
Molecular Mechanism of Food-Derived Polyphenols on PD-L1 Dimerization: A Molecular Dynamics Simulation Study


Abstract
:1. Introduction
2. Results and Discussion
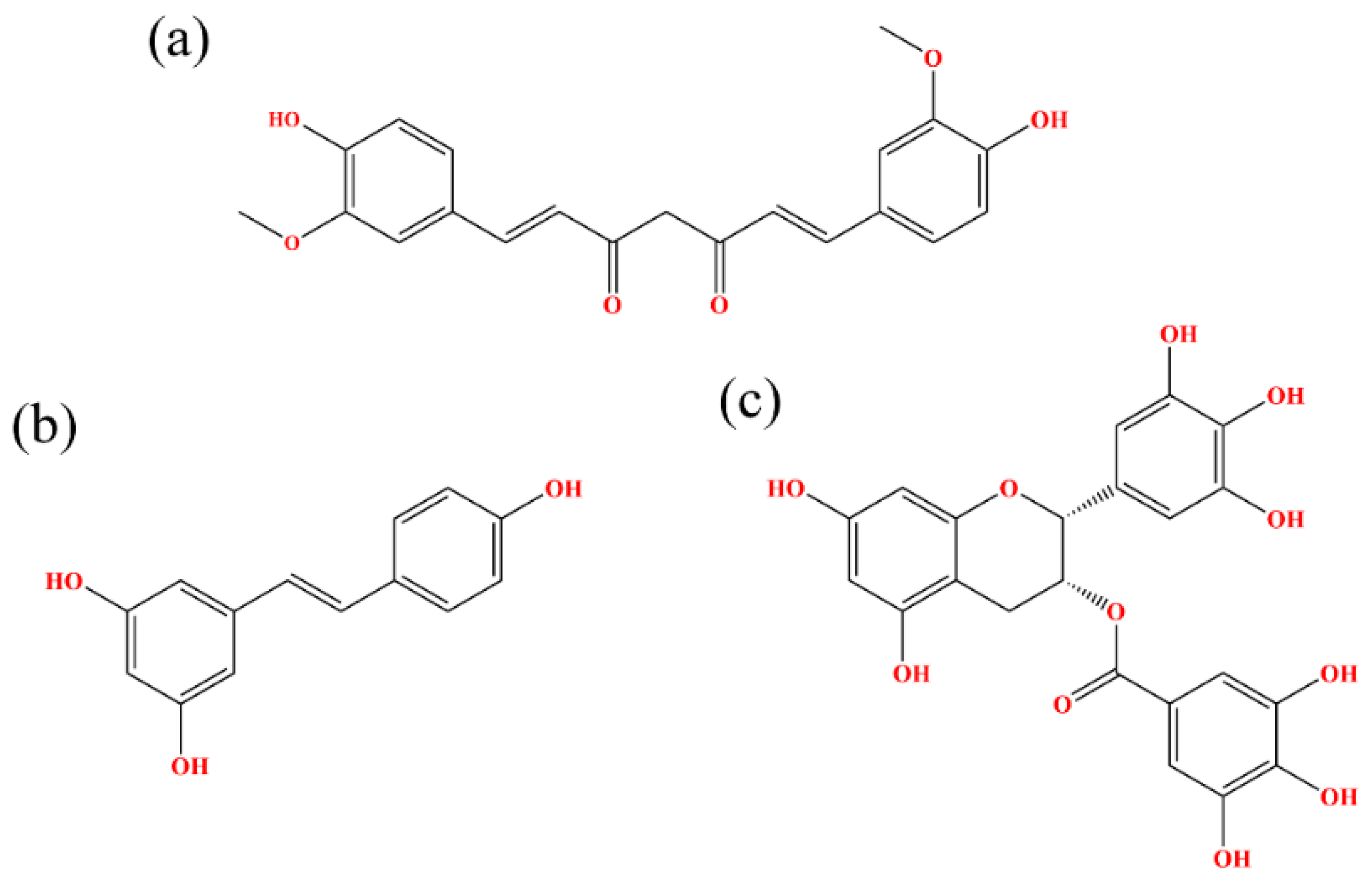
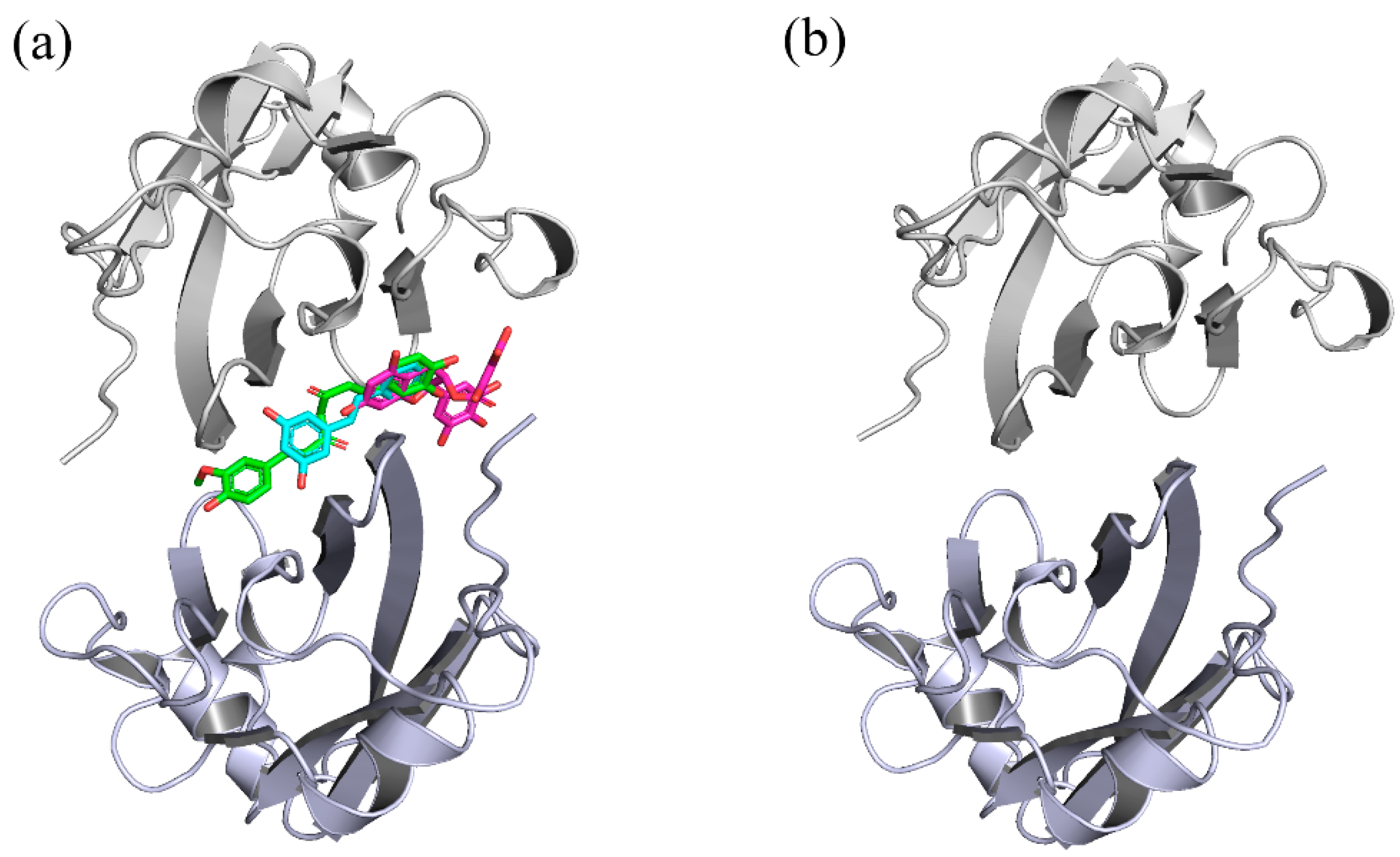
2.1. Docking
2.2. RMSD
2.3. RMSF
2.4. Binding Free Energy
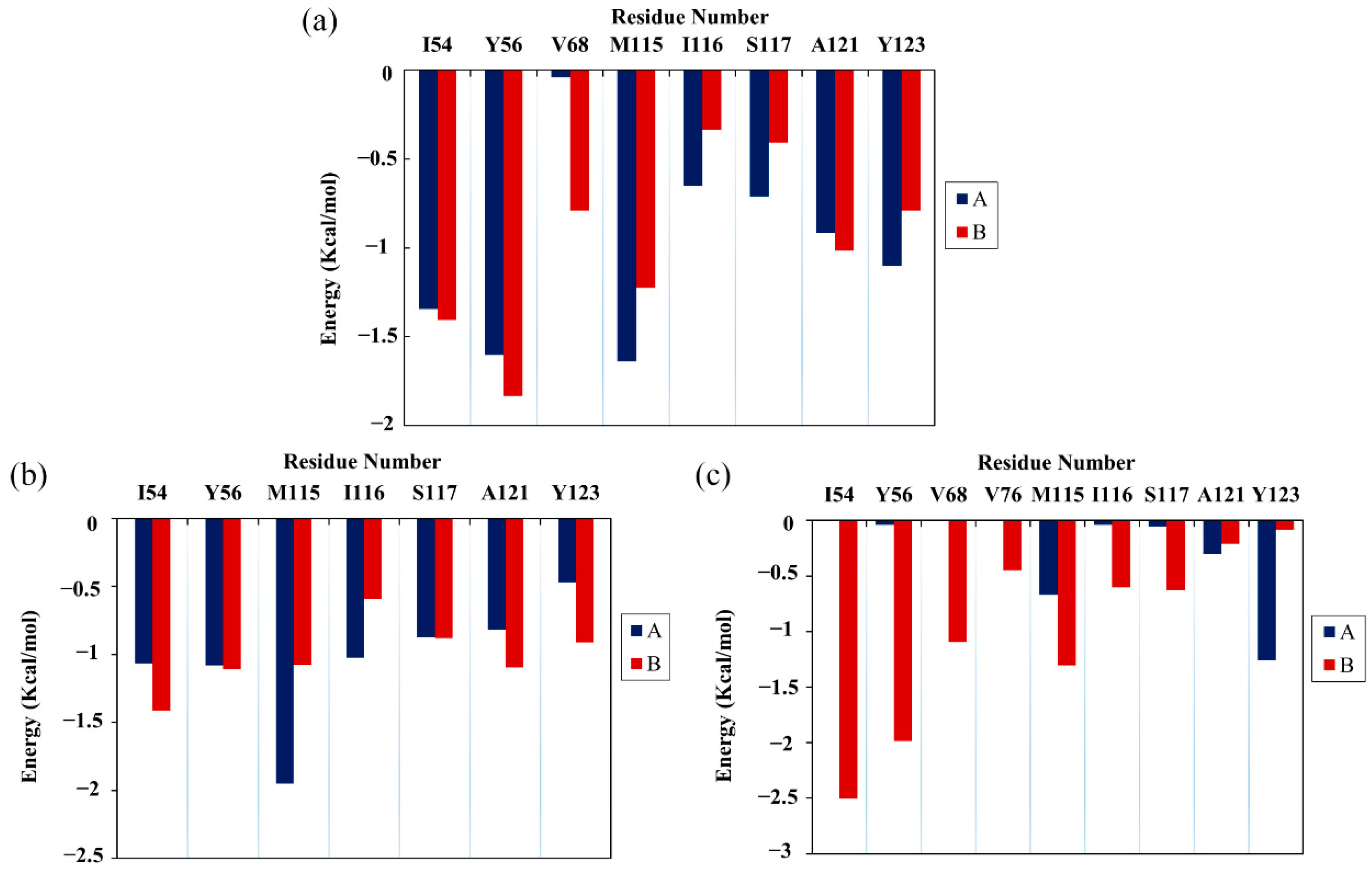
2.5. Per-Residue Energy Decomposition
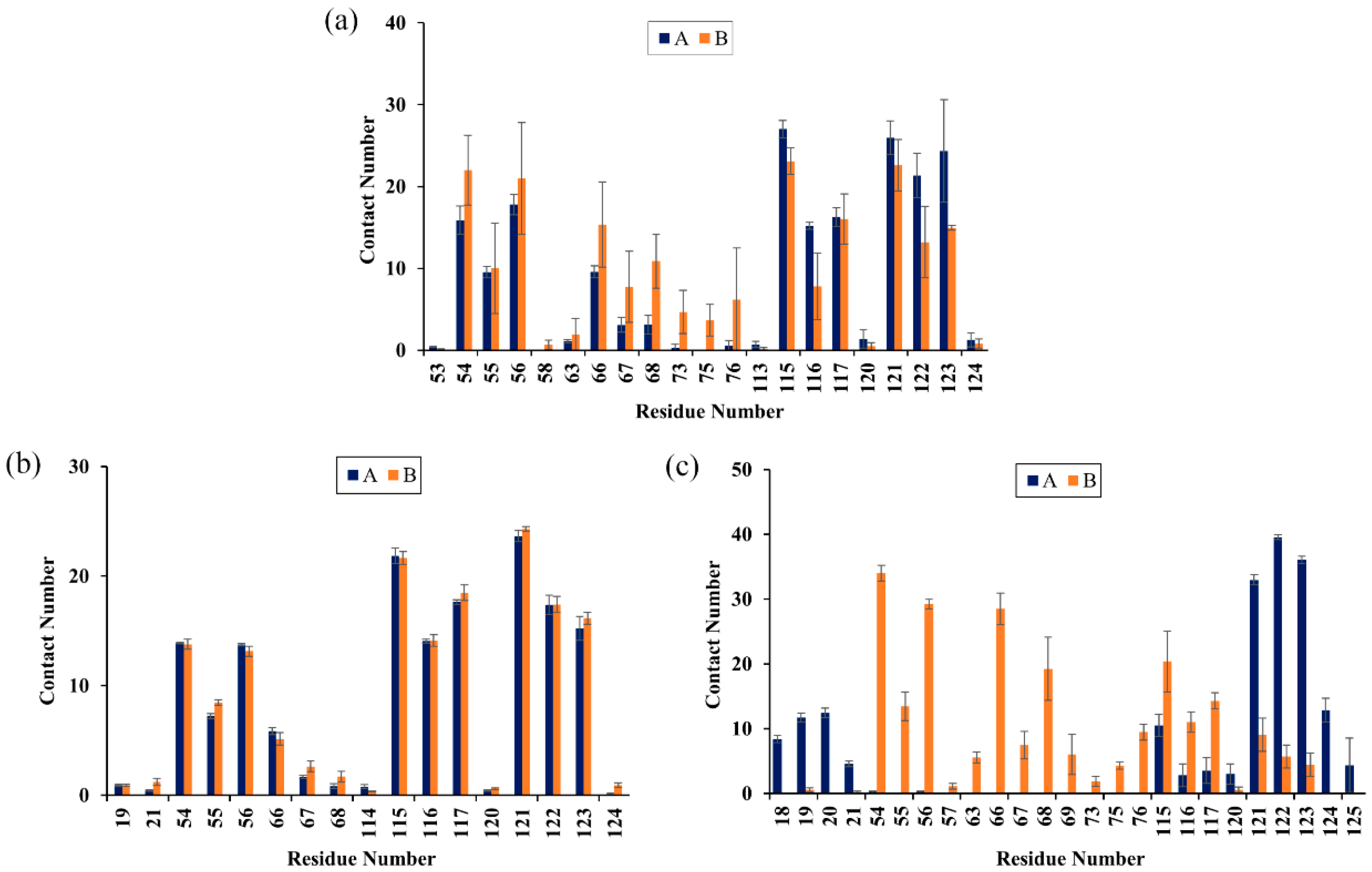
2.6. Contact Numbers
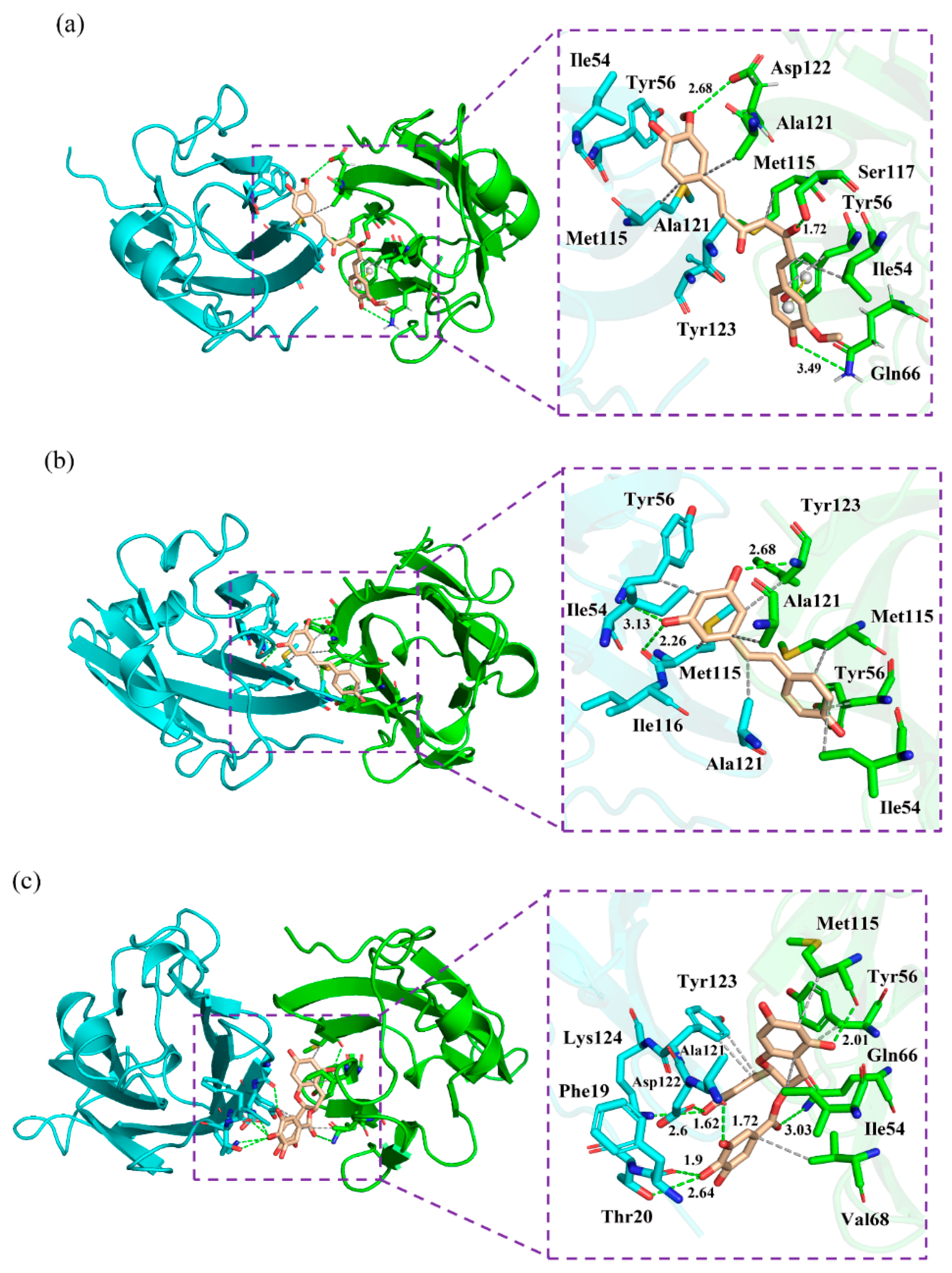
2.7. Nonbonded Interactions
2.8. Cross-Correlation Matrix Analysis
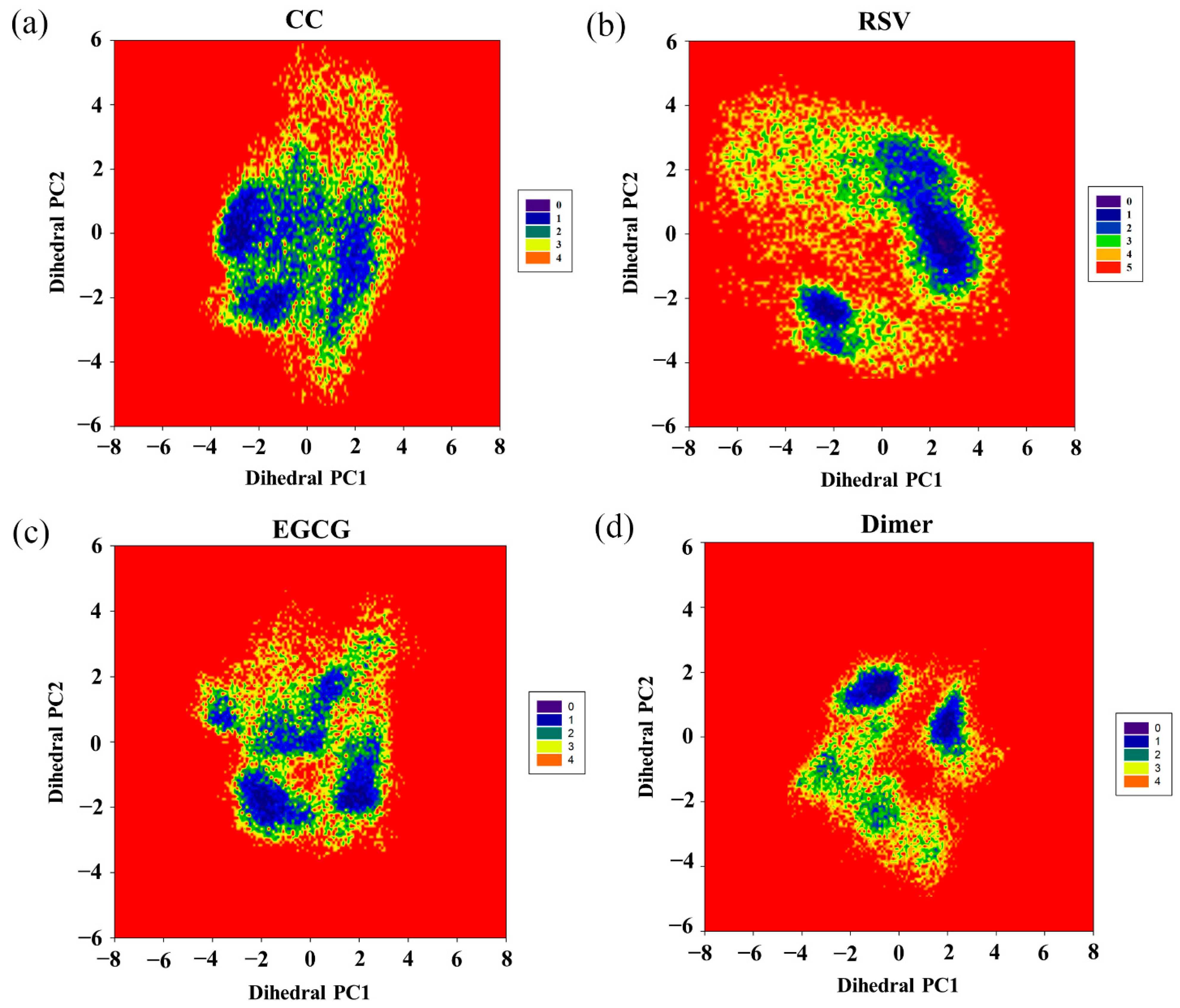
2.9. Free Energy Landscape
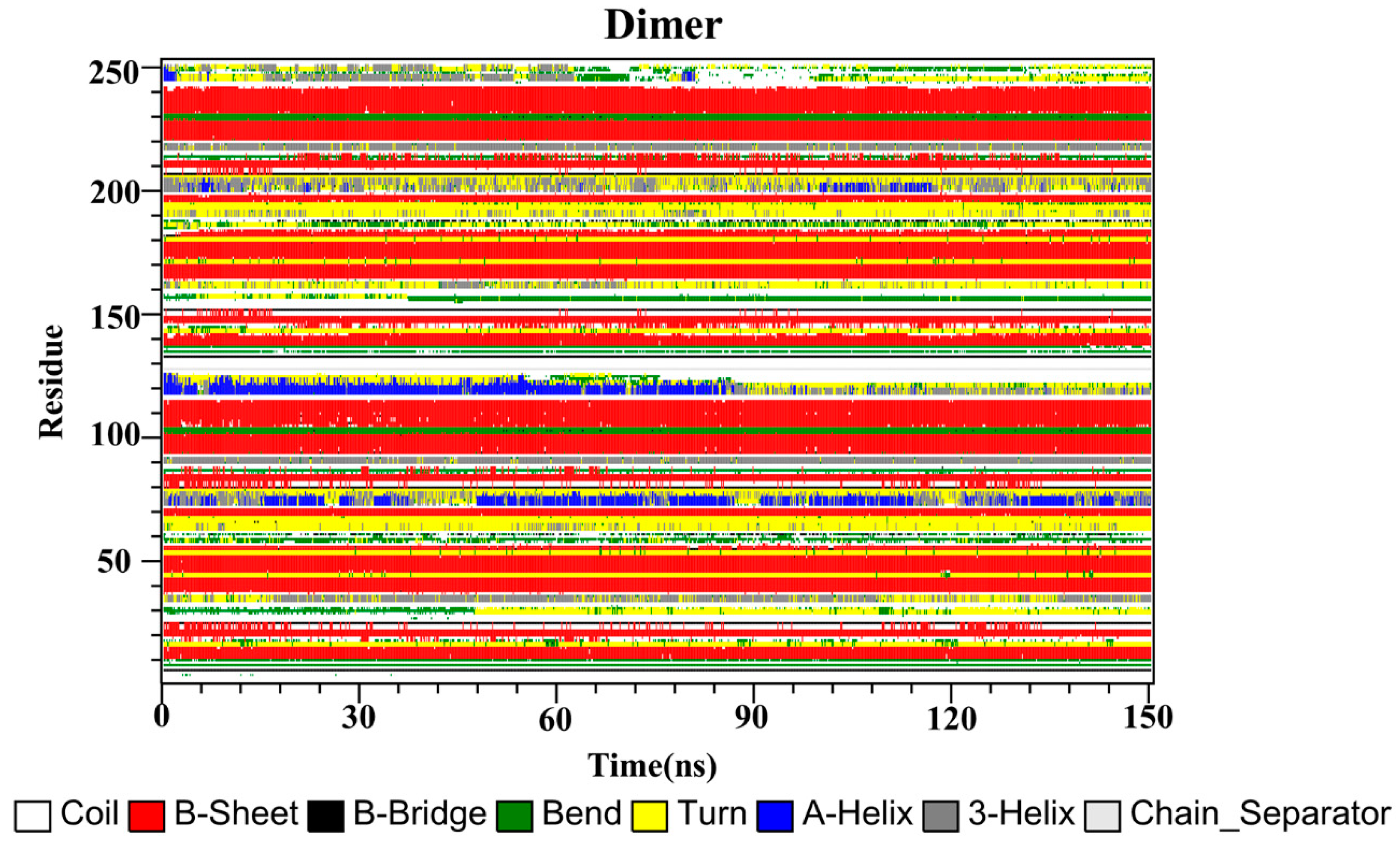
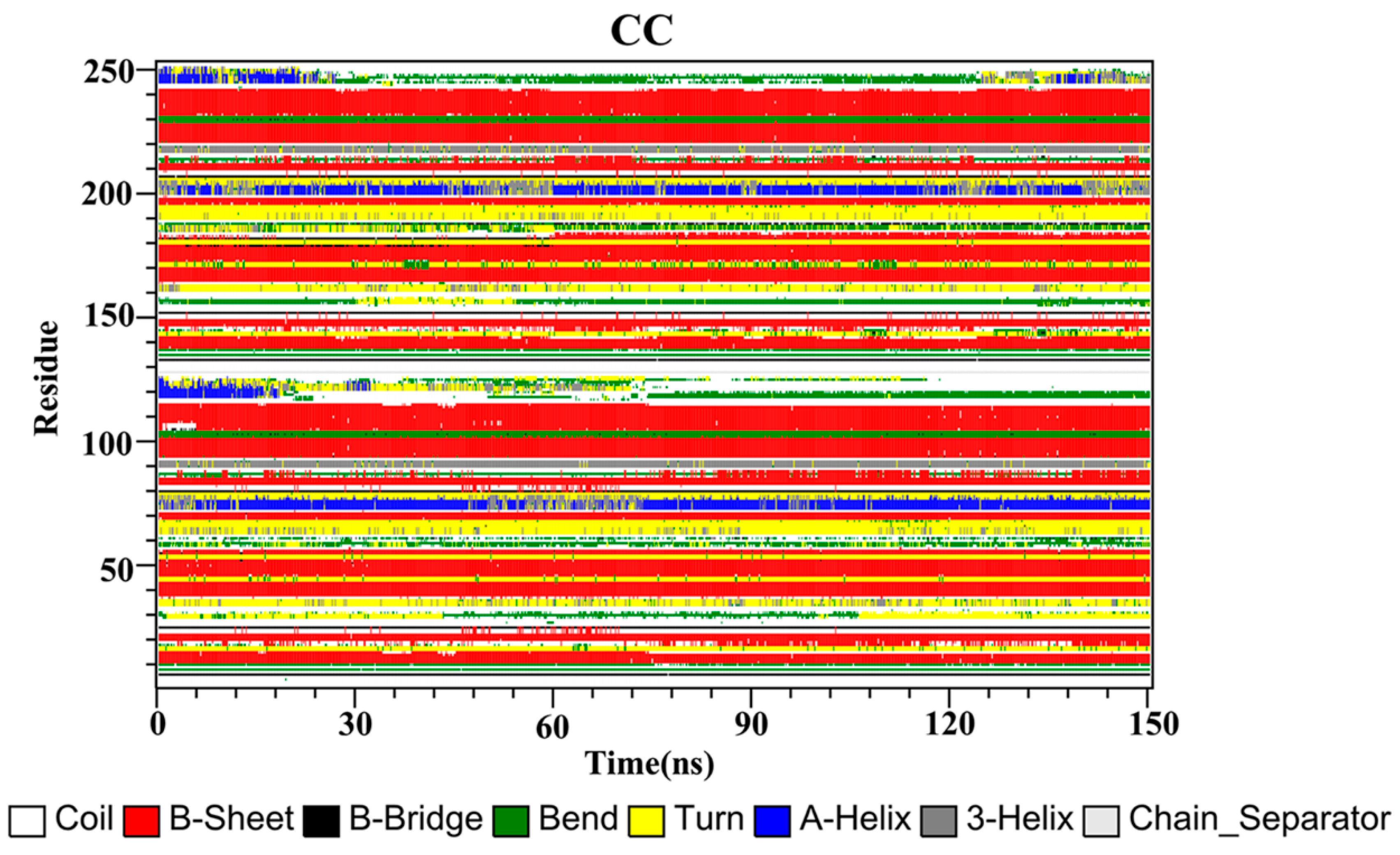
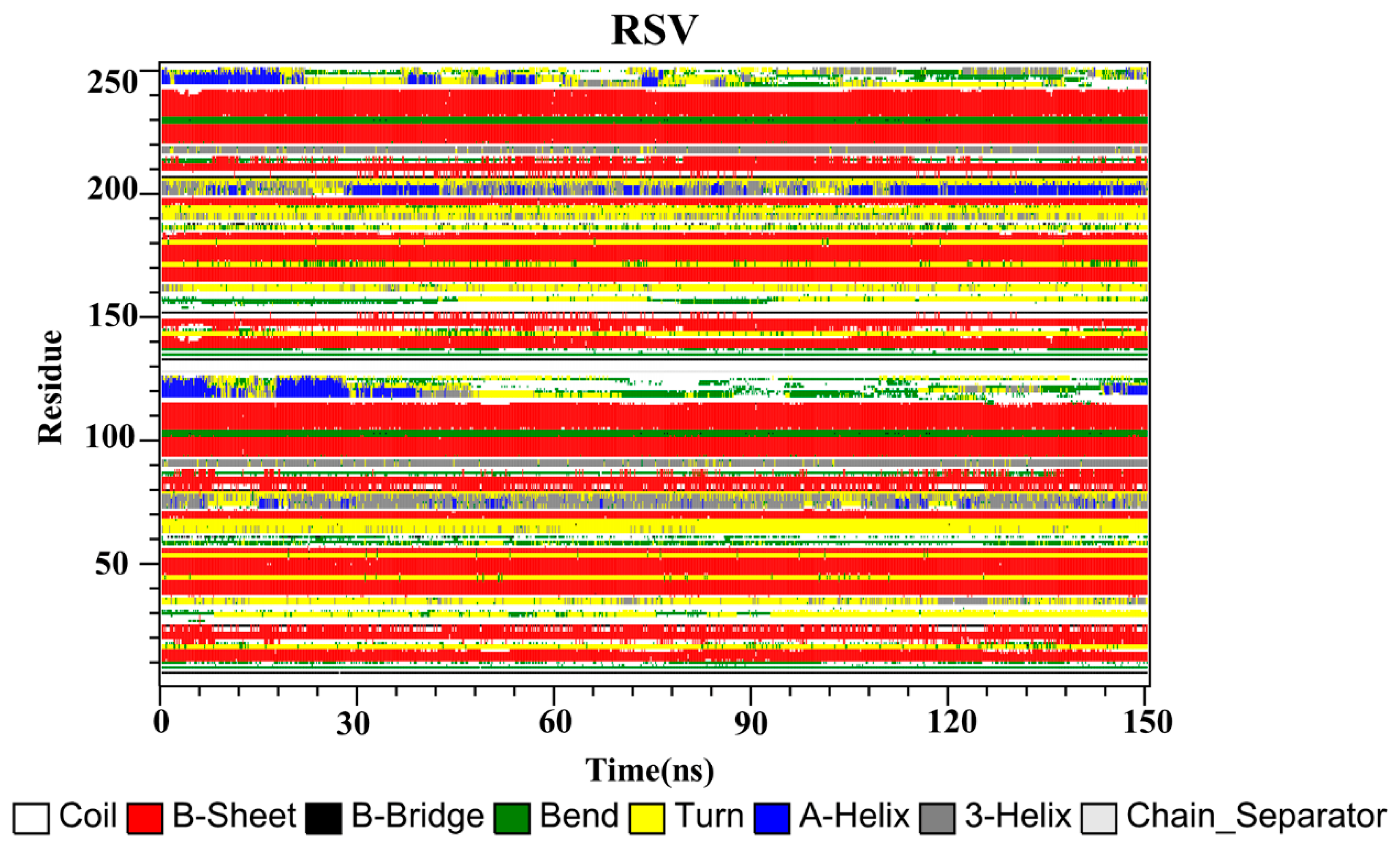
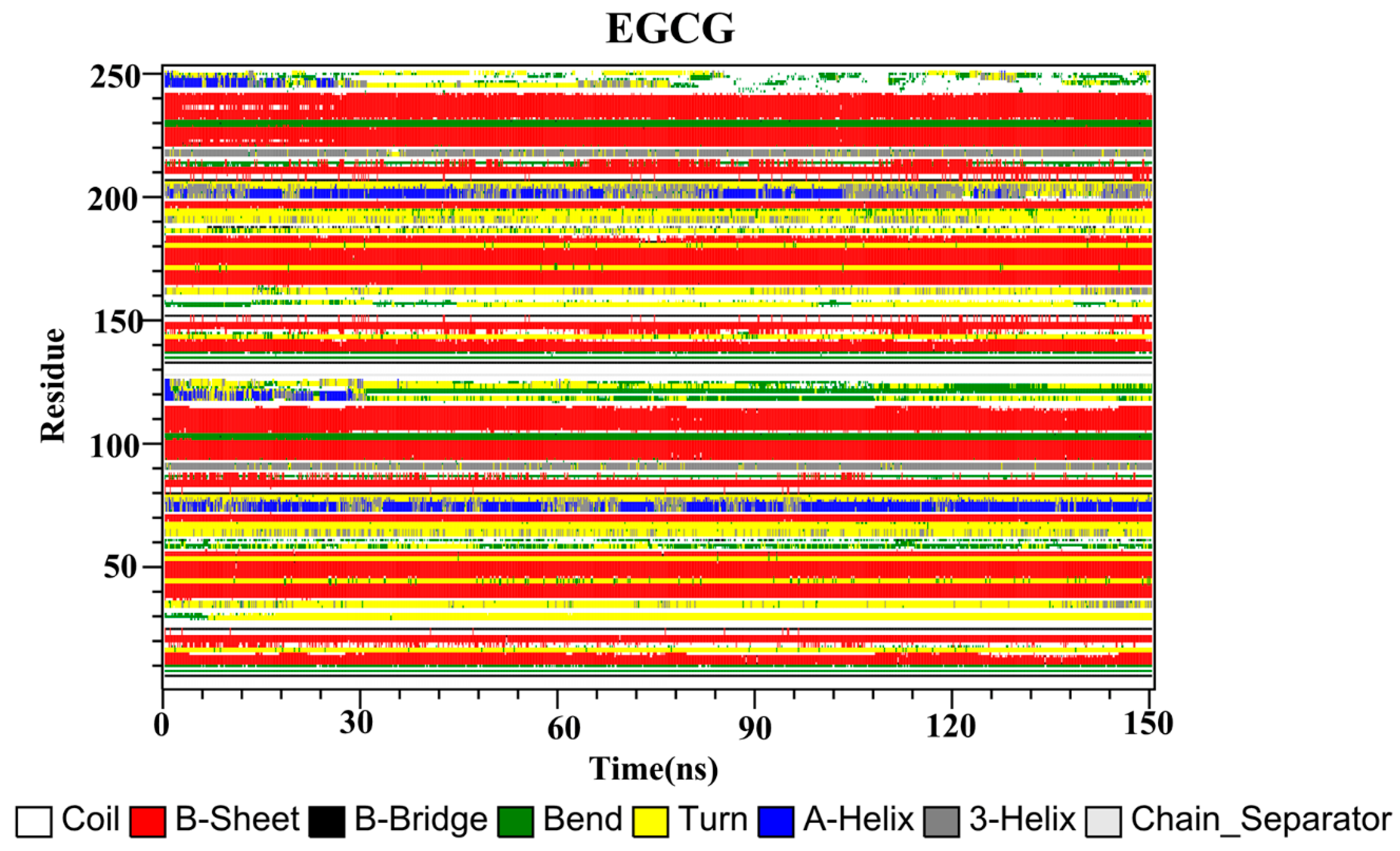
2.10. Secondary Structure
3. Materials and Methods
3.1. Molecular Docking
3.2. Molecular Dynamics Simulation
3.3. Binding Free Energy Calculation
3.4. Simulation Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef]
- Gianchecchi, E.; Delfino, D.V.; Fierabracci, A. Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmun. Rev. 2013, 12, 1091–1100. [Google Scholar] [CrossRef]
- Fife, B.T.; Pauken, K.E. The role of the PD-1 pathway in autoimmunity and peripheral tolerance. Ann. N. Y. Acad. Sci. 2011, 1217, 45–59. [Google Scholar] [CrossRef]
- Wu, Q.; Jiang, L.; Li, S.-C.; He, Q.-J.; Yang, B.; Cao, J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharmacol. Sin. 2021, 42, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Page, D.B.; Li, B.; Connell, L.C.; Schindler, K.; Lacouture, M.; Postow, M.A.; Wolchok, J.D. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 2015, 26, 2375–2391. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of Anti–PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Philips, G.K.; Atkins, M.B. Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. Int. Immunol. 2014, 27, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Perez, H.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody-drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.-M.; Hu, X.-Q.; Liu, X.-X.; Ruan, B.-F.; Xu, J.; Liao, C. From monoclonal antibodies to small molecules: The development of inhibitors targeting the PD-1/PD-L1 pathway. Drug Discov. Today 2016, 21, 1027–1036. [Google Scholar] [CrossRef]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Torner, R.; Skalniak, L.; Domling, A.; Dubin, G.; Holak, T.A. Small-molecule inhibitors of the programmed cell death-1/programmed death-ligand 1 (PD-1/PD-L1) interaction via. transiently induced protein states and simerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Domling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalniak, L.; Zak, K.M.; Guzik, K.; Magiera-Mularz, K.; Musielak, B.; Pachota, M.; Szelazek, B.; Kocik, J.; Grudnik, P.; Tomala, M.; et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 2017, 8, 72167–72181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, T.; Sa, G.; Saha, B.; Das, K. Multifocal signal modulation therapy of cancer: Ancient weapon, modern targets. Mol. Cell. Biochem. 2009, 336, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Varalakshmi, C.; Ali, A.M.; Pardhasaradhi, B.; Srivastava, R.M.; Singh, S.; Khar, A. Immunomodulatory effects of curcumin: In-Vivo. Int. Immunopharmacol. 2008, 8, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Iranshahi, M.; Sahebkar, A.; Takasaki, M.; Konoshima, T.; Tokuda, H. Cancer chemopreventive activity of the prenylated coumarin, umbelliprenin, In Vivo. Eur. J. Cancer Prev. 2009, 18, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Rawangkan, A.; Wongsirisin, P.; Namiki, K.; Iida, K.; Kobayashi, Y.; Shimizu, Y.; Fujiki, H.; Suganuma, M. Green tea catechin is an alternative immune checkpoint inhibitor that inhibits PD-L1 expression and lung tumor growth. Molecules 2018, 23, 2071. [Google Scholar] [CrossRef] [Green Version]
- Panda, A.K.; Chakraborty, D.; Sarkar, I.; Khan, T.; Sa, G. New insights into therapeutic activity and anticancer properties of curcumin. J. Exp. Pharmacol. 2017, 9, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Hesari, A.; Azizian, M.; Sheikhi, A.; Nesaei, A.; Sanaei, S.; Mahinparvar, N.; Derakhshani, M.; Hedayt, P.; Ghasemi, F.; Mirzaei, H. Chemopreventive and therapeutic potential of curcumin in esophageal cancer: Current and future status. Int. J. Cancer 2019, 144, 1215–1226. [Google Scholar] [CrossRef]
- Mirzaei, H.; Naseri, G.; Rezaee, R.; Mohammadi, M.; Banikazemi, Z.; Mirzaei, H.R.; Salehi, H.; Peyvandi, M.; Pawelek, J.M.; Sahebkar, A. Curcumin: A new candidate for melanoma therapy? Int. J. Cancer 2016, 139, 1683–1695. [Google Scholar] [CrossRef]
- Sahebkar, A.; Serban, M.-C.; Ursoniu, S.; Banach, M. Effect of curcuminoids on oxidative stress: A systematic review and meta-analysis of randomized controlled trials. J. Funct. Foods 2015, 18, 898–909. [Google Scholar] [CrossRef]
- Abdollahi, E.; Momtazi, A.A.; Johnston, T.P.; Sahebkar, A. Therapeutic effects of curcumin in inflammatory and immune-mediated diseases: A nature-made jack-of-all-trades? J. Cell. Physiol. 2018, 233, 830–848. [Google Scholar] [CrossRef]
- Shafabakhsh, R.; Pourhanifeh, M.H.; Mirzaei, H.R.; Sahebkar, A.; Asemi, Z.; Mirzaei, H. Targeting regulatory T cells by curcumin: A potential for cancer immunotherapy. Pharmacol. Res. 2019, 147, 104353. [Google Scholar] [CrossRef]
- Liao, F.; Liu, L.; Luo, E.; Hu, J. Curcumin enhances anti-tumor immune response in tongue squamous cell carcinoma. Arch. Oral Biol. 2018, 92, 32–37. [Google Scholar] [CrossRef]
- Lim, S.-O.; Li, C.-W.; Xia, W.; Cha, J.-H.; Chan, L.-C.; Wu, Y.; Chang, S.-S.; Lin, W.-C.; Hsu, J.-M.; Hsu, Y.-H.; et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-R.; Chen, Y.-S.; Chin, Y.-T.; Li, Z.-L.; Shih, Y.-J.; Yang, Y.-C.S.; ChangOu, C.A.; Su, P.-Y.; Wang, S.-H.; Wu, Y.-H.; et al. Thyroid hormone-induced expression of inflammatory cytokines interfere with resveratrol-induced anti-proliferation of oral cancer cells. Food Chem. Toxicol. 2019, 132, 110693. [Google Scholar] [CrossRef]
- Chin, Y.-T.; Wei, P.-L.; Ho, Y.; Nana, A.W.; Changou, C.A.; Chen, Y.-R.; Yang, Y.-C.S.; Hsieh, M.-T.; Hercbergs, A.; Davis, P.J.; et al. Thyroxine inhibits resveratrol-caused apoptosis by PD-L1 in ovarian cancer cells. Endocr.-Relat. Cancer 2018, 25, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Verdura, S.; Cuyàs, E.; Cortada, E.; Brunet, J.; Lopez-Bonet, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Resveratrol targets PD-L1 glycosylation and dimerization to enhance antitumor T-cell immunity. Aging 2020, 12, 8–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Quan, L.; Lyu, J.; He, Z.; Wang, X.; Meng, J.; Zhao, Z.; Zhu, L.; Liu, X.; Li, H. Discovery of peptide inhibitors targeting human programmed death 1 (PD-1) receptor. Oncotarget 2016, 7, 64967–64976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Qin, Y.; Wu, Y.; Zhao, W.; Zhai, W.; Qi, Y.; Wang, C.; Gao, Y. The design of high affinity human PD-1 mutants by using molecular dynamics simulations (MD). Cell Commun. Signal. 2018, 16, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altis, A.; Nguyen, P.H.; Hegger, R.; Stock, G. Dihedral angle principal component analysis of molecular dynamics simulations. J. Chem. Phys. 2007, 126, 244111. [Google Scholar] [CrossRef] [Green Version]
- Guan, S.; Wang, T.; Kuai, Z.; Qian, M.; Tian, X.; Zhang, X.; Yu, Y.; Wang, S.; Zhang, H.; Li, H.; et al. Exploration of binding and inhibition mechanism of a small molecule inhibitor of influenza virus H1N1 hemagglutinin by molecular dynamics simulation. Sci. Rep. 2017, 7, 3786. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. G_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, J.; Zhao, X.; Zhao, Y.; Zhu, S. Network pharmacology and molecular docking study on the active ingredients of qidengmingmu capsule for the treatment of diabetic retinopathy. Sci. Rep. 2021, 11, 7382. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Shi, D.; Li, L.; Guo, J.; Liu, H.; Yao, X. Revealing inhibition difference between PFI-2 enantiomers against SETD7 by molecular dynamics simulations, binding free energy calculations and unbinding pathway analysis. Sci. Rep. 2017, 7, 46547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.; Chun, J.; Jin, X.; Kim, J.; Yoon, J.; No, K.T. Investigation of protein-protein interactions and hot spot region between PD-1 and PD-L1 by fragment molecular orbital method. Sci. Rep. 2019, 9, 16727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zak, K.M.; Grudnik, P.; Magiera-Mularz, K.; Dömling, A.; Dubin, G.; Holak, T.A. Structural biology of the immune checkpoint receptor PD-1 and its ligands PD-L1/PD-L2. Structure 2017, 25, 1163–1174. [Google Scholar] [CrossRef]
- Ahmed, M.; Barakat, K. The too many faces of PD-L1: A comprehensive conformational analysis study. Biochemistry 2017, 56, 5428–5439. [Google Scholar] [CrossRef]
- Tu, Y.; Ma, S.; Liu, F.; Sun, Y.; Dong, X. Hematoxylin inhibits amyloid beta-protein fibrillation and alleviates amyloid-induced cytotoxicity. J. Phys. Chem. B 2016, 120, 11360–11368. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Jin, Y.; Wang, B.; Liu, B. Molecular mechanism of small-molecule inhibitors in blocking the PD-1/PD-L1 pathway through PD-L1 dimerization. Int. J. Mol. Sci. 2021, 22, 4766. [Google Scholar] [CrossRef]
- Shi, D.; An, X.; Bai, Q.; Bing, Z.; Zhou, S.; Liu, H.; Yao, X. Computational insight into the small molecule intervening PD-L1 dimerization and the potential structure-activity relationship. Front. Chem. 2019, 7, 764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almahmoud, S.; Zhong, H.A. Molecular modeling studies on the binding mode of the PD-1/PD-L1 complex inhibitors. Int. J. Mol. Sci. 2019, 20, 4654. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, A.; Ahmed, M.; Okoye, I.; Arutyunova, E.; Babu, D.; Turnbull, W.L.; Kundu, J.K.; Shields, J.; Agopsowicz, K.C.; Xu, L.; et al. Comprehensive in vitro characterization of PD-L1 small molecule inhibitors. Sci. Rep. 2019, 9, 12392. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liang, L.; Gu, J.; Zhuo, W.; Yan, X.; Xie, T.; Wu, Z.; Liu, X.; Gou, X.; Liu, W.; et al. Inhibition of programmed cell death protein ligand-1 (PD-L1) by benzyl ether derivatives: Analyses of conformational change, molecular recognition and binding free energy. J. Biomol. Struct. Dyn. 2019, 37, 4801–4812. [Google Scholar] [CrossRef] [PubMed]
- Soremekun, O.S.; Olotu, F.A.; Agoni, C.; Soliman, M.E.S. Recruiting monomer for dimer formation: Resolving the antagonistic mechanisms of novel immune check point inhibitors against programmed death ligand-1 in cancer immunotherapy. Mol. Simul. 2019, 45, 777–789. [Google Scholar] [CrossRef]
- Zhan, D.; Guan, S.; Jin, H.; Han, W.; Wang, S. Stereoselectivity of phosphotriesterase with paraoxon derivatives: A computational study. J. Biomol. Struct. Dyn. 2016, 34, 600–611. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2010, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Su, P.C.; Tsai, C.C.; Mehboob, S.; Hevener, K.E.; Johnson, M.E. Comparison of radii sets, entropy, QM methods, and sampling on MM-PBSA, MM-GBSA, and QM/MM-GBSA ligand binding energies of F. tularensis enoyl-ACP reductase (FabI). J. Comput. Chem. 2015, 36, 1859–1873. [Google Scholar] [CrossRef] [Green Version]
- Dragic, T.; Trkola, A.; Thompson, D.A.D.; Cormier, E.G.; Kajumo, F.A.; Maxwell, E.; Lin, S.W.; Ying, W.; Smith, S.O.; Sakmar, T.P.; et al. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc. Natl. Acad. Sci. USA 2000, 97, 5639–5644. [Google Scholar] [CrossRef] [Green Version]
- Swain, S.S.; Paidesetty, S.K.; Dehury, B.; Sahoo, J.; Vedithi, S.C.; Mahapatra, N.; Hussain, T.; Padhy, R.N. Molecular docking and simulation study for synthesis of alternative dapsone derivative as a newer antileprosy drug in multidrug therapy. J. Cell. Biochem. 2018, 119, 9838–9852. [Google Scholar] [CrossRef]
- Dehury, B.; Behera, S.K.; Mahapatra, N. Structural dynamics of casein kinase I (CKI) from malarial parasite Plasmodium falciparum (Isolate 3D7): Insights from theoretical modelling and molecular simulations. J. Mol. Graph. Model. 2017, 71, 154–166. [Google Scholar] [CrossRef]
- Wang, Q.; Ning, L.; Niu, Y.; Liu, H.; Yao, X. Molecular mechanism of the inhibition and remodeling of human islet amyloid polypeptide (hIAPP1–37) oligomer by resveratrol from molecular dynamics simulation. J. Phys. Chem. B 2015, 119, 15–24. [Google Scholar] [CrossRef]
- Zhou, R.; Berne, B.J.; Germain, R. The free energy landscape for β hairpin folding in explicit water. Proc. Natl. Acad. Sci. USA 2001, 98, 14931–14936. [Google Scholar] [CrossRef] [Green Version]
- Garcıa, A.E.; Sanbonmatsu, K.Y. Exploring the energy landscape of a β hairpin in explicit solvent. Proteins 2001, 42, 345–354. [Google Scholar] [CrossRef]
- Marzinek, J.; Lakshminarayanan, R.; Goh, E.; Huber, R.; Panzade, S.; Verma, C.; Bond, P.J. Characterizing the conformational landscape of flavivirus fusion peptides via. simulation and experiment. Sci. Rep. 2016, 6, 19160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaleo, E.; Mereghetti, P.; Fantucci, P.; Grandori, R.; De Gioia, L. Free-energy landscape, principal component analysis, and structural clustering to identify representative conformations from molecular dynamics simulations: The myoglobin case. J. Mol. Graph. Model. 2009, 27, 889–899. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contribution | CC | RSV | EGCG | Dimer |
|---|---|---|---|---|
| ΔEvdw a | −57.28 ± 3.27 | −42.96 ± 0.40 | −42.88 ± 1.70 | −44.59 ± 9.85 |
| ΔEele b | −3.83 ± 0.76 | −5.16 ± 0.20 | −23.14 ± 3.14 | −124.35 ± 23.36 |
| ΔEPB c | 32.33 ± 3.63 | 23.02 ± 0.22 | 50.43 ± 3.76 | 211.28 ± 17.07 |
| ΔESA d | −4.95 ± 0.14 | −3.39 ± 0.03 | −4.73 ± 0.16 | −6.23 ± 0.37 |
| ΔEpolar,total e | 28.50 ± 3.18 | 17.86 ± 0.42 | 27.29 ± 1.42 | 86.94 ± 10.00 |
| ΔEnonpolar,total f | −62.23 ± 3.35 | −46.35 ± 0.43 | −47.61 ± 1.75 | −50.82 ± 10.06 |
| ΔG g | −33.72 ± 0.23 | −28.49 ± 0.40 | −20.31 ± 0.35 | 36.11 ± 0.89 |
| Inhibitor | N-Terminal | C Sheet | C’ Sheet | F Sheet | G Sheet | Total Sheet | Total |
|---|---|---|---|---|---|---|---|
| CC | 3 | 96 | 50 | 105 | 124 | 376 | 379 |
| RSV | 1 | 70 | 18 | 108 | 115 | 311 | 312 |
| EGCG | 32 | 77 | 55 | 63 | 141 | 335 | 368 |
| Donor | Donor H | Acceptor | Occupancy (%) |
|---|---|---|---|
| BSer117@OG | HG | CC@O1 | 76.74 |
| CC@O2 | H7 | BGln66@OE1 | 87.04 |
| RSV@O3 | HO3 | AMet115@O | 76.74 |
| EGCG@O50 | H50 | AAla121@O | 80.40 |
| EGCG@O10 | H10 | AAsp122@N | 59.80 |
| EGCG@O47 | H47 | APhe19@O | 59.47 |
| EGCG@O10 | H10 | AAsp122@OD1 | 39.53 |
| EGCG@O03 | H03 | BMet115@O | 43.52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Liang, J.; Liu, B.; Jin, Y. Molecular Mechanism of Food-Derived Polyphenols on PD-L1 Dimerization: A Molecular Dynamics Simulation Study. Int. J. Mol. Sci. 2021, 22, 10924. https://doi.org/10.3390/ijms222010924
Guo Y, Liang J, Liu B, Jin Y. Molecular Mechanism of Food-Derived Polyphenols on PD-L1 Dimerization: A Molecular Dynamics Simulation Study. International Journal of Molecular Sciences. 2021; 22(20):10924. https://doi.org/10.3390/ijms222010924
Chicago/Turabian StyleGuo, Yan, Jianhuai Liang, Boping Liu, and Yulong Jin. 2021. "Molecular Mechanism of Food-Derived Polyphenols on PD-L1 Dimerization: A Molecular Dynamics Simulation Study" International Journal of Molecular Sciences 22, no. 20: 10924. https://doi.org/10.3390/ijms222010924