
Butyrate Alters Pyruvate Flux and Induces Lipid Accumulation in Cultured Colonocytes

 , and
, and
Abstract
:1. Introduction
2. Results
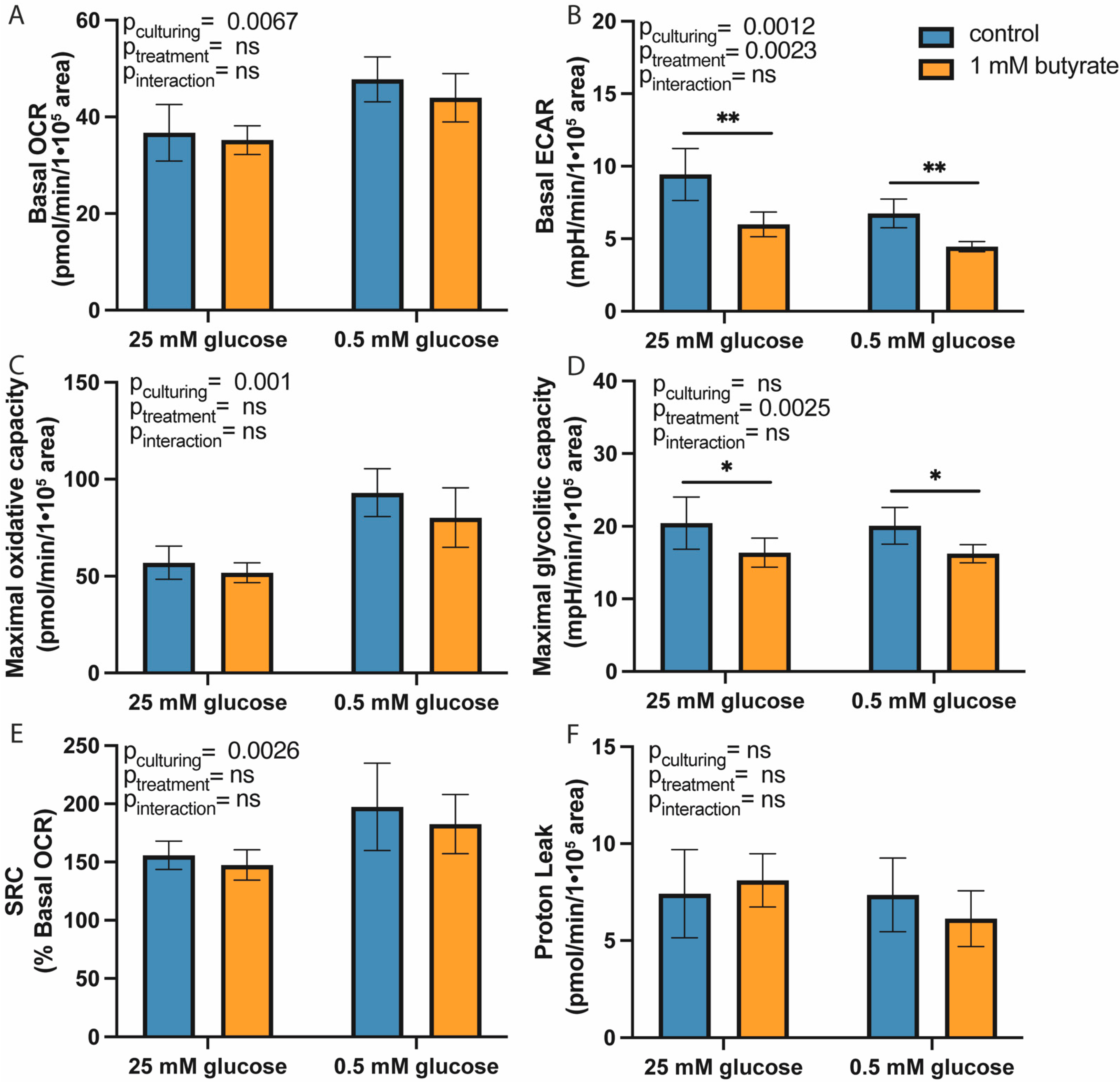
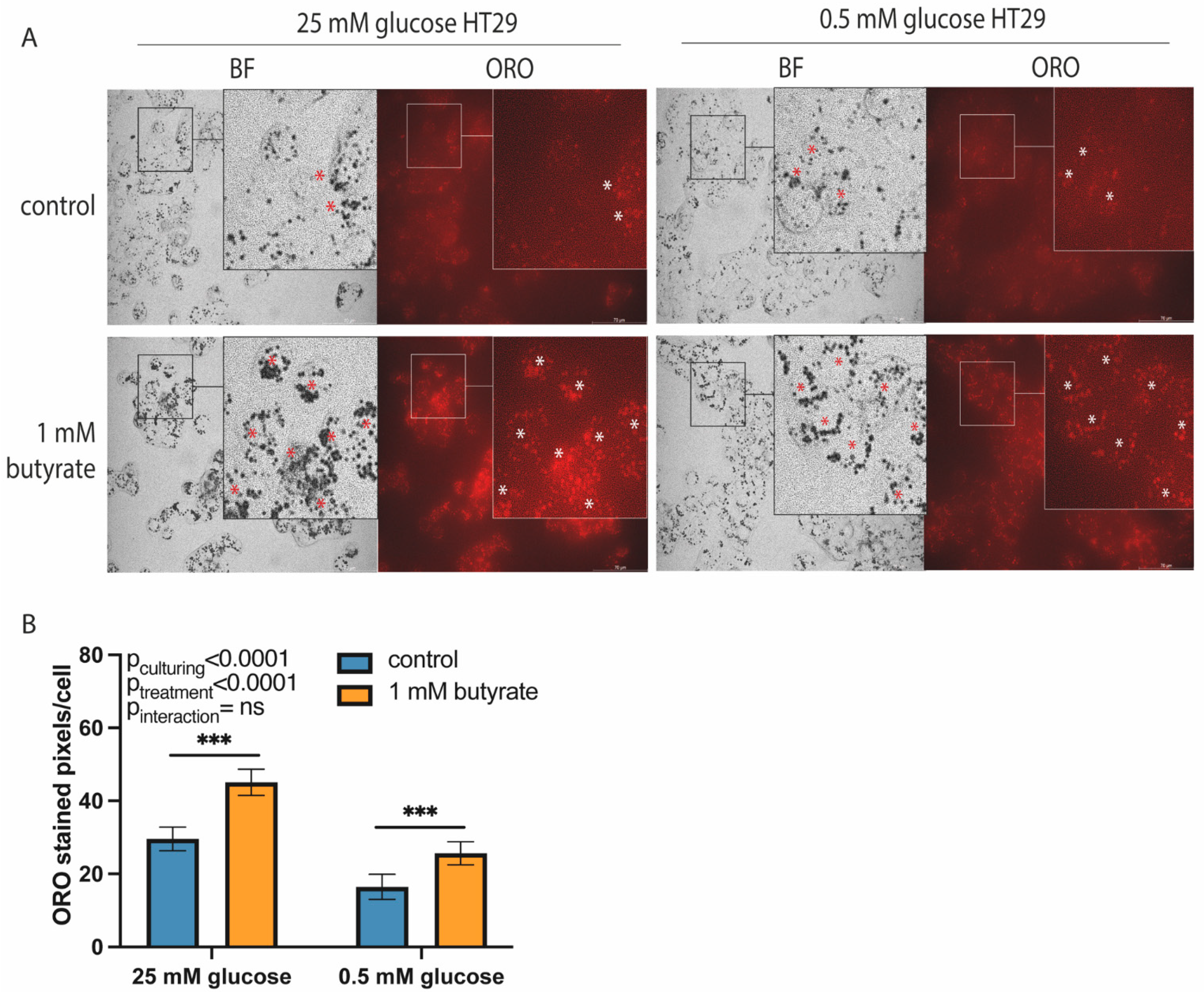
2.1. Long-Term Exposure to Butyrate Decreases Glycolytic Function of Both High- and Low-Glucose-Cultured HT29 Cells
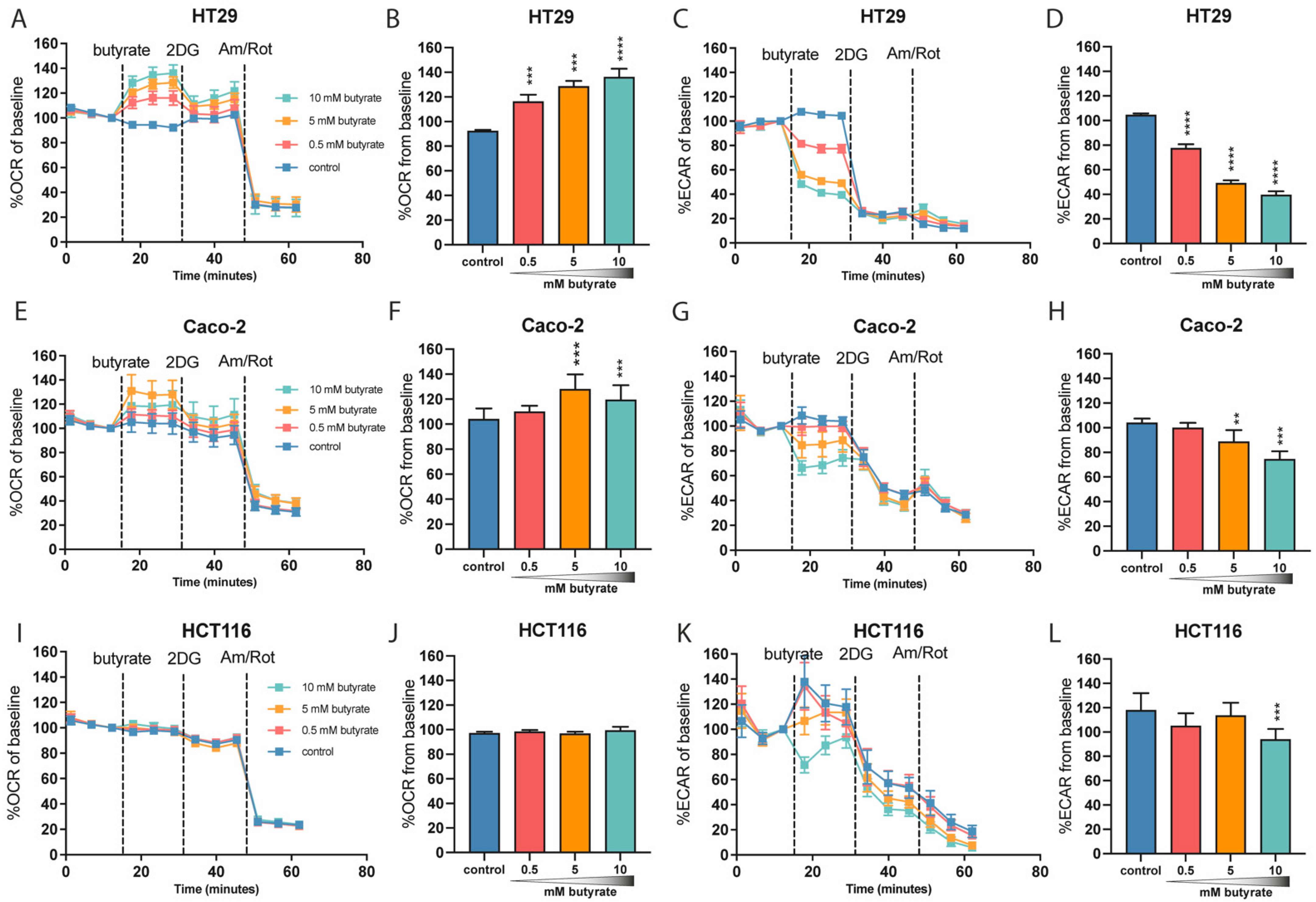
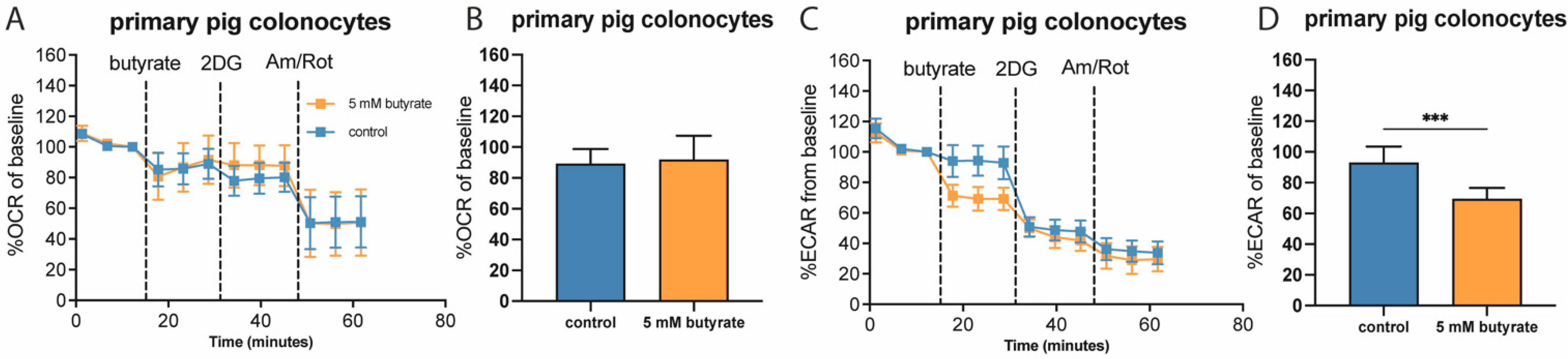
2.2. Butyrate Acutely Lowers Glycolytic Function in Multiple Colon-Derived Cell Lines
2.3. Butyrate Alters Pyruvate Flux in Cultured Colon Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Intestinal Colon Cell Isolation
4.3. Metabolic Flux Analysis with Seahorse XFe96 Analyzer
4.4. Imaging Procedure
4.5. Oil-Red-O Staining and Image Processing
4.6. WST-1 Cell Viability Assay
4.7. RNA Extraction and Semi-Quantative Real-Time Polymerase Chain Reation (qPCR)
4.8. Statistical Analysis and Data Representation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roediger, W.E. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology 1982, 83, 424–429. [Google Scholar] [CrossRef]
- Sengupta, S.; Muir, J.G.; Gibson, P.R. Does butyrate protect from colorectal cancer? J. Gastroenterol. Hepatol. 2006, 21, 209–218. [Google Scholar] [CrossRef]
- Chen, J.; Vitetta, L. Butyrate in Inflammatory Bowel Disease Therapy. Gastroenterology 2020, 158, 1511. [Google Scholar] [CrossRef] [PubMed]
- Krokowicz, L.; Kaczmarek, B.F.; Krokowicz, P.; Stojcev, Z.; Mackiewicz, J.; Walkowiak, J.; Drews, M.; Banasiewicz, T. Sodium butyrate and short chain fatty acids in prevention of travellers’ diarrhoea: A randomized prospective study. Travel Med. Infect. Dis. 2014, 12, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Van der Beek, C.M.; Canfora, E.E.; Lenaerts, K.; Troost, F.J.; Olde Damink, S.W.; Holst, J.J.; Masclee, A.A.; Dejong, C.H.; Blaak, E.E. Distal, not proximal, colonic acetate infusions promote fat oxidation and improve metabolic markers in overweight/obese men. Clin. Sci. 2016, 130, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, C.-D.; Li, H.-F.; Tian, M.-L.; Pan, J.-Q.; Shu, G.; Jiang, Q.-Y.; Yin, Y.-L.; Zhang, L. LSD1 mediates microbial metabolite butyrate-induced thermogenesis in brown and white adipose tissue. Metabolism 2020, 102, 154011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; de Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dähling, S.; Kastenmüller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8+ T Cells. Immunity 2019, 51, 285–297.e5. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, C.; Jiang, Q.; Yin, Y. Butyrate in Energy Metabolism: There Is Still More to Learn. Trends Endocrinol. Metab. 2021, 32, 159–169. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Den Hond, E.; Hiele, M.; Evenepoel, P.; Peeters, M.; Ghoos, Y.; Rutgeerts, P. In vivo butyrate metabolism and colonic permeability in extensive ulcerative colitis. Gastroenterology 1998, 115, 584–590. [Google Scholar] [CrossRef]
- Ahmad, M.S.; Krishnan, S.; Ramakrishna, B.S.; Mathan, M.; Pulimood, A.B.; Murthy, S.N. Butyrate and glucose metabolism by colonocytes in experimental colitis in mice. Gut 2000, 46, 493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, M.A.; Grahn, M.F.; Boyle, M.A.; Hutton, M.; Rogers, J.; Williams, N.S. Butyrate oxidation is impaired in the colonic mucosa of sufferers of quiescent ulcerative colitis. Gut 1994, 35, 73–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer-Picón, E.; Dotti, I.; Corraliza, A.M.; Mayorgas, A.; Esteller, M.; Perales, J.C.; Ricart, E.; Masamunt, M.C.; Carrasco, A.; Tristán, E.; et al. Intestinal Inflammation Modulates the Epithelial Response to Butyrate in Patients With Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 26, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309. [Google Scholar] [CrossRef]
- Fleming, S.E.; Gill, R. Aging Stimulates Fatty Acid Oxidation in Rat Colonocytes But Does Not Influence the Response to Dietary Fiber. J. Gerontol. Ser. A 1997, 52, B318–B330. [Google Scholar] [CrossRef] [Green Version]
- Hofmanová, J.; Vaculová, A.; Koubková, Z.; Hýžd’alová, M.; Kozubík, A. Human fetal colon cells and colon cancer cells respond differently to butyrate and PUFAs. Mol. Nutr. Food Res. 2009, 53, S102–S113. [Google Scholar] [CrossRef]
- Zweibaum, A.; Pinto, M.; Chevalier, G.; Dussaulx, E.; Triadou, N.; Lacroix, B.; Haffen, K.; Brun, J.L.; Rousset, M. Enterocytic differentiation of a subpopulation of the human colon tumor cell line HT-29 selected for growth in sugar-free medium and its inhibition by glucose. J. Cell. Physiol. 1985, 122, 21–29. [Google Scholar] [CrossRef]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy Substrate Modulates Mitochondrial Structure and Oxidative Capacity in Cancer Cells. Cancer Res. 2004, 64, 985. [Google Scholar] [CrossRef] [Green Version]
- Kase, E.T.; Nikolić, N.; Bakke, S.S.; Bogen, K.K.; Aas, V.; Thoresen, G.H.; Rustan, A.C. Remodeling of Oxidative Energy Metabolism by Galactose Improves Glucose Handling and Metabolic Switching in Human Skeletal Muscle Cells. PLoS ONE 2013, 8, e59972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- JanssenDuijghuijsen, L.M.; Grefte, S.; de Boer, V.C.J.; Zeper, L.; van Dartel, D.A.M.; van der Stelt, I.; Bekkenkamp-Grovenstein, M.; van Norren, K.; Wichers, H.J.; Keijer, J. Mitochondrial ATP Depletion Disrupts Caco-2 Monolayer Integrity and Internalizes Claudin 7. Front. Physiol. 2017, 8, 794. [Google Scholar] [CrossRef] [Green Version]
- Leschelle, X.; Delpal, S.; Goubern, M.; Blottiere, H.M.; Blachier, F. Butyrate metabolism upstream and downstream acetyl-CoA synthesis and growth control of human colon carcinoma cells. Eur. J. Biochem. 2000, 267, 6435–6442. [Google Scholar] [CrossRef] [Green Version]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.-Y.; Xu, Z.; Liu, X.; Tan, M.; Ye, B.-C. Protein Acetylation and Butyrylation Regulate the Phenotype and Metabolic Shifts of the Endospore-forming Clostridium acetobutylicum. Mol. Cell. Proteom. MCP 2018, 17, 1156–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Halestrap, A.P.; Paraskeva, C. Butyrate can act as a stimulator of growth or inducer of apoptosis in human colonic epithelial cell lines depending on the presence of alternative energy sources. Carcinogenesis 1997, 18, 1265–1270. [Google Scholar] [CrossRef] [Green Version]
- Marcil, V.; Delvin, E.; Seidman, E.; Poitras, L.; Zoltowska, M.; Garofalo, C.; Levy, E. Modulation of lipid synthesis, apolipoprotein biogenesis, and lipoprotein assembly by butyrate. Am. J. Physiol.-Gastrointest. Liver Physiol. 2002, 283, G340–G346. [Google Scholar] [CrossRef] [Green Version]
- Boren, J.; Lee, W.-N.P.; Bassilian, S.; Centelles, J.J.; Lim, S.; Ahmed, S.; Boros, L.G.; Cascante, M. The Stable Isotope-based Dynamic Metabolic Profile of Butyrate-induced HT29 Cell Differentiation. J. Biol. Chem. 2003, 278, 28395–28402. [Google Scholar] [CrossRef] [Green Version]
- Geng, H.-W.; Yin, F.-Y.; Zhang, Z.-F.; Gong, X.; Yang, Y. Butyrate Suppresses Glucose Metabolism of Colorectal Cancer Cells via GPR109a-AKT Signaling Pathway and Enhances Chemotherapy. Front. Mol. Biosci. 2021, 8, 634874. [Google Scholar] [CrossRef]
- Tan, H.T.; Tan, S.; Lin, Q.; Lim, T.K.; Hew, C.L.; Chung, M.C.M. Quantitative and Temporal Proteome Analysis of Butyrate-treated Colorectal Cancer Cells. Mol. Cell. Proteom. 2008, 7, 1174–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghorbaniaghdam, A.; Henry, O.; Jolicoeur, M. A kinetic-metabolic model based on cell energetic state: Study of CHO cell behavior under Na-butyrate stimulation. Bioprocess Biosyst. Eng. 2013, 36, 469–487. [Google Scholar] [CrossRef]
- Matthews, G.M.; Howarth, G.S.; Butler, R.N. Short-chain fatty acids induce apoptosis in colon cancer cells associated with changes to intracellular redox state and glucose metabolism. Chemotherapy 2012, 58, 102–109. [Google Scholar] [CrossRef]
- Butler, R.N.; Stafford, I.; Triantafillos, E.; O’Dee, C.D.; Jarrett, I.G.; Fettman, M.J.; Roberts-Thomson, I.C. Pyruvate sparing by butyrate and propionate in proliferating colonic epithelium. Comp. Biochem. Physiol. B 1990, 97, 333–337. [Google Scholar] [CrossRef]
- Tabuchi, Y.; Arai, Y.; Kondo, T.; Takeguchi, N.; Asano, S. Identification of genes responsive to sodium butyrate in colonic epithelial cells. Biochem. Biophys. Res. Commun. 2002, 293, 1287–1294. [Google Scholar] [CrossRef]
- Han, A.; Bennett, N.; MacDonald, A.; Johnstone, M.; Whelan, J.; Donohoe, D.R. Cellular Metabolism and Dose Reveal Carnitine-Dependent and -Independent Mechanisms of Butyrate Oxidation in Colorectal Cancer Cells. J. Cell. Physiol. 2016, 231, 1804–1813. [Google Scholar] [CrossRef] [PubMed]
- Long, R.W.; Porter, J.W. Incorporation of the Butyryl Unit of Butyryl Coenzyme A into Palmitic Acid by the Pigeon Liver System. J. Biol. Chem. 1959, 234, 1406–1410. [Google Scholar] [CrossRef]
- Blouin, J.-M.; Penot, G.; Collinet, M.; Nacfer, M.; Forest, C.; Laurent-Puig, P.; Coumoul, X.; Barouki, R.; Benelli, C.; Bortoli, S. Butyrate elicits a metabolic switch in human colon cancer cells by targeting the pyruvate dehydrogenase complex. Int. J. Cancer 2011, 128, 2591–2601. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Liu, C.-X.; Xu, W.; Huang, L.; Zhao, J.-Y.; Zhao, S.-M. Butyrate induces apoptosis by activating PDC and inhibiting complex I through SIRT3 inactivation. Signal Transduct. Target. Ther. 2017, 2, 16035. [Google Scholar] [CrossRef] [PubMed]
- Fung, K.Y.C.; Lewanowitsch, T.; Henderson, S.T.; Priebe, I.; Hoffmann, P.; McColl, S.R.; Lockett, T.; Head, R.; Cosgrove, L.J. Proteomic Analysis of Butyrate Effects and Loss of Butyrate Sensitivity in HT29 Colorectal Cancer Cells. J. Proteome Res. 2009, 8, 1220–1227. [Google Scholar] [CrossRef]
- Roediger, W.E.W.; Kapaniris, O.; Millard, S. Lipogenesis from n-butyrate in colonocytes. Mol. Cell. Biochem. 1992, 116, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Zambell, K.L.; Fitch, M.D.; Fleming, S.E. Acetate and Butyrate Are the Major Substrates for De Novo Lipogenesis in Rat Colonic Epithelial Cells. J. Nutr. 2003, 133, 3509–3515. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, G.; Bleeker, A.; Gerding, A.; van Eunen, K.; Havinga, R.; van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.-J.; et al. Short-Chain Fatty Acids Protect Against High-Fat Diet–Induced Obesity via a PPARγ-Dependent Switch from Lipogenesis to Fat Oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Li, X.; Su, C.; Xi, M.; Zhang, X.; Jiang, Z.; Wang, L.; Hong, B. Butyrate protects against high-fat diet-induced atherosclerosis via up-regulating ABCA1 expression in apolipoprotein E-deficiency mice. Br. J. Pharmacol. 2020, 177, 1754–1772. [Google Scholar] [CrossRef]
- Yang, T.; Yang, H.; Heng, C.; Wang, H.; Chen, S.; Hu, Y.; Jiang, Z.; Yu, Q.; Wang, Z.; Qian, S.; et al. Amelioration of non-alcoholic fatty liver disease by sodium butyrate is linked to the modulation of intestinal tight junctions in db/db mice. Food Funct. 2020, 11, 10675–10689. [Google Scholar] [CrossRef]
- Zhang, H.; Ren, E.; Xu, R.; Su, Y. Transcriptomic Responses Induced in Muscle and Adipose Tissues of Growing Pigs by Intravenous Infusion of Sodium Butyrate. Biology 2021, 10, 559. [Google Scholar] [CrossRef]
- Rumberger, J.M.; Arch, J.R.S.; Green, A. Butyrate and other short-chain fatty acids increase the rate of lipolysis in 3T3-L1 adipocytes. PeerJ 2014, 2, e611. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Liu, S.; Zhang, Z.; Zhang, J.; Jin, X.; Zhang, J.; Jiang, W.; Li, H.; Lin, H. Low and high concentrations of butyrate regulate fat accumulation in chicken adipocytes via different mechanisms. Adipocyte 2020, 9, 120–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | RefSeq | Forward Primer * | Reverse Primer * | bp |
|---|---|---|---|---|
| RSP15 | NM_003194.5 | AGAAGCCGGAAGTGGTGAAGAC | AGAGGGATGAAGCGGGAGGAG | 220 |
| B2M | NM_004048.4 | TGCCGTGTGAACCATGTG | GCGGCATCTTCAAACCTC | 92 |
| ACLY | NM_001096.3 | GGACTTCGGCAGAGGTAGAG | TGATCAGCTGGTCTGGCTTG | 227 |
| ACACA | NM_198834.3 | GGGCTAGGTCTTTTTGGAAGTG | GGCCAAGGGAGATGGTTCAT | 104 |
| G6PD | NM_001360016.2 | AAGCGCAGACAGCGTCAT | TGAAGGTGTTTTCGGGCAGA | 215 |
| PGD | NM_002631.4 | TGTGACTGGGTGGGAGATGA | TCCCTGATCTTTGGCAGCAG | 257 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bekebrede, A.F.; Deuren, T.v.; Gerrits, W.J.J.; Keijer, J.; Boer, V.C.J.d. Butyrate Alters Pyruvate Flux and Induces Lipid Accumulation in Cultured Colonocytes. Int. J. Mol. Sci. 2021, 22, 10937. https://doi.org/10.3390/ijms222010937
Bekebrede AF, Deuren Tv, Gerrits WJJ, Keijer J, Boer VCJd. Butyrate Alters Pyruvate Flux and Induces Lipid Accumulation in Cultured Colonocytes. International Journal of Molecular Sciences. 2021; 22(20):10937. https://doi.org/10.3390/ijms222010937
Chicago/Turabian StyleBekebrede, Anna F., Thirza van Deuren, Walter J. J. Gerrits, Jaap Keijer, and Vincent C. J. de Boer. 2021. "Butyrate Alters Pyruvate Flux and Induces Lipid Accumulation in Cultured Colonocytes" International Journal of Molecular Sciences 22, no. 20: 10937. https://doi.org/10.3390/ijms222010937