
OSMI-1 Enhances TRAIL-Induced Apoptosis through ER Stress and NF-κB Signaling in Colon Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
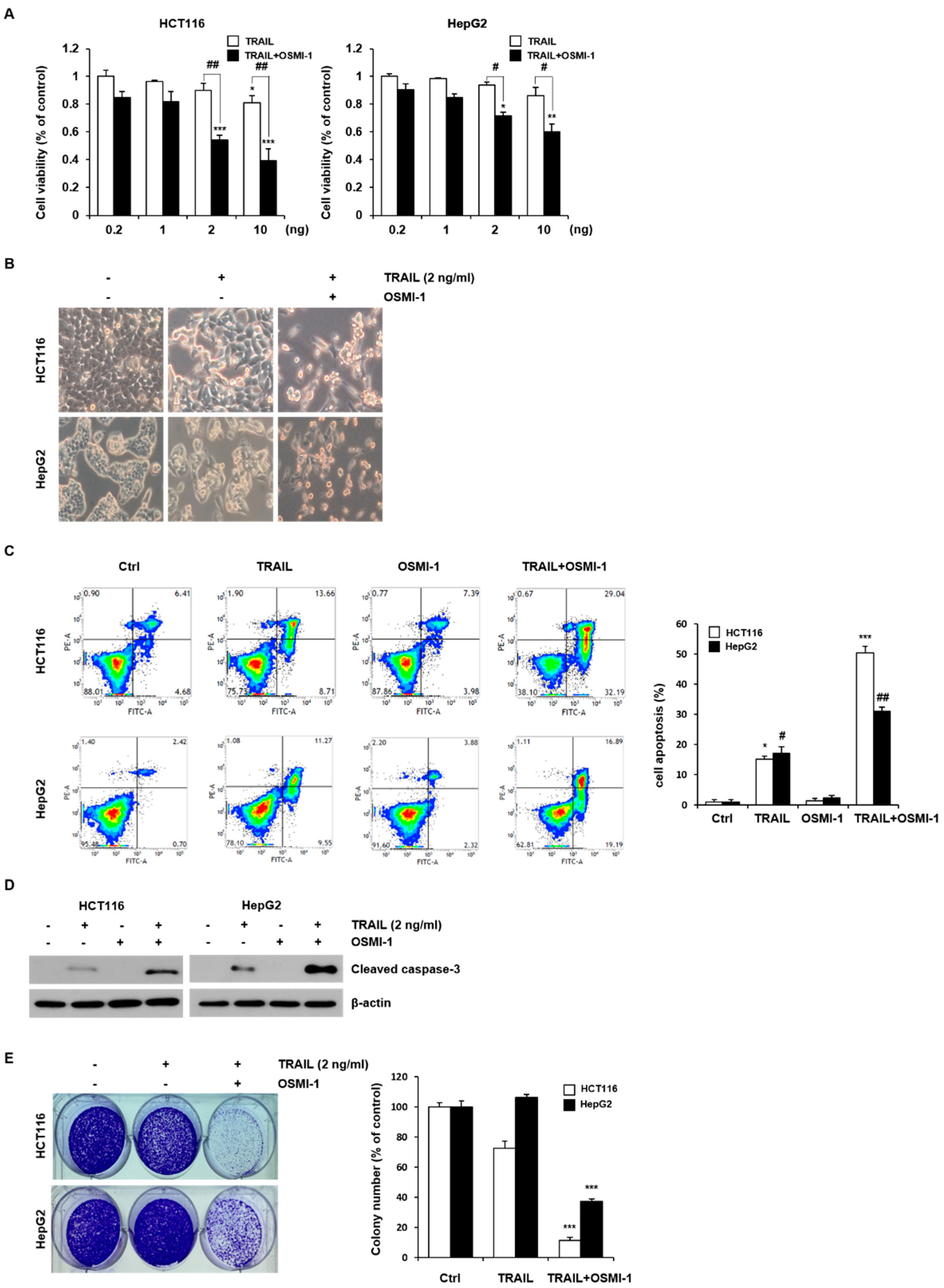
2.1. Combination Treatment with TRAIL and OSMI-1 in HCT116 and HepG2 Cells
2.2. TRAIL-Induced Apoptosis via Caspase-8 Is Enhanced by OSMI-1
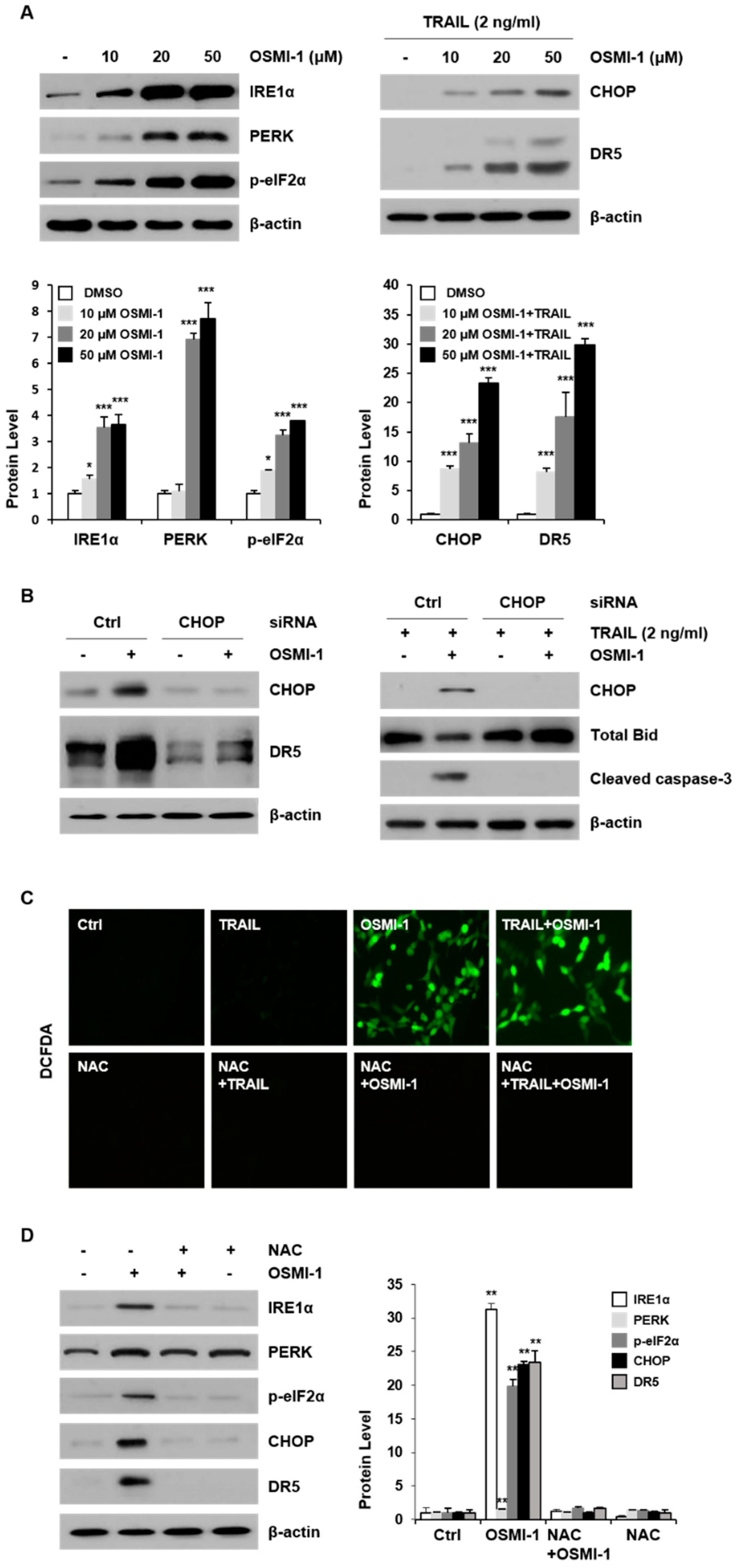
2.3. OSMI-1 Enhances TRAIL-Induced Apoptosis through ER Stress-Mediated CHOP/DR5
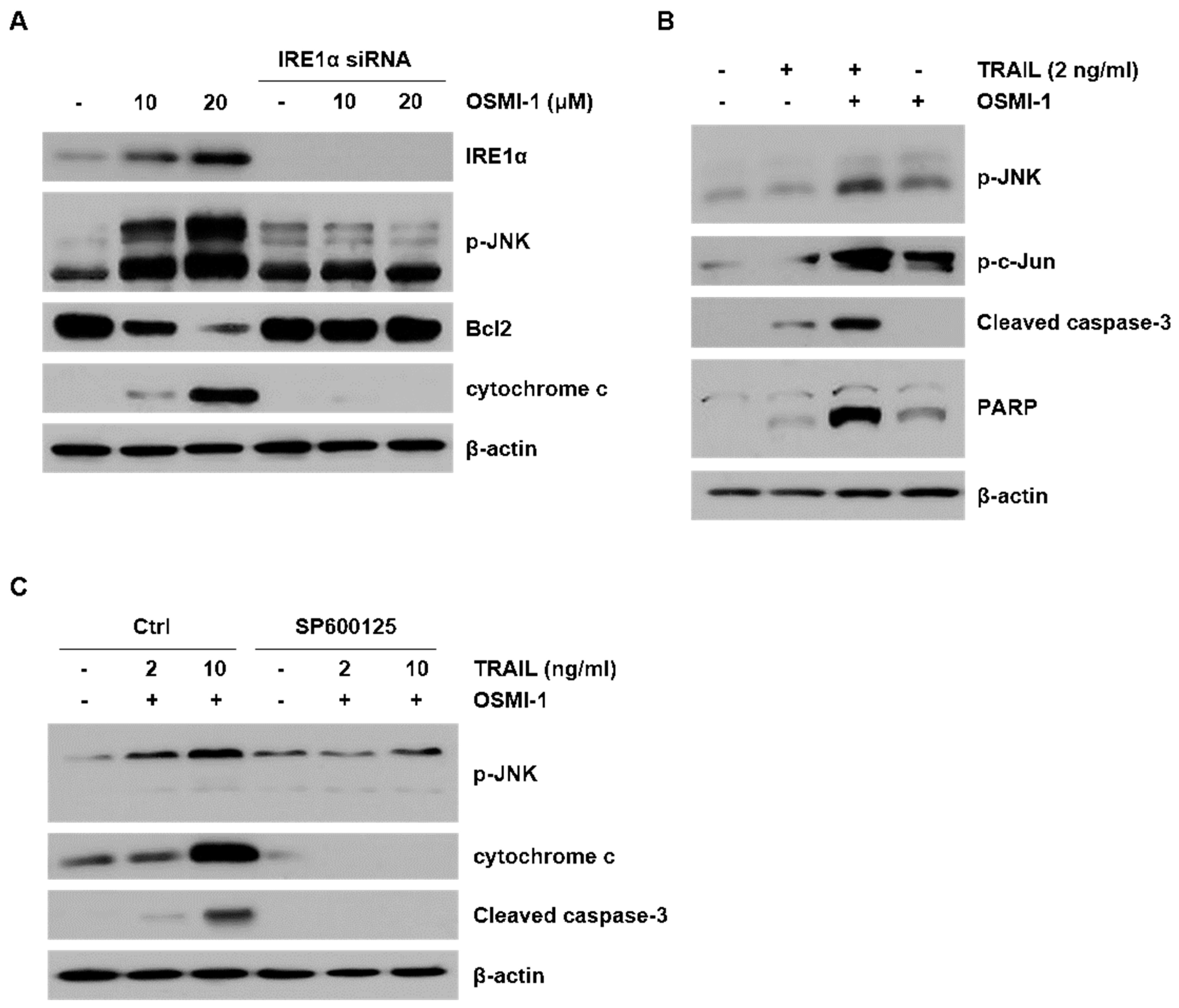
2.4. ER Stress-Induced Apoptosis though JNK Activation by OSMI-1
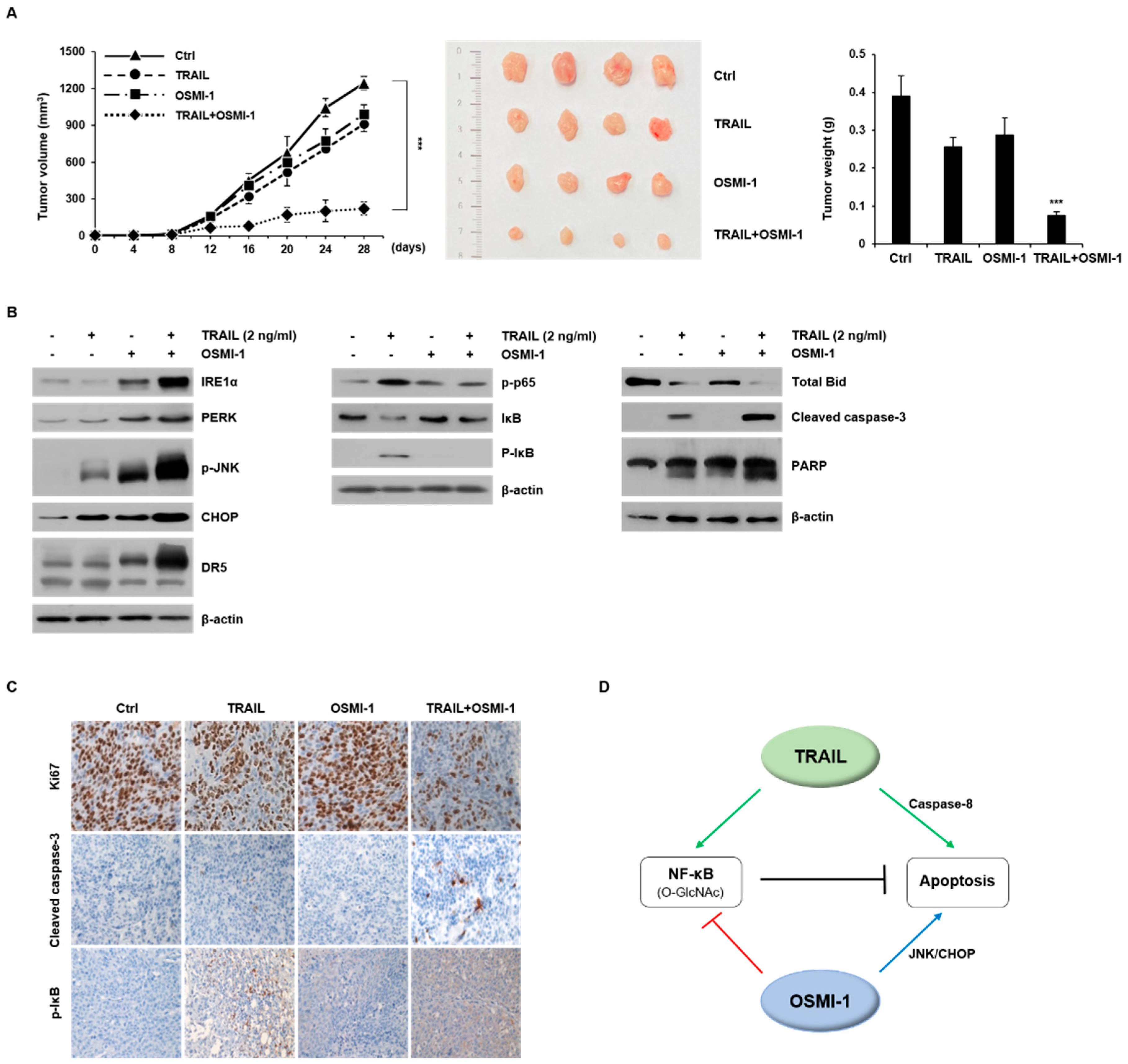
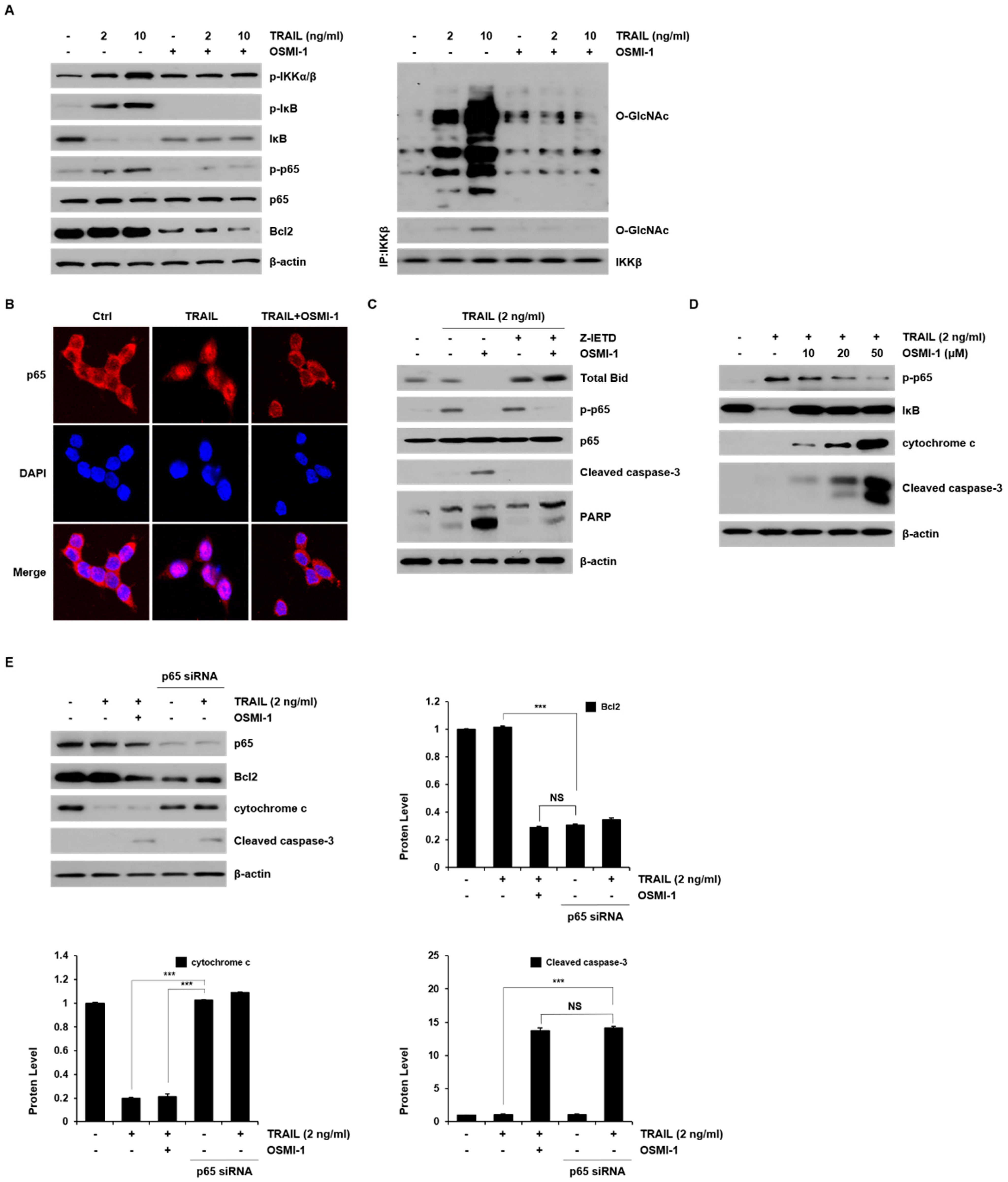
2.5. Modulation of TRAIL-Induced NF-κB Signaling by OSMI-1
2.6. Combination Treatment Synergistically Enhances Anticancer Activity on HCT116 Xenograft in Nude Mice
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Preparation of Cell Lysates and Western Blot Analysis
4.3. Cell Viability Assay
4.4. Annexin V/Fluorescein Isothiocyanate (FITC) Flow Cytometric Assay
4.5. Measurement of the Accumulation of ROS
4.6. Immunoprecipitation (IP)
4.7. Colony Formation Assay
4.8. Tumor Implantation and Growth
4.9. Immunohistochemistry
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef]
- Walczak, H.; Degli-Esposti, M.A.; Johnson, R.S.; Smolak, P.J.; Waugh, J.Y.; Boiani, N.; Timour, M.S.; Gerhart, M.J.; Schooley, K.A.; Smith, C.A.; et al. TRAIL-R2: A novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997, 16, 5386–5397. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Pop, C.; Tremblay, A.G.; Blais, V.; Denault, J.B.; Salvesen, G.S.; Green, D.R. Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J. Biol. Chem. 2010, 285, 16632–16642. [Google Scholar] [CrossRef] [PubMed]
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Kristensson, M.; Melander, F.; Leandersson, K.; Ronnstrand, L.; Wernstedt, C.; Andersson, T. p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J. Exp. Med. 2004, 199, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Billen, L.P.; Shamas-Din, A.; Andrews, D.W. Bid: A Bax-like BH3 protein. Oncogene 2008, 27 (Suppl. 1), S93–S104. [Google Scholar] [CrossRef]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94, 481–490. [Google Scholar] [CrossRef]
- Wei, M.C.; Lindsten, T.; Mootha, V.K.; Weiler, S.; Gross, A.; Ashiya, M.; Thompson, C.B.; Korsmeyer, S.J. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genome Res. 2000, 14, 2060–2071. [Google Scholar]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, J.Y.; Wei, W.Z.; Wu, G.S. Activation of the Akt survival pathway contributes to TRAIL resistance in cancer cells. PLoS ONE 2010, 5, e10226. [Google Scholar] [CrossRef] [PubMed]
- Goncharenko-Khaider, N.; Lane, D.; Matte, I.; Rancourt, C.; Piche, A. The inhibition of Bid expression by Akt leads to resistance to TRAIL-induced apoptosis in ovarian cancer cells. Oncogene 2010, 29, 5523–5536. [Google Scholar] [CrossRef] [PubMed]
- Cardoso Alves, L.; Corazza, N.; Micheau, O.; Krebs, P. The multifaceted role of TRAIL signaling in cancer and immunity. FEBS J. 2020, 288, 5530–5554. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Scheidereit, C. The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Mizrachi, L.; Lovly, C.M.; Ratner, L. The role of NF-{kappa}B-1 and NF-{kappa}B-2-mediated resistance to apoptosis in lymphomas. Proc. Natl. Acad. Sci. USA 2006, 103, 9220–9225. [Google Scholar] [CrossRef]
- Yang, W.H.; Park, S.Y.; Nam, H.W.; Kim, D.H.; Kang, J.G.; Kang, E.S.; Kim, Y.S.; Lee, H.C.; Kim, K.S.; Cho, J.W. NFkappaB activation is associated with its O-GlcNAcylation state under hyperglycemic conditions. Proc. Natl. Acad. Sci. USA 2008, 105, 17345–17350. [Google Scholar] [CrossRef]
- Ngoh, G.A.; Facundo, H.T.; Zafir, A.; Jones, S.P. O-GlcNAc signaling in the cardiovascular system. Circ. Res. 2010, 107, 171–185. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell. Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell. Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Luna, C.; Mendoza, N.; Casao, A.; Perez-Pe, R.; Cebrian-Perez, J.A.; Muino-Blanco, T. c-Jun N-terminal kinase and p38 mitogen-activated protein kinase pathways link capacitation with apoptosis and seminal plasma proteins protect sperm by interfering with both routesdagger. Biol. Reprod. 2017, 96, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Chou, C.W.; Chang, Y.F.; Chen, C.C. Proteasome inhibitors enhance TRAIL-induced apoptosis through the intronic regulation of DR5: Involvement of NF-kappa B and reactive oxygen species-mediated p53 activation. J. Immunol. 2008, 180, 8030–8039. [Google Scholar] [CrossRef]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. Loss of p53 enhances catalytic activity of IKKbeta through O-linked beta-N-acetyl glucosamine modification. Proc. Natl. Acad. Sci. USA 2009, 106, 3431–3436. [Google Scholar] [CrossRef]
- Yamanaka, T.; Shiraki, K.; Sugimoto, K.; Ito, T.; Fujikawa, K.; Ito, M.; Takase, K.; Moriyama, M.; Nakano, T.; Suzuki, A. Chemotherapeutic agents augment TRAIL-induced apoptosis in human hepatocellular carcinoma cell lines. Hepatology 2000, 32, 482–490. [Google Scholar] [CrossRef]
- Vignati, S.; Codegoni, A.; Polato, F.; Broggini, M. Trail activity in human ovarian cancer cells: Potentiation of the action of cytotoxic drugs. Eur. J. Cancer 2002, 38, 177–183. [Google Scholar] [CrossRef]
- Clarke, P.; Meintzer, S.M.; Spalding, A.C.; Johnson, G.L.; Tyler, K.L. Caspase 8-dependent sensitization of cancer cells to TRAIL-induced apoptosis following reovirus-infection. Oncogene 2001, 20, 6910–6919. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.C.; Ruiz-Magana, M.J.; Ruiz-Ruiz, C. Regulation of the resistance to TRAIL-induced apoptosis in human primary T lymphocytes: Role of NF-kappaB inhibition. Mol. Immunol. 2007, 44, 2587–2597. [Google Scholar] [CrossRef]
- Kim, Y.S.; Schwabe, R.F.; Qian, T.; Lemasters, J.J.; Brenner, D.A. TRAIL-mediated apoptosis requires NF-kappaB inhibition and the mitochondrial permeability transition in human hepatoma cells. Hepatology 2002, 36, 1498–1508. [Google Scholar] [CrossRef]
- Karacay, B.; Sanlioglu, S.; Griffith, T.S.; Sandler, A.; Bonthius, D.J. Inhibition of the NF-kappaB pathway enhances TRAIL-mediated apoptosis in neuroblastoma cells. Cancer. Gene. Ther. 2004, 11, 681–690. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar] [CrossRef]
- Junjappa, R.P.; Patil, P.; Bhattarai, K.R.; Kim, H.R.; Chae, H.J. IRE1alpha Implications in Endoplasmic Reticulum Stress-Mediated Development and Pathogenesis of Autoimmune Diseases. Front. Immunol. 2018, 9, 1289. [Google Scholar] [CrossRef]
- Kato, H.; Nakajima, S.; Saito, Y.; Takahashi, S.; Katoh, R.; Kitamura, M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 2012, 19, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes. Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef]
- James, L.R.; Tang, D.; Ingram, A.; Ly, H.; Thai, K.; Cai, L.; Scholey, J.W. Flux through the hexosamine pathway is a determinant of nuclear factor kappaB- dependent promoter activation. Diabetes 2002, 51, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Golks, A.; Tran, T.T.; Goetschy, J.F.; Guerini, D. Requirement for O-linked N-acetylglucosaminyltransferase in lymphocytes activation. EMBO J. 2007, 26, 4368–4379. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.-J.; Lee, D.-E.; Choi, S.-Y.; Kwon, O.-S. OSMI-1 Enhances TRAIL-Induced Apoptosis through ER Stress and NF-κB Signaling in Colon Cancer Cells. Int. J. Mol. Sci. 2021, 22, 11073. https://doi.org/10.3390/ijms222011073
Lee S-J, Lee D-E, Choi S-Y, Kwon O-S. OSMI-1 Enhances TRAIL-Induced Apoptosis through ER Stress and NF-κB Signaling in Colon Cancer Cells. International Journal of Molecular Sciences. 2021; 22(20):11073. https://doi.org/10.3390/ijms222011073
Chicago/Turabian StyleLee, Su-Jin, Da-Eun Lee, Soo-Young Choi, and Oh-Shin Kwon. 2021. "OSMI-1 Enhances TRAIL-Induced Apoptosis through ER Stress and NF-κB Signaling in Colon Cancer Cells" International Journal of Molecular Sciences 22, no. 20: 11073. https://doi.org/10.3390/ijms222011073
APA StyleLee, S.-J., Lee, D.-E., Choi, S.-Y., & Kwon, O.-S. (2021). OSMI-1 Enhances TRAIL-Induced Apoptosis through ER Stress and NF-κB Signaling in Colon Cancer Cells. International Journal of Molecular Sciences, 22(20), 11073. https://doi.org/10.3390/ijms222011073