
Alterations in ACE and ACE2 Activities and Cardiomyocyte Signaling Underlie Improved Myocardial Function in a Rat Model of Repeated Remote Ischemic Conditioning

 , , ,
, , ,  , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Results
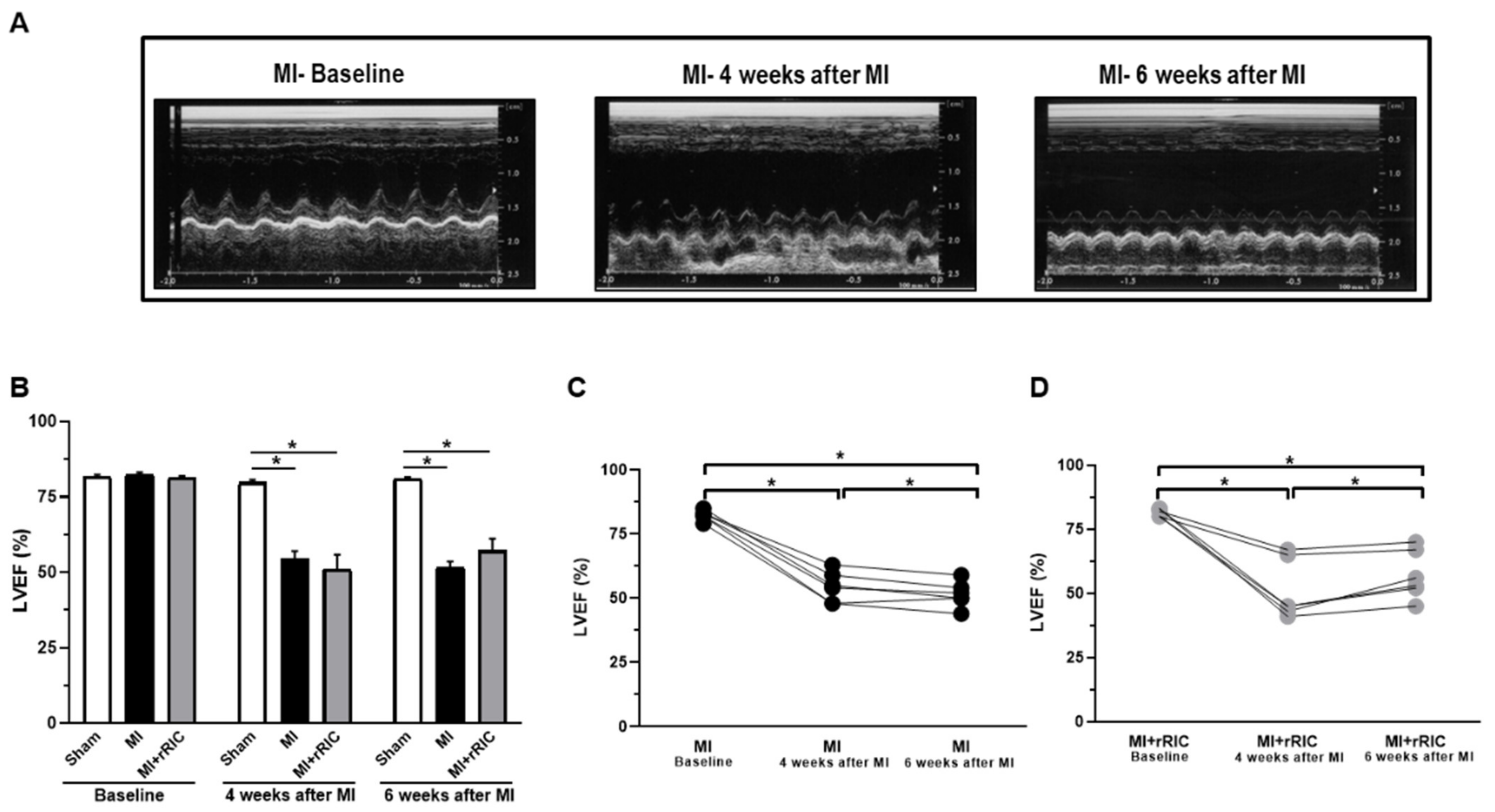
2.1. rRIC Improved LV Systolic Function
2.2. ACE and ACE2 Activities in the Anterior and Inferior LV Areas
2.3. Differences in Ca2+-Regulated Cardiomyocyte Force Production upon rRIC
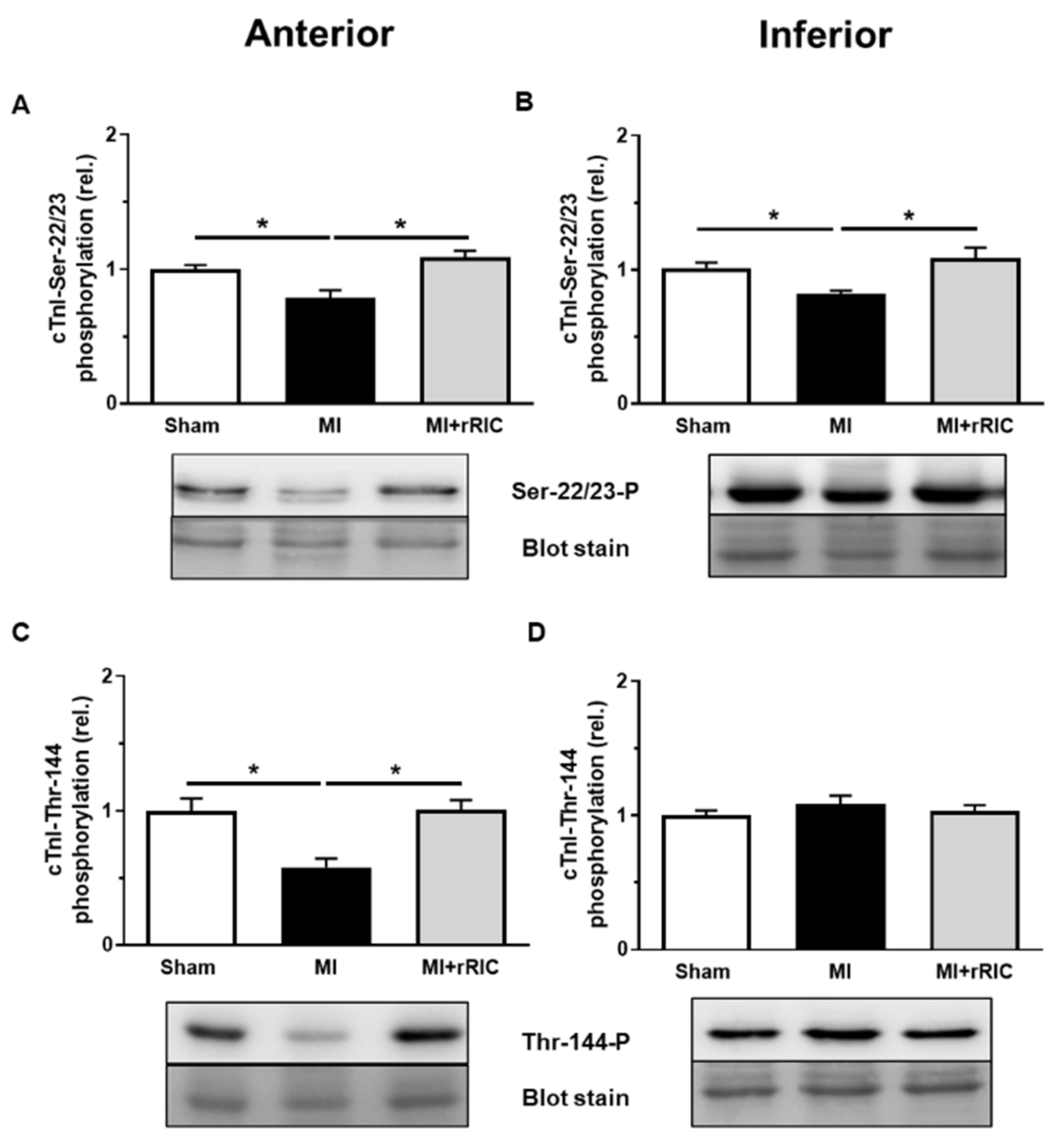
2.4. rRIC Influenced Myofilament Protein Phosphorylation after Ischemic Injury
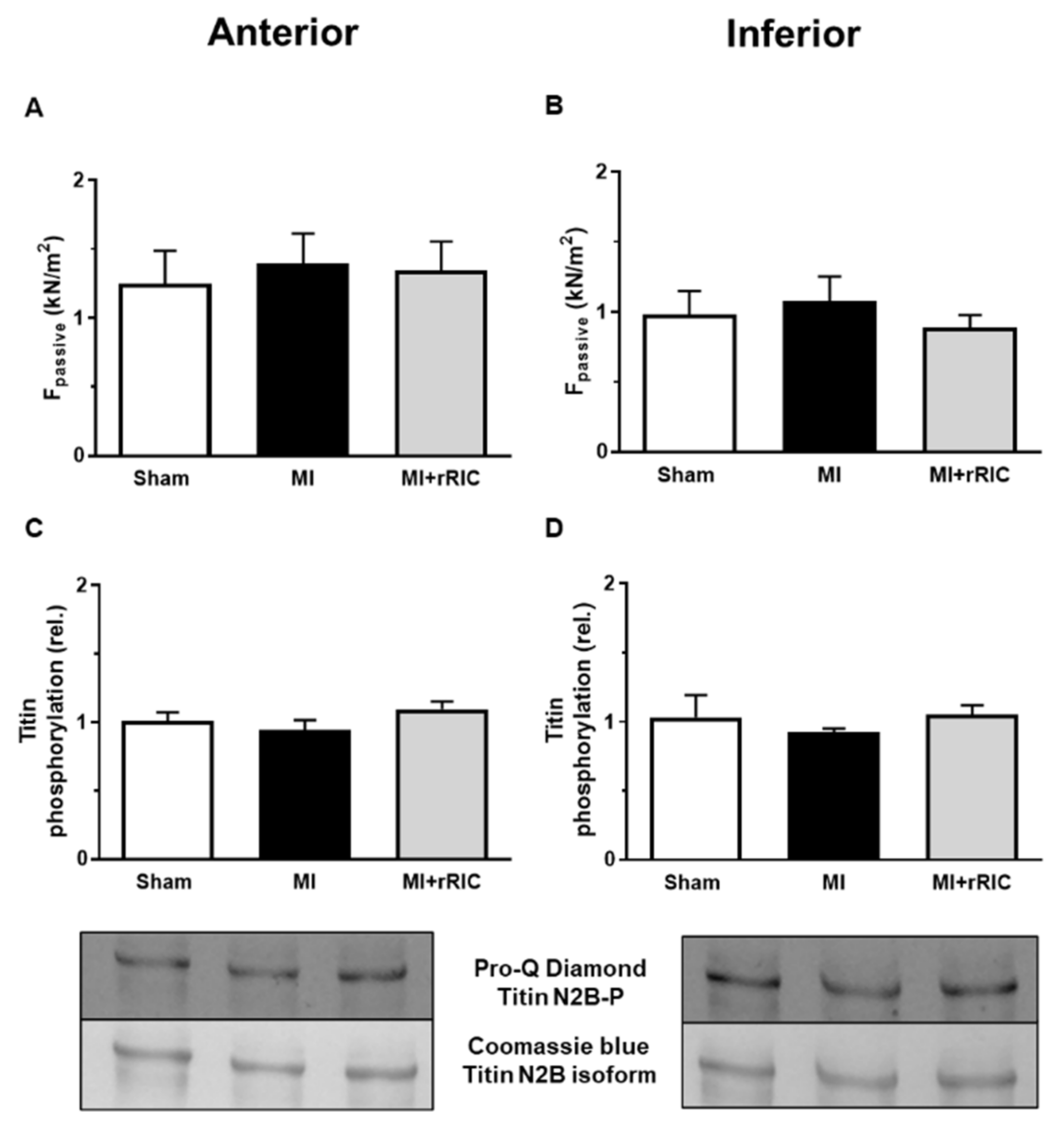
2.5. Titin-Dependent Stiffness of LV Cardiomyocytes Was Not Changed after Myocardial Infarction
3. Discussion
4. Materials and Methods
4.1. Experimental Specimens and Ethical Statement
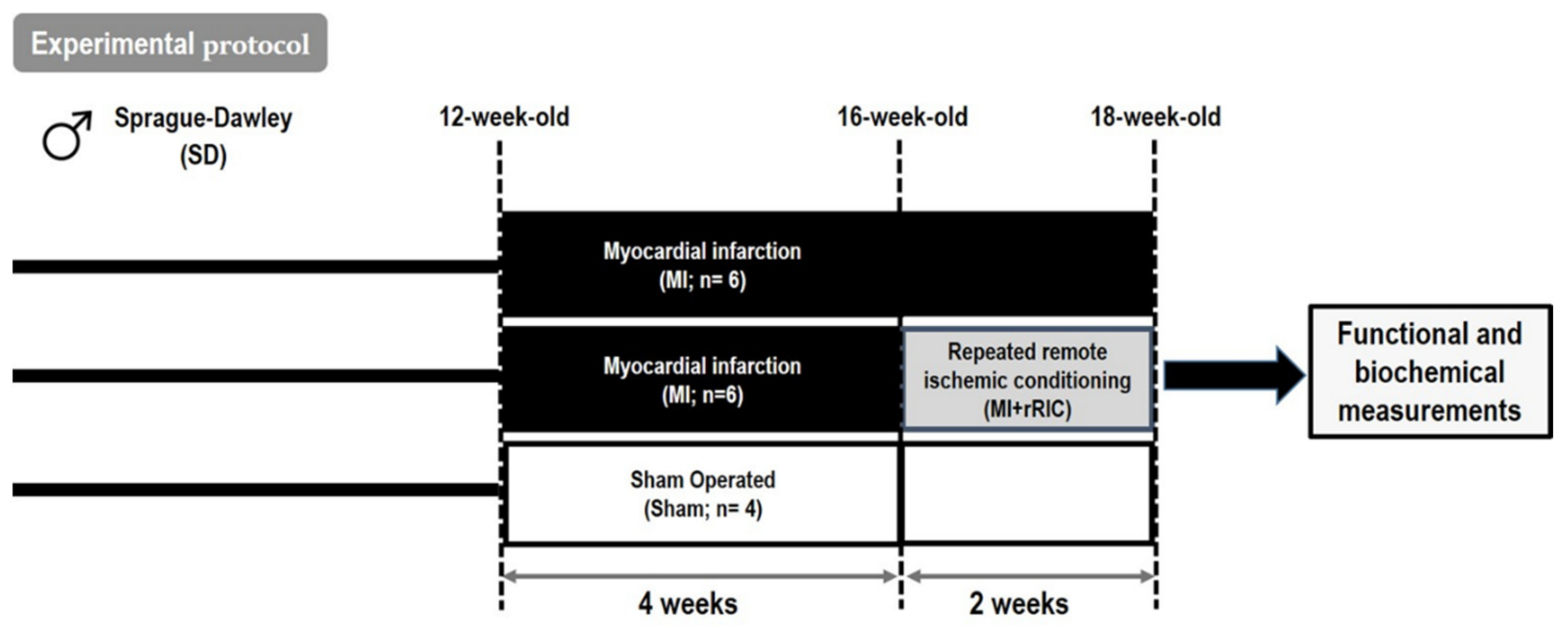
4.2. Experimental Protocol for rRIC
4.3. Transthoracic Echocardiography
4.4. Histological Analyses
4.5. Assessment of ACE and ACE2 Activities
4.6. Contractile Force Measurements in Permeabilized Cardiomyocytes
4.7. Biochemical Analysis of Myofilament Proteins
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bahit, M.C.; Kochar, A.; Granger, C.B. Post-Myocardial Infarction Heart Failure. JACC Heart Fail. 2018, 6, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Sutton, M.G.; Sharpe, N. Left ventricular remodeling after myocardial infarction: Pathophysiology and therapy. Circulation 2000, 101, 2981–2988. [Google Scholar] [CrossRef] [PubMed]
- Velden, V.D.J.; Papp, Z.; Zaremba, R.; Boontje, N.M.; De Jong, J.W.; Owen, V.J.; Burton, P.; Goldmann, P.; Jaquet, K.; Stienen, G. Increased Ca-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc. Res. 2003, 57, 37–47. [Google Scholar] [CrossRef]
- Ruppert, M.; Bódi, B.; Korkmaz-Icöz, S.; Loganathan, S.; Jiang, W.; Lehmann, L.; Oláh, A.; Barta, B.A.; Sayour, A.A.; Merkely, B.; et al. Myofilament Ca2+ sensitivity correlates with left ventricular contractility during the progression of pressure overload-induced left ventricular myocardial hypertrophy in rats. J. Mol. Cell. Cardiol. 2019, 129, 208–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borbeély, A.; van der Velden, J.; Papp, Z.; Bronzwaer, J.G.; Edes, I.; Stienen, G.; Paulus, W.J. Cardiomyocyte Stiffness in Diastolic Heart Failure. Circulation 2005, 111, 774–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heusch, G.; Bøtker, H.E.; Przyklenk, K.; Redington, A.; Yellon, D. Remote ischemic conditioning. J. Am. Coll. Cardiol. 2015, 65, 177–195. [Google Scholar] [CrossRef] [Green Version]
- Kleinbongard, P.; Skyschally, A.; Heusch, G. Cardioprotection by remote ischemic conditioning and its signal transduction. Pflug. Arch. Eur. J. Phy. 2017, 469, 843. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Kharbanda, R.K.; Møller, U.K.; Ramlall, M.; Aarøe, J.; Butler, R.; Bulluck, H.; Clayton, T.; Dana, A.; Dodd, M.; et al. Effect of remote ischaemic conditioning on clinical outcomes in patients with acute myocardial infarction (CONDI-2/ERIC-PPCI): A single-blind randomised controlled trial. Lancet 2019, 394, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Pilz, P.M.; Hamza, O.; Gidlöf, O.; Gonçalves, I.F.; Tretter, E.V.; Trojanek, S.; Abraham, D.; Heber, S.; Haller, P.M.; Podesser, B.K.; et al. Remote ischemic perconditioning attenuates adverse cardiac remodeling and preserves left ventricular function in a rat model of reperfused myocardial infarction. Int. J. Cardiol. 2019, 285, 72–79. [Google Scholar] [CrossRef]
- Ikonomidis, I.; Vlastos, D.; Andreadou, I.; Gazouli, M.; Efentakis, P.; Varoudi, M.; Makavos, G.; Kapelouzou, A.; Lekakis, J.; Parissis, J.; et al. Vascular conditioning prevents adverse left ventricular remodelling after acute myocardial infarction: A randomised remote conditioning study. Basic Res. Cardiol. 2021, 116, 1–14. [Google Scholar] [CrossRef]
- Heusch, G. Molecular basis of cardioprotection: Signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef]
- Fukuda, S.; Kono, Y.; Hanatani, A.; Nakanishi, K.; Otsuka, K.; Taguchi, H.; Shimada, K. Remote ischemic conditioning improves coronary microcirculation in healthy subjects and patients with heart failure. Drug Des. Dev. Ther. 2014, 8, 1175–1181. [Google Scholar] [CrossRef] [Green Version]
- Portes, L.A.; Saraiva, R.M.; Dos Santos, A.A.; Tucci, P.J. Swimming Training Attenuates Remodeling, Contractile Dysfunction and Congestive Heart Failure in Rats with Moderate and Large Myocardial Infarctions. Clin. Exp. Pharmacol. Physiol. 2009, 36, 394–399. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Schuijt, M.P.; Danser, A.H.J. Cardiac angiotensin II: An intracrine hormone? Am. J. Hypertens. 2002, 15, 1109–1116. [Google Scholar] [CrossRef] [Green Version]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- He, Q.; Wang, F.; Ryan, T.D.; Chalasani, M.; Redington, A.N. Repeated Remote Ischemic Conditioning Reduces Doxorubicin-Induced Cardiotoxicity. JACC CardioOncol. 2020, 2, 41–52. [Google Scholar] [CrossRef]
- Regulski, M.; Greenwood, T.; Leschinsky, B. Impact of repeated remote ischemic conditioning on diabetic foot ulcers: A proof-of-concept study. Wound Repair Regen. 2021, 29, 853–858. [Google Scholar] [CrossRef]
- Schirone, L.; Forte, M.; Palmerio, S.; Yee, D.; Nocella, C.; Angelini, F.; Pagano, F.; Schiavon, S.; Bordin, A.; Carrizzo, A.; et al. A Review of the Molecular Mechanisms Underlying the Development and Progression of Cardiac Remodeling. Oxidative Med. Cell. Longev. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Traverse, J.H.; Swingen, C.M.; Henry, T.D.; Fox, J.; Wang, Y.L.; Chavez, I.J.; Lips, D.L.; Lesser, J.R.; Pedersen, W.R.; Burke, N.M.; et al. NHLBI-Sponsored Randomized Trial of Postconditioning During Primary Percutaneous Coronary Intervention for ST-Elevation Myocardial Infarction. Circ. Res. 2019, 124, 769–778. [Google Scholar] [CrossRef]
- Li, Y.; Xu, Q.; Ma, L.; Wu, D.; Gao, J.; Chen, G.; Li, H. Systematic profiling of ACE2 expression in diverse physiological and pathological conditions for COVID-19/SARS-CoV-2. J. Cell. Mol. Med. 2020, 24, 9478–9482. [Google Scholar] [CrossRef]
- Úri, K.; Fagyas, M.; Kertész, A.; Borbély, A.; Jenei, C.; Bene, O.; Csanádi, Z.; Paulus, W.J.; Édes, I.; Papp, Z.; et al. Circulating ACE2 activity correlates with cardiovascular disease development. J. Renin-Angiotensin-Aldosterone Syst. 2016, 17, 4. [Google Scholar] [CrossRef] [Green Version]
- Santer, D.; Nagel, F.; Gonçalves, I.F.; Kaun, C.; Wojta, J.; Fagyas, M.; Krššák, M.; Balogh, Á.; Papp, Z.; Tóth, A.; et al. Tenascin-C aggravates ventricular dilatation and angiotensin-converting enzyme activity after myocardial infarction in mice. ESC Heart Fail. 2020, 7, 2113–2122. [Google Scholar] [CrossRef]
- Fagyas, M.; Kertész, A.; Siket, I.M.; Bánhegyi, V.; Kracskó, B.; Szegedi, A.; Szokol, M.; Vajda, G.; Rácz, I.; Gulyás, H.; et al. Level of the SARS-CoV-2 receptor ACE2 activity is highly elevated in old-aged patients with aortic stenosis: Implications for ACE2 as a biomarker for the severity of COVID-19. Geroscience 2021, 43, 19–29. [Google Scholar] [CrossRef]
- Úri, K.; Fagyas, M.; Mányiné Siket, I.; Kertész, A.; Csanádi, Z.; Sándorfi, G.; Clemens, M.; Fedor, R.; Papp, Z.; Édes, I.; et al. New perspectives in the renin-angiotensin-aldosterone system (RAAS) IV: Circulating ACE2 as a biomarker of systolic dysfunction in human hypertension and heart failure. PLoS ONE 2014, 9, e87845. [Google Scholar] [CrossRef]
- Bánhegyi, V.; Enyedi, A.; Fülöp, G.; Oláh, A.; Siket, I.; Váradi, C.; Bottyán, K.; Lódi, M.; Csongrádi, A.; Umar, A.; et al. Human Tissue Angiotensin Converting Enzyme (ACE) Activity Is Regulated by Genetic Polymorphisms, Posttranslational Modifications, Endogenous Inhibitors and Secretion in the Serum, Lungs and Heart. Cells 2021, 10, 1708. [Google Scholar] [CrossRef] [PubMed]
- Fagyas, M.; Bánhegyi, V.; Úri, K.; Enyedi, A.; Lizanecz, E.; Mányiné Siket, I.; Mártha, L.; Fülöp, G.Á.; Radovits, T.; Pólos, M.; et al. Changes in the SARS-CoV-2 cellular receptor ACE2 levels in cardiovascular patients: A potential biomarker for the stratification of COVID-19 patients. GeroScience 2021. accepted by publisher. [Google Scholar]
- Messer, A.E.; Jacques, A.M.; Marston, S.B. Troponin phosphorylation and regulatory function in human heart muscle: Dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J. Mol. Cell. Cardiol. 2007, 42, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Kunst, G.; Kress, K.R.; Gruen, M.; Uttenweiler, D.; Gautel, M.; Fink, R.H.A. Myosin Binding Protein C, a Phosphorylation-Dependent Force Regulator in Muscle That Controls the Attachment of Myosin Heads by Its Interaction With Myosin S2. Circ. Res. 2000, 86, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnam, S.; Sevrieva, I.; Sun, Y.-B.; Irving, M.; Kampourakis, T. Site-specific phosphorylation of myosin binding protein-C coordinates thin and thick filament activation in cardiac muscle. Proc. Natl. Acad. Sci. USA 2019, 116, 15485–15494. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Grant, J.E.; Doede, C.M.; Sadayappan, S.; Robbins, J.; Walker, J.W. PKC-betaII sensitizes cardiac myofilaments to Ca2+ by phosphorylating troponin I on threonine-144. J. Mol. Cell. Cardiol. 2006, 41, 823–833. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Gedik, N.; Kirca, M.; Stoian, L.; Frey, U.; Zandi, A.; Thielmann, M.; Jakob, H.; Peters, J.; Kamler, M.; et al. Mitochondrial and Contractile Function of Human Right Atrial Tissue in Response to Remote Ischemic Conditioning. J. Am. Heart Assoc. 2018, 7, e009540. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.; Mehr, A.P.; Kreutz, R. Physiology of Local Renin-Angiotensin Systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef]
- Hirsch, A.T.; Talsness, C.E.; Schunkert, H.; Paul, M.; Dzau, V.J. Tissue-specific activation of cardiac angiotensin converting enzyme in experimental heart failure. Circ. Res. 1991, 69, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Inserte, J.; Hernando, V.; Ruiz-Meana, M.; Poncelas-Nozal, M.; Fernández, C.; Agulló, L.; Sartorio, C.; Vilardosa, Ú.; Garcia-Dorado, D. Delayed phospholamban phosphorylation in post-conditioned heart favours Ca2+ normalization and contributes to protection. Cardiovasc. Res. 2014, 103, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandy, P.; Hausenloy, D.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of Risk Factors, Comorbidities, and Comedications with Ischemia/Reperfusion Injury and Cardioprotection by Preconditioning, Postconditioning, and Remote Conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef] [PubMed]
- Osmanagic-Myers, S.; Kiss, A.; Manakanatas, C.; Hamza, O.; Sedlmayer, F.; Szabo, P.L.; Fischer, I.; Fichtinger, P.; Podesser, B.K.; Eriksson, M.; et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J. Clin. Investig. 2018, 129, 531–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves, I.F.; Acar, E.; Costantino, S.; Szabo, P.L.; Hamza, O.; Tretter, E.V.; Klein, K.U.; Trojanek, S.; Abraham, D.; Paneni, F.; et al. Epigenetic modulation of tenascin C in the heart: Implications on myocardial ischemia, hypertrophy and metabolism. J. Hypertens. 2019, 37, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Ghotbi, A.A.; Clemmensen, A.; Kyhl, K.; Follin, B.; Hasbak, P.; Engstrøm, T.; Ripa, R.S.; Kjaer, A. Rubidium-82 PET imaging is feasible in a rat myocardial infarction model. J. Nucl. Cardiol. 2017, 26, 798–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabó, P.L.; Ebner, J.; Koenig, X.; Hamza, O.; Watzinger, S.; Trojanek, S.; Abraham, D.; Todt, H.; Kubista, H.; Schicker, K.; et al. Cardiovascular phenotype of the Dmd(mdx) rat—A suitable animal model for Duchenne muscular dystrophy. Dis. Model. Mech. 2021, 14, dmm047704. [Google Scholar] [CrossRef]
- Fagyas, M.; Úri, K.; Siket, I.M.; Fülöp, G.Á.; Csató, V.; Daragó, A.; Boczán, J.; Bányai, E.; Szentkirályi, I.E.; Maros, T.M.; et al. New Perspectives in the Renin-Angiotensin-Aldosterone System (RAAS) II: Albumin Suppresses Angiotensin Converting Enzyme (ACE) Activity in Human. PLoS ONE 2014, 9, e87844. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter/Group | Sham | MI | MI + rRIC |
|---|---|---|---|
| n | 4 | 6 | 6 |
| BW (g) | 448 ± 13 | 448 ± 5 | 439 ± 5 |
| HW (g) | 1.37 ± 0.02 | 1.78 ± 0.06 * | 1.62 ± 0.02 * |
| HW/BW (g) | 2.97 ± 0.10 | 3.80 ± 0.15 * | 3.70 ± 0.05 * |
| infarct size (%) | n.a. | 21.72 ± 1.17 * | 22.02 ± 1.78 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bódi, B.; Pilz, P.M.; Mártha, L.; Lang, M.; Hamza, O.; Fagyas, M.; Szabó, P.L.; Abraham, D.; Tóth, A.; Podesser, B.K.; et al. Alterations in ACE and ACE2 Activities and Cardiomyocyte Signaling Underlie Improved Myocardial Function in a Rat Model of Repeated Remote Ischemic Conditioning. Int. J. Mol. Sci. 2021, 22, 11064. https://doi.org/10.3390/ijms222011064
Bódi B, Pilz PM, Mártha L, Lang M, Hamza O, Fagyas M, Szabó PL, Abraham D, Tóth A, Podesser BK, et al. Alterations in ACE and ACE2 Activities and Cardiomyocyte Signaling Underlie Improved Myocardial Function in a Rat Model of Repeated Remote Ischemic Conditioning. International Journal of Molecular Sciences. 2021; 22(20):11064. https://doi.org/10.3390/ijms222011064
Chicago/Turabian StyleBódi, Beáta, Patrick M. Pilz, Lilla Mártha, Miriam Lang, Ouafa Hamza, Miklós Fagyas, Petra L. Szabó, Dietmar Abraham, Attila Tóth, Bruno K. Podesser, and et al. 2021. "Alterations in ACE and ACE2 Activities and Cardiomyocyte Signaling Underlie Improved Myocardial Function in a Rat Model of Repeated Remote Ischemic Conditioning" International Journal of Molecular Sciences 22, no. 20: 11064. https://doi.org/10.3390/ijms222011064
APA StyleBódi, B., Pilz, P. M., Mártha, L., Lang, M., Hamza, O., Fagyas, M., Szabó, P. L., Abraham, D., Tóth, A., Podesser, B. K., Kiss, A., & Papp, Z. (2021). Alterations in ACE and ACE2 Activities and Cardiomyocyte Signaling Underlie Improved Myocardial Function in a Rat Model of Repeated Remote Ischemic Conditioning. International Journal of Molecular Sciences, 22(20), 11064. https://doi.org/10.3390/ijms222011064