Two Novel Hydroxymethylbilane Synthase Splicing Mutations Predispose to Acute Intermittent Porphyria


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification of Novel HMBS Mutations
2.2. Corroboration of HMBS Mutations as the Determinant of Alternative Splicing in Splicing Assay
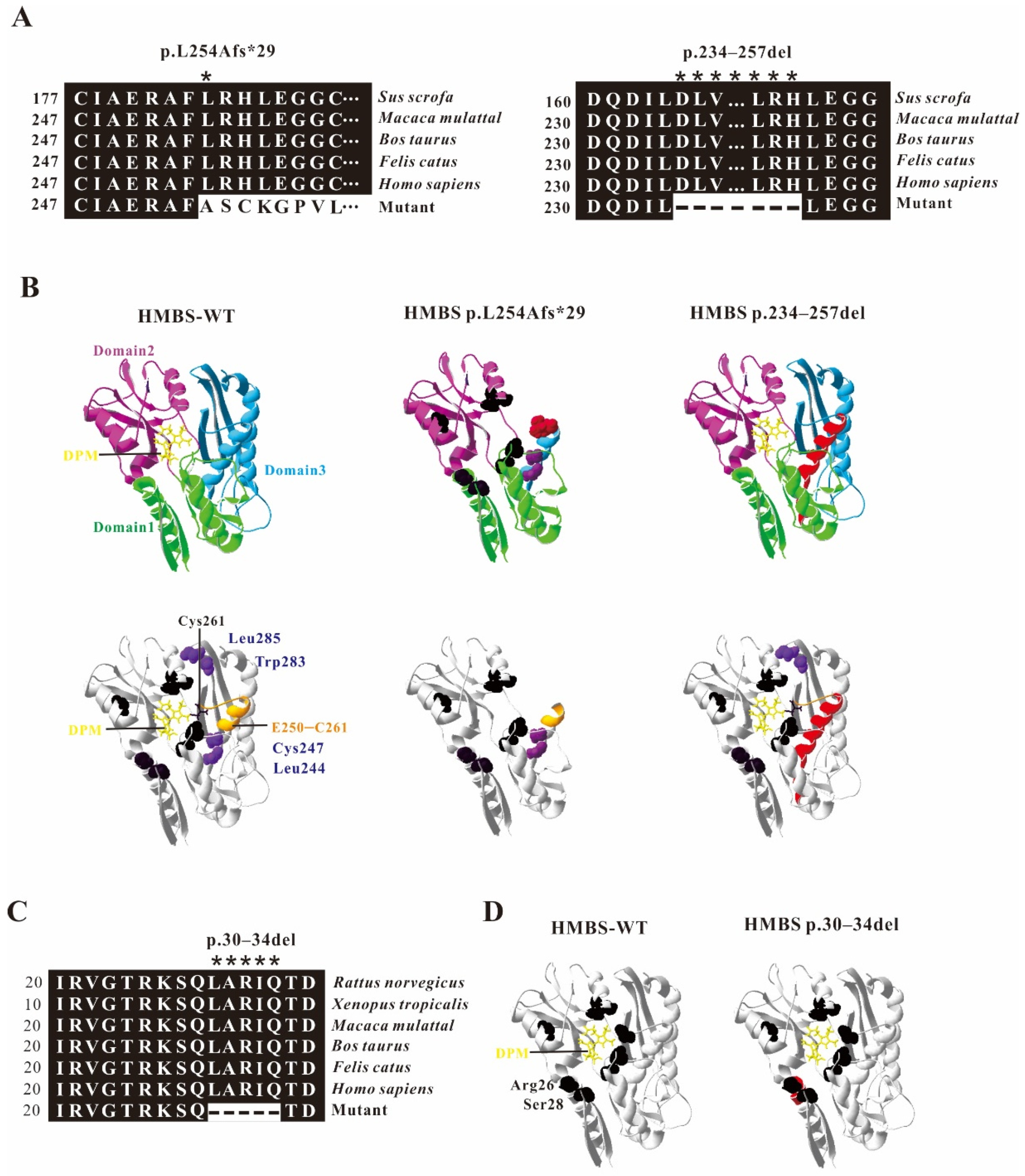
2.3. Model Analysis of Abnormal Structure of HMBS Caused by Mutations
2.4. Verification of Abnormal HMBS Enzyme Activities Caused by Mutations
3. Discussion
4. Materials and Methods
4.1. Ethical Compliance and Patient Information
4.2. DNA Extraction and Sequencing
4.3. Construction of In Vitro Expression Vector
4.4. Transient Transfection
4.5. RT-PCR, PCR and Sequencing
4.6. Western Blotting
4.7. The Evolutionary Conservation Analysis of Amino Acid Residues and the Structure Prediction of Mutant Proteins
4.8. Determination of HMBS Enzyme Activity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bustad, H.J.; Kallio, J.P.; Vorland, M.; Fiorentino, V.; Sandberg, S.; Schmitt, C.; Aarsand, A.K.; Martinez, A. Acute Intermittent Porphyria: An Overview of Therapy Developments and Future Perspectives Focusing on Stabilisation of HMBS and Proteostasis Regulators. Int. J. Mol. Sci. 2021, 22, 675. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Mosquera, L.F.; Sonthalia, S. Acute Intermittent Porphyria. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Besur, S.; Hou, W.; Schmeltzer, P.; Bonkovsky, H.L. Clinically important features of porphyrin and heme metabolism and the porphyrias. Metabolites 2014, 4, 977–1006. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Rudnick, S.; Cengia, B.; Bonkovsky, H.L. Acute Hepatic Porphyrias: Review and Recent Progress. Hepatol. Commun. 2019, 3, 193–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, M.; Chen, B.; Desnick, R.J. Recent advances on porphyria genetics: Inheritance, penetrance & molecular heterogeneity, including new modifying/causative genes. Mol. Genet. Metab. 2019, 128, 320–331. [Google Scholar] [PubMed]
- Norman, G.; Egger, C.L.; Karl, E. Anderson. Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias, in: The Online Metabolic and Molecular Bases of Inherited Disease. In The Online Metabolic and Molecular Bases of Inherited Disease; McGraw Hill: New York, NY, USA, 2001; p. 463. [Google Scholar]
- Bissell, D.M.; Anderson, K.E.; Bonkovsky, H.L. Porphyria. N. Engl. J. Med. 2017, 377, 2100–2101. [Google Scholar] [CrossRef] [PubMed]
- Besur, S.; Schmeltzer, P.; Bonkovsky, H.L. Acute Porphyrias. J. Emerg. Med. 2015, 49, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Duque-Serrano, L.; Patarroyo-Rodriguez, L.; Gotlib, D.; Molano-Eslava, J.C. Psychiatric Aspects of Acute Porphyria: A Comprehensive Review. Curr. Psychiatry Rep. 2018, 20, 5. [Google Scholar] [CrossRef]
- Floderus, Y.; Sardh, E.; Moller, C.; Andersson, C.; Rejkjaer, L.; Andersson, D.E.; Harper, P. Variations in porphobilinogen and 5-aminolevulinic acid concentrations in plasma and urine from asymptomatic carriers of the acute intermittent porphyria gene with increased porphyrin precursor excretion. Clin. Chem. 2006, 52, 701–707. [Google Scholar] [CrossRef] [Green Version]
- Naik, H.; Stoecker, M.; Sanderson, S.C.; Balwani, M.; Desnick, R.J. Experiences and concerns of patients with recurrent attacks of acute hepatic porphyria: A qualitative study. Mol. Genet. Metab. 2016, 119, 278–283. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, R.; von und zu Fraunberg, M. Molecular and Biochemical Studies of Acute Intermittent Porphyria in 196 Patients and Their Families. Mol. Diagn. Genet. 2002, 48, 1891–1900. [Google Scholar] [CrossRef]
- Schuurmans, M.M.; Schneider-Yin, X.; Rüfenacht, U.B.; Schnyder, C.; Minder, C.E.; Puy, H.; Deybach, J.C.; Minder, E.I. Influence of Age and Gender on the Clinical Expression of Acute Intermittent Porphyria Based on Molecular Study of Porphobilinogen Deaminase Gene Among Swiss Patients. Mol. Med. 2001, 7, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Fraunberg, M.; Pischik, E.; Udd, L.; Kauppinen, R. Clinical and biochemical characteristics and genotype-phenotype correlation in 143 Finnish and Russian patients with acute intermittent porphyria. Medicine 2005, 84, 35–47. [Google Scholar] [CrossRef]
- Kauppinen, R.; Mustajoki, P. Prognosis of acute porphyria: Occurrence of acute attacks, precipitating factors, and associated diseases. Medicine 1992, 71, 1–13. [Google Scholar] [CrossRef]
- Grandchamp, B.; De Verneuil, H.; Beaumont, C.; Chretien, S.; Walter, O.; Nordmann, Y. Tissue-specific expression of porphobilinogen deaminase. Two isoenzymes from a single gene. Eur. J. Biochem. 1987, 162, 105–110. [Google Scholar] [CrossRef]
- Chretien, S.; Dubart, A.; Beaupain, D.; Raich, N.; Grandchampt, B.; Rosa, J.; Goossens, M.; Romeo, P. Alternative transcription and splicing of the human porphobilinogen deaminase gene result either in tissue-specific or in housekeeping expression. Biochemistry 1988, 85, 6–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bung, N.; Roy, A.; Chen, B.; Das, D.; Pradhan, M.; Yasuda, M.; New, M.I.; Desnick, R.J.; Bulusu, G. Human hydroxymethylbilane synthase: Molecular dynamics of the pyrrole chain elongation identifies step-specific residues that cause AIP. Proc. Natl. Acad. Sci. USA 2018, 115, E4071–E4080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, R.; Kolstoe, S.E.; Mohammed, F.; Al, D.B.A.; Mosely, J.E.; Sarwar, M.; Cooper, J.B.; Wood, S.P.; Shoolingin-Jordan, P.M. Structure of human porphobilinogen deaminase at 2.8 A: The molecular basis of acute intermittent porphyria. Biochem. J. 2009, 420, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Hastings, M.L.; Krainer, A.R. Pre-mRNA splicing in the new millennium. Curr. Opin. Cell Biol. 2001, 13, 302–309. [Google Scholar] [CrossRef]
- Maquat, L.E. Defects in RNA Splicing and the Consequence of Shortened Translational Reading Frames. Am. J. Hum. Genet. 1996, 59, 279–286. [Google Scholar]
- Patel, A.A.; Steitz, J.A. Splicing double: Insights from the second spliceosome. Nat. Rev. Mol. Cell Biol. 2003, 4, 960–970. [Google Scholar] [CrossRef]
- Burset, M.; Seledtsov, I.A.; Solovyev, V.V. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000, 4364–4375. [Google Scholar] [CrossRef]
- Maquat, L.E. When cells stop making sense: Effects of nonsense codons on RNA metabolism in vertebrate cells. RNA 1995, 1, 453–465. [Google Scholar]
- Song, G.; Li, Y.; Cheng, C.; Zhao, Y.; Gao, A.; Zhang, R.; Joachimiak, A.; Shaw, N.; Liu, Z.J. Structural insight into acute intermittent porphyria. FASEB J. 2009, 23, 396–404. [Google Scholar] [CrossRef]
- Di Maio, A.; Skuba, A.; Himes, B.T.; Bhagat, S.L.; Hyun, J.K.; Tessler, A.; Bishop, D.; Son, Y.J. In vivo imaging of dorsal root regeneration: Rapid immobilization and presynaptic differentiation at the CNS/PNS border. J. Neurosci. 2011, 31, 4569–4582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, P.M.; Warren, M.J. Evidence for a dipyrromethane cofactor at the catalytic site of E. coli porphobilinogen deaminase. FEBS Lett. 1987, 225, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Pluta, P.; Roversi, P.; Bernardo-Seisdedos, G.; Rojas, A.; Cooper, J.; Gu, S.; Pickersgill, R.; Millet, O. Structural basis of pyrrole polymerization in human porphobilinogen deaminase. Biochim. Biophys. Acta. Gen. Subj. 2018, 1862, 1948–1955. [Google Scholar] [CrossRef] [PubMed]
- Di Pierro, E.; De Canio, M.; Mercadante, R.; Savino, M.; Granata, F.; Tavazzi, D.; Nicolli, A.M.; Trevisan, A.; Marchini, S.; Fustinoni, S. Laboratory Diagnosis of Porphyria. Diagnostics 2021, 11, 1343. [Google Scholar] [CrossRef]
- Unzu, C.; Sampedro, A.; Mauleon, I.; Alegre, M.; Beattie, S.G.; De Salamanca, R.E.; Snapper, J.; Twisk, J.; Petry, H.; Gonzalez-Aseguinolaza, G.; et al. Sustained enzymatic correction by rAAV-mediated liver gene therapy protects against induced motor neuropathy in acute porphyria mice. Mol. Ther. 2011, 19, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, X.; Wu, H.; Peng, H.; Sun, W.; He, B.; Yuan, Z. A novel heterozygous mutation in the HMBS gene in a patient with acute intermittent porphyria and posterior reversible encephalopathy syndrome. Mol. Med. Rep. 2020, 22, 516–524. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Xiao, H.; Xiong, Q.; Wu, C.; Li, P. Two Novel Hydroxymethylbilane Synthase Splicing Mutations Predispose to Acute Intermittent Porphyria. Int. J. Mol. Sci. 2021, 22, 11008. https://doi.org/10.3390/ijms222011008
Zhang Y, Xiao H, Xiong Q, Wu C, Li P. Two Novel Hydroxymethylbilane Synthase Splicing Mutations Predispose to Acute Intermittent Porphyria. International Journal of Molecular Sciences. 2021; 22(20):11008. https://doi.org/10.3390/ijms222011008
Chicago/Turabian StyleZhang, Yanping, Han Xiao, Qiuhong Xiong, Changxin Wu, and Ping Li. 2021. "Two Novel Hydroxymethylbilane Synthase Splicing Mutations Predispose to Acute Intermittent Porphyria" International Journal of Molecular Sciences 22, no. 20: 11008. https://doi.org/10.3390/ijms222011008