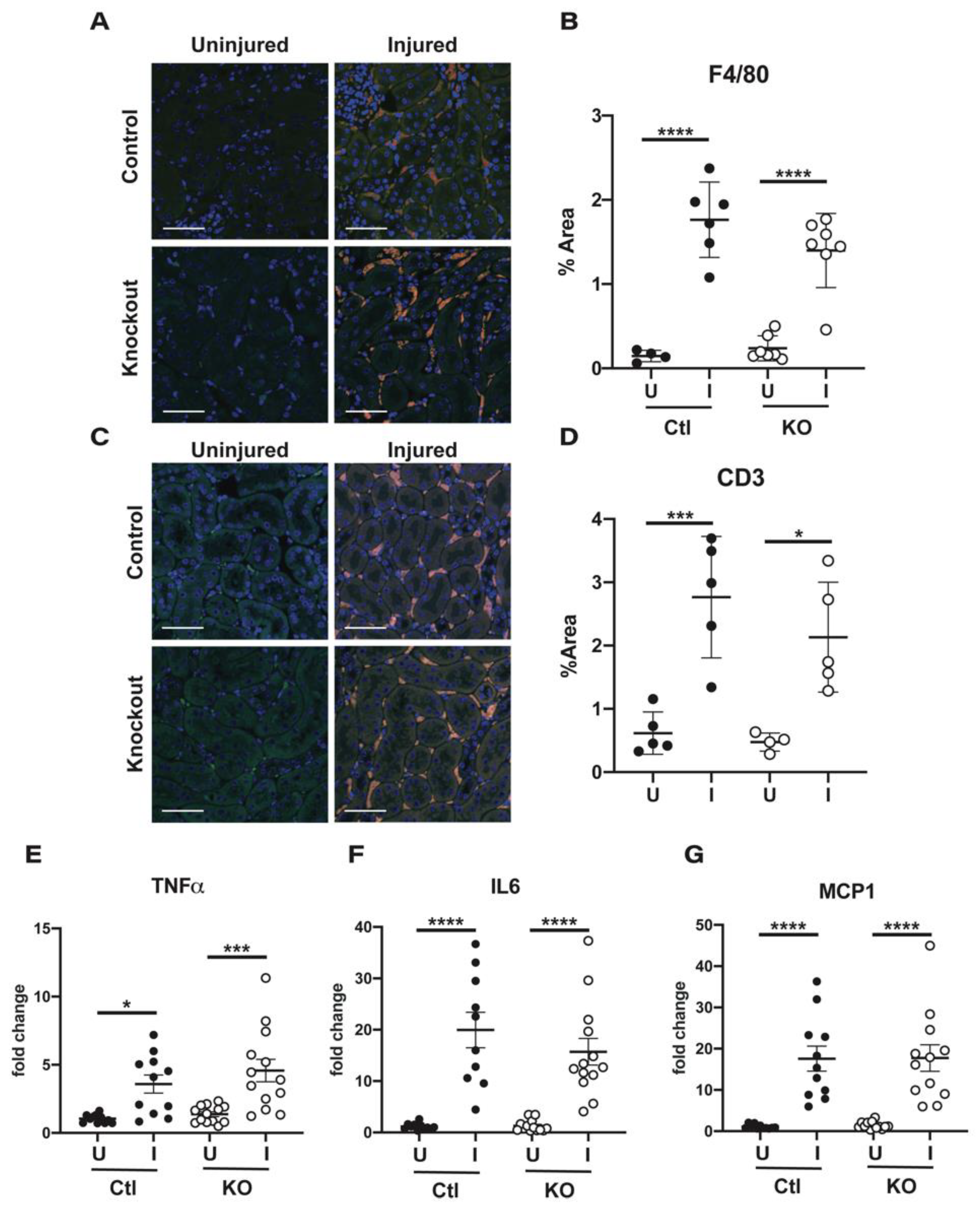
2.5. Immune Cell Infiltration and Cytokine Production Are Similar in Kidneys from Control and Knockout Animals
3. Discussion
4. Materials and Methods
4.1. Animals
Generation of γ ENaC Floxed Mice
4.2. IRI Surgery
4.3. Measure of Renal Dysfunction and Injury
4.4. Quantitative, Real-Time, Reverse-Transcriptase Polymerase Chain Reaction
4.5. SDS Page Electrophoresis
4.6. Immunofluorescent Analysis
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Han, S.J.; Lee, H.T. Mechanisms and therapeutic targets of ischemic acute kidney injury. Kidney Res. Clin. Pract. 2019, 38, 427–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nourbakhsh, N.; Singh, P. Role of renal oxygenation and mitochondrial function in the pathophysiology of acute kidney injury. Nephron Clin. Pract. 2014, 127, 149–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Yoder, M.C. Renal endothelial dysfunction in acute kidney ischemia reperfusion injury. Cardiovasc Hematol. Disord Drug Targets 2014, 14, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.; Miyagawa, I. Real-time monitoring of nitric oxide in ischemia-reperfusion rat kidney. Urol. Res. 2000, 28, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Katsumi, H.; Takashima, R.; Suzuki, H.; Hirai, N.; Matsuura, S.; Kimura, H.; Morishita, M.; Yamamoto, A. S-nitrosylated l-serine-modified dendrimer as a kidney-targeting nitric oxide donor for prevention of renal ischaemia/reperfusion injury. Free Radic. Res. 2020, 54, 841–847. [Google Scholar] [CrossRef]
- Milsom, A.B.; Patel, N.S.; Mazzon, E.; Tripatara, P.; Storey, A.; Mota-Filipe, H.; Sepodes, B.; Webb, A.J.; Cuzzocrea, S.; Hobbs, A.J.; et al. Role for endothelial nitric oxide synthase in nitrite-induced protection against renal ischemia-reperfusion injury in mice. Nitric Oxide 2010, 22, 141–148. [Google Scholar] [CrossRef]
- Yang, T.; Zhang, X.M.; Tarnawski, L.; Peleli, M.; Zhuge, Z.; Terrando, N.; Harris, R.A.; Olofsson, P.S.; Larsson, E.; Persson, A.E.G.; et al. Dietary nitrate attenuates renal ischemia-reperfusion injuries by modulation of immune responses and reduction of oxidative stress. Redox Biol. 2017, 13, 320–330. [Google Scholar] [CrossRef]
- Mutchler, S.M.; Straub, A.C. Compartmentalized nitric oxide signaling in the resistance vasculature. Nitric Oxide 2015, 49, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Fleming, I. Molecular mechanisms underlying the activation of eNOS. Pflugers Arch. 2010, 459, 793–806. [Google Scholar] [CrossRef]
- Ghimire, K.; Zaric, J.; Alday-Parejo, B.; Seebach, J.; Bousquenaud, M.; Stalin, J.; Bieler, G.; Schnittler, H.J.; Ruegg, C. MAGI1 Mediates eNOS Activation and NO Production in Endothelial Cells in Response to Fluid Shear Stress. Cells 2019, 8, 388. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lee, T.S.; Kolb, E.M.; Sun, K.; Lu, X.; Sladek, F.M.; Kassab, G.S.; Garland, T., Jr.; Shyy, J.Y. AMP-activated protein kinase is involved in endothelial NO synthase activation in response to shear stress. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1281–1287. [Google Scholar] [CrossRef] [Green Version]
- Fels, J.; Callies, C.; Kusche-Vihrog, K.; Oberleithner, H. Nitric oxide release follows endothelial nanomechanics and not vice versa. Pflugers Arch. 2010, 460, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Mutchler, S.M.; Kirabo, A.; Kleyman, T.R. Epithelial Sodium Channel and Salt-Sensitive Hypertension. Hypertension 2021, 77, 759–767. [Google Scholar] [CrossRef]
- Jeggle, P.; Callies, C.; Tarjus, A.; Fassot, C.; Fels, J.; Oberleithner, H.; Jaisser, F.; Kusche-Vihrog, K. Epithelial sodium channel stiffens the vascular endothelium in vitro and in Liddle mice. Hypertension 2013, 61, 1053–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Habibi, J.; Aroor, A.R.; Hill, M.A.; Yang, Y.; Whaley-Connell, A.; Jaisser, F.; Sowers, J.R. Epithelial Sodium Channel in Aldosterone-Induced Endothelium Stiffness and Aortic Dysfunction. Hypertension 2018, 72, 731–738. [Google Scholar] [CrossRef]
- Kusche-Vihrog, K.; Tarjus, A.; Fels, J.; Jaisser, F. The epithelial Na+ channel: A new player in the vasculature. Curr. Opin. Nephrol. Hypertens 2014, 23, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Tarjus, A.; Gonzalez-Rivas, C.; Amador-Martinez, I.; Bonnard, B.; Lopez-Marure, R.; Jaisser, F.; Barrera-Chimal, J. The Absence of Endothelial Sodium Channel alpha (alphaENaC) Reduces Renal Ischemia/Reperfusion Injury. Int. J. Mol. Sci. 2019, 20, 3132. [Google Scholar] [CrossRef] [Green Version]
- Kashlan, O.B.; Kleyman, T.R. ENaC structure and function in the wake of a resolved structure of a family member. Am. J. Physiol. Renal. Physiol. 2011, 301, F684–F696. [Google Scholar] [CrossRef] [Green Version]
- Jeggle, P.; Smith, E.S.; Stewart, A.P.; Haerteis, S.; Korbmacher, C.; Edwardson, J.M. Atomic force microscopy imaging reveals the formation of ASIC/ENaC cross-clade ion channels. Biochem. Biophys. Res. Commun. 2015, 464, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Czikora, I.; Alli, A.A.; Sridhar, S.; Matthay, M.A.; Pillich, H.; Hudel, M.; Berisha, B.; Gorshkov, B.; Romero, M.J.; Gonzales, J.; et al. Epithelial Sodium Channel-alpha Mediates the Protective Effect of the TNF-Derived TIP Peptide in Pneumolysin-Induced Endothelial Barrier Dysfunction. Front. Immunol. 2017, 8, 842. [Google Scholar] [CrossRef] [Green Version]
- Westhoff, J.H.; Seibert, F.S.; Waldherr, S.; Bauer, F.; Tonshoff, B.; Fichtner, A.; Westhoff, T.H. Urinary calprotectin, kidney injury molecule-1, and neutrophil gelatinase-associated lipocalin for the prediction of adverse outcome in pediatric acute kidney injury. Eur. J. Pediatr 2017, 176, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Fels, J.; Oberleithner, H.; Kusche-Vihrog, K. Menage a trois: Aldosterone, sodium and nitric oxide in vascular endothelium. Biochim. Biophys. Acta 2010, 1802, 1193–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, F.R.; Venegas, F.; Gonzalez, M.; Andres, S.; Vallejos, C.; Riquelme, G.; Sierralta, J.; Michea, L. Endothelial epithelial sodium channel inhibition activates endothelial nitric oxide synthase via phosphoinositide 3-kinase/Akt in small-diameter mesenteric arteries. Hypertension 2009, 53, 1000–1007. [Google Scholar] [CrossRef] [Green Version]
- Sternak, M.; Bar, A.; Adamski, M.G.; Mohaissen, T.; Marczyk, B.; Kieronska, A.; Stojak, M.; Kus, K.; Tarjus, A.; Jaisser, F.; et al. The Deletion of Endothelial Sodium Channel alpha (alphaENaC) Impairs Endothelium-Dependent Vasodilation and Endothelial Barrier Integrity in Endotoxemia in Vivo. Front. Pharmacol. 2018, 9, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrypnyk, N.I.; Harris, R.C.; de Caestecker, M.P. Ischemia-reperfusion model of acute kidney injury and post injury fibrosis in mice. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat, Z.; Kadkhodaee, M.; Seifi, B.; Salehi, E. Inducible and endothelial nitric oxide synthase distribution and expression with hind limb per-conditioning of the rat kidney. Arch. Med. Sci. 2019, 15, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.K.; Patel, N.S.; Kvale, E.O.; Cuzzocrea, S.; Brown, P.A.; Stewart, K.N.; Mota-Filipe, H.; Thiemermann, C. Inhibition of inducible nitric oxide synthase reduces renal ischemia/reperfusion injury. Kidney Int. 2002, 61, 862–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Sun, L.; Zhang, W.; Tang, Y.; Li, X.; Jing, R.; Liu, T. Limb ischemic preconditioning ameliorates renal microcirculation through activation of PI3K/Akt/eNOS signaling pathway after acute kidney injury. Eur. J. Med. Res. 2020, 25, 10. [Google Scholar] [CrossRef] [Green Version]
- Soliman, E.; Shewaikh, S.M.; Fahmy, A.; Elshazly, S. Entacapone scavenges peroxynitrite and protects against kidney and liver injuries induced by renal ischemia/reperfusion in rats. Int. Urol. Nephrol. 2021. [Google Scholar] [CrossRef]
- Vo, P.A.; Lad, B.; Tomlinson, J.A.; Francis, S.; Ahluwalia, A. autoregulatory role of endothelium-derived nitric oxide (NO) on Lipopolysaccharide-induced vascular inducible NO synthase expression and function. J. Biol. Chem. 2005, 280, 7236–7243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trac, P.T.; Thai, T.L.; Linck, V.; Zou, L.; Greenlee, M.; Yue, Q.; Al-Khalili, O.; Alli, A.A.; Eaton, A.F.; Eaton, D.C. Alveolar nonselective channels are ASIC1a/alpha-ENaC channels and contribute to AFC. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L797–L811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, N.; Bartoszewski, R.; Qadri, Y.J.; Bebok, Z.; Bubien, J.K.; Fuller, C.M.; Benos, D.J. Knockdown of ASIC1 and epithelial sodium channel subunits inhibits glioblastoma whole cell current and cell migration. J. Biol. Chem. 2009, 284, 24526–24541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, N.; Lee, W.; Clark, E.; Bartoszewski, R.; McNicholas, C.M.; Latham, C.B.; Bebok, Z.; Parpura, V.; Fuller, C.M.; Palmer, C.A.; et al. Interaction of ASIC1 and ENaC subunits in human glioma cells and rat astrocytes. Am. J. Physiol. Cell Physiol. 2011, 300, C1246–C1259. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lemus, L.A.; Aroor, A.R.; Ramirez-Perez, F.I.; Jia, G.; Habibi, J.; DeMarco, V.G.; Barron, B.; Whaley-Connell, A.; Nistala, R.; Sowers, J.R. Amiloride Improves Endothelial Function and Reduces Vascular Stiffness in Female Mice Fed a Western Diet. Front. Physiol. 2017, 8, 456. [Google Scholar] [CrossRef] [Green Version]
- Sowers, J.R.; Habibi, J.; Aroor, A.R.; Yang, Y.; Lastra, G.; Hill, M.A.; Whaley-Connell, A.; Jaisser, F.; Jia, G. Epithelial sodium channels in endothelial cells mediate diet-induced endothelium stiffness and impaired vascular relaxation in obese female mice. Metabolism 2019, 99, 57–66. [Google Scholar] [CrossRef]
- Bruns, J.B.; Hu, B.; Ahn, Y.J.; Sheng, S.; Hughey, R.P.; Kleyman, T.R. Multiple epithelial Na+ channel domains participate in subunit assembly. Am. J. Physiol. Renal. Physiol. 2003, 285, F600–F609. [Google Scholar] [CrossRef] [Green Version]
- Rasband, W.S. ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, USA. 2011. Available online: https://www.semanticscholar.org/paper/ImageJ%2C-U.S.-National-Institutes-of-Health%2C-USA-Rasband/034dbc2e4c735500c519183180f8cf6033fcb28d (accessed on 15 September 2021).
- GraphPad Prism, version 9.2. software for statistical analysis. GraphPad Software, LLC: San Diego, CA, USA, 2021.


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mutchler, S.M.; Hasan, M.; Kohan, D.E.; Kleyman, T.R.; Tan, R.J. Deletion of the Gamma Subunit of ENaC in Endothelial Cells Does Not Protect against Renal Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2021, 22, 10914. https://doi.org/10.3390/ijms222010914
Mutchler SM, Hasan M, Kohan DE, Kleyman TR, Tan RJ. Deletion of the Gamma Subunit of ENaC in Endothelial Cells Does Not Protect against Renal Ischemia Reperfusion Injury. International Journal of Molecular Sciences. 2021; 22(20):10914. https://doi.org/10.3390/ijms222010914
Chicago/Turabian StyleMutchler, Stephanie M., Mahpara Hasan, Donald E. Kohan, Thomas R. Kleyman, and Roderick J. Tan. 2021. "Deletion of the Gamma Subunit of ENaC in Endothelial Cells Does Not Protect against Renal Ischemia Reperfusion Injury" International Journal of Molecular Sciences 22, no. 20: 10914. https://doi.org/10.3390/ijms222010914
APA StyleMutchler, S. M., Hasan, M., Kohan, D. E., Kleyman, T. R., & Tan, R. J. (2021). Deletion of the Gamma Subunit of ENaC in Endothelial Cells Does Not Protect against Renal Ischemia Reperfusion Injury. International Journal of Molecular Sciences, 22(20), 10914. https://doi.org/10.3390/ijms222010914