
An Open Question: Is Non-Ionizing Radiation a Tool for Controlling Apoptosis-Induced Proliferation?


Abstract
:1. Introduction
2. Non-Ionizing Radiation and Control of Cellular Processes
3. Apoptosis and Control of Tissue Growth
4. Potentials of Non-Ionizing Radiation to Control Tissue Growth
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Belpomme, D.; Hardell, L.; Belyaev, I.; Burgio, E.; Carpenter, D.O. Thermal and non-thermal health effects of low intensity non-ionizing radiation: An international perspective. Environ. Pollut. 2018, 242, 643–658. [Google Scholar] [CrossRef]
- Gupta, S.; Sharma, R.S.; Singh, R. Non-ionizing radiation as possible carcinogen. Int. J. Environ. Health Res. 2020, 1–25. [Google Scholar] [CrossRef]
- Zablotskii, V.; Syrovets, T.; Schmidt, Z.W.; Dejneka, A.; Simmet, T. Modulation of monocytic leukemia cell function and survival by high gradient magnetic fields and mathematical modeling studies. Biomaterials 2014, 35, 3164–3171. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X. Magnetic Fields and Reactive Oxygen Species. Int. J. Mol. Sci. 2017, 18, 2175. [Google Scholar] [CrossRef] [Green Version]
- Novoselova, E.G.; Novikov, V.V.; Lunin, S.M.; Glushkova, O.V.; Novoselova, T.V.; Parfenyuk, S.B.; Novoselov, S.V.; Khrenov, M.O.; Fesenko, E.E. Effects of low-level combined static and weak low-frequency alternating magnetic fields on cytokine production and tumor development in mice. Electromagn. Biol. Med. 2019, 38, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Free radicals and antioxidants—Quo vadis? Trends Pharmacol. Sci. 2011, 32, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, F.S.; Greenebaum, B. The effects of weak magnetic fields on radical pairs. Bioelectromagnetics 2015, 36, 45–54. [Google Scholar] [CrossRef]
- Gurhan, H.; Bruzon, R.; Kandala, S.; Greenebaum, B.; Barnes, F. Effects Induced by a Weak Static Magnetic Field of Different Intensities on HT-1080 Fibrosarcoma Cells. Bioelectromagnetics 2021, 42, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Van Huizen, A.V.; Morton, J.M.; Kinsey, L.J.; Von Kannon, D.G.; Saad, M.A.; Birkholz, T.R.; Czajka, J.M.; Cyrus, J.; Barnes, F.S.; Beane, W.S. Weak magnetic fields alter stem cell-mediated growth. Sci. Adv. 2019, 5, eaau7201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.Q.; Wang, C.; Lu, D.F.; Zhao, X.D.; Tan, L.H.; Chen, X. Induction of apoptosis and ferroptosis by a tumor suppressing magnetic field through ROS-mediated DNA damage. Aging 2020, 12, 3662–3681. [Google Scholar] [CrossRef]
- Lee, S.K.; Park, S.; Gimm, Y.M.; Kim, Y.W. Extremely low frequency magnetic fields induce spermatogenic germ cell apoptosis: Possible mechanism. Biomed. Res. Int. 2014, 2014, 567183. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Chi, H.; Chang, H.Y.; Sang, T.K. Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3082. [Google Scholar] [CrossRef] [Green Version]
- Malemud, C.J. Defective T-Cell Apoptosis and T-Regulatory Cell Dysfunction in Rheumatoid Arthritis. Cells 2018, 7, 223. [Google Scholar] [CrossRef] [Green Version]
- Oyaizu, N.; Pahwa, S. Role of apoptosis in HIV disease pathogenesis. J. Clin. Immunol. 1995, 15, 217–231. [Google Scholar] [CrossRef]
- Fung, T.S.; Liu, D.X. Coronavirus infection, ER stress, apoptosis and innate immunity. Front. Microbiol. 2014, 5, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Wang, Y.; Liu, L.; Zhang, X.; Jiang, S.; Wang, B. Apoptosis Disorder, a Key Pathogenesis of HCMV-Related Diseases. Int. J. Mol. Sci. 2021, 22, 4106. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayers, T.J. Targeting the extrinsic apoptosis signaling pathway for cancer therapy. Cancer Immunol. Immunother. 2011, 60, 1173–1180. [Google Scholar] [CrossRef]
- Fulda, S. Targeting extrinsic apoptosis in cancer: Challenges and opportunities. Semin. Cell Dev. Biol. 2015, 39, 20–25. [Google Scholar] [CrossRef]
- Kiran Kumar, K.M.; Naveen Kumar, M.; Patil, R.H.; Nagesh, R.; Hegde, S.M.; Kavya, K.; Babu, R.L.; Ramesh, G.T.; Sharma, S.C. Cadmium induces oxidative stress and apoptosis in lung epithelial cells. Toxicol. Mech. Methods 2016, 26, 658–666. [Google Scholar] [CrossRef] [Green Version]
- Kusaczuk, M.; Krętowski, R.; Naumowicz, M.; Stypułkowska, A.; Cechowska-Pasko, M. Silica nanoparticle-induced oxidative stress and mitochondrial damage is followed by activation of intrinsic apoptosis pathway in glioblastoma cells. Int. J. Nanomed. 2018, 13, 2279–2294. [Google Scholar] [CrossRef] [Green Version]
- Guerin, D.J.; Kha, C.X.; Tseng, K.A. From Cell Death to Regeneration: Rebuilding After Injury. Front. Cell Dev. Biol. 2021, 9, 655048. [Google Scholar] [CrossRef]
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front. Cell Dev. Biol. 2021, 9, 714370. [Google Scholar] [CrossRef]
- Ryoo, H.D.; Bergmann, A. The role of apoptosis-induced proliferation for regeneration and cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008797. [Google Scholar] [CrossRef] [Green Version]
- Fogarty, C.E.; Bergmann, A. Killers creating new life: Caspases drive apoptosis-induced proliferation in tissue repair and disease. Cell Death Differ. 2017, 24, 1390–1400. [Google Scholar] [CrossRef] [Green Version]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Huang, Q.; Chen, J.; Peng, Y.; Roop, D.R.; Bedford, J.S.; Li, C.Y. Apoptotic cells activate the "phoenix rising" pathway to promote wound healing and tissue regeneration. Sci. Signal. 2010, 3, ra13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chera, S.; Ghila, L.; Dobretz, K.; Wenger, Y.; Bauer, C.; Buzgariu, W.; Martinou, J.C.; Galliot, B. Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev. Cell 2009, 17, 279–289. [Google Scholar] [CrossRef]
- Pérez-Garijo, A.; Shlevkov, E.; Morata, G. The role of Dpp and Wg in compensatory proliferation and in the formation of hyperplastic overgrowths caused by apoptotic cells in the Drosophila wing disc. Development 2009, 136, 1169–1177. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Wang, S.; Hernandez, J.; Yenigun, V.B.; Hertlein, G.; Fogarty, C.E.; Lindblad, J.L.; Bergmann, A. Genetic models of apoptosis-induced proliferation decipher activation of JNK and identify a requirement of EGFR signaling for tissue regenerative responses in Drosophila. PLoS Genet. 2014, 10, e1004131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Bergmann, A. Multiple mechanisms modulate distinct cellular susceptibilities toward apoptosis in the developing Drosophila eye. Dev. Cell 2014, 30, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryoo, H.D.; Gorenc, T.; Steller, H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev. Cell 2004, 7, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Diwanji, N.; Bergmann, A. The beneficial role of extracellular reactive oxygen species in apoptosis-induced compensatory proliferation. Fly 2017, 11, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diwanji, N.; Bergmann, A. An unexpected friend—ROS in apoptosis-induced compensatory proliferation: Implications for regeneration and cancer. Semin. Cell Dev. Biol. 2018, 80, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999, 6, 1028–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Miki, Y. The cell death machinery governed by the p53 tumor suppressor in response to DNA damage. Cancer Sci. 2010, 101, 831–835. [Google Scholar] [CrossRef]
- Luna-Vargas, M.P.; Chipuk, J.E. The deadly landscape of pro-apoptotic BCL-2 proteins in the outer mitochondrial membrane. FEBS J. 2016, 283, 2676–2689. [Google Scholar] [CrossRef] [PubMed]
- Pinal, N.; Calleja, M.; Morata, G. Pro-apoptotic and pro-proliferation functions of the JNK pathway of Drosophila: Roles in cell competition, tumorigenesis and regeneration. Open Biol. 2019, 9, 180256. [Google Scholar] [CrossRef] [Green Version]
- Pinal, N.; Martín, M.; Medina, I.; Morata, G. Short-term activation of the Jun N-terminal kinase pathway in apoptosis-deficient cells of Drosophila induces tumorigenesis. Nat. Commun. 2018, 9, 1541. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Gao, X.; Liu, B.; He, X.; Xu, J.; Qiang, J.; Wu, Q.; Liu, S. NMT1 inhibition modulates breast cancer progression through stress-triggered JNK pathway. Cell Death Dis. 2018, 9, 1143. [Google Scholar] [CrossRef] [Green Version]
- Ramiro-Cortés, Y.; Morán, J. Role of oxidative stress and JNK pathway in apoptotic death induced by potassium deprivation and staurosporine in cerebellar granule neurons. Neurochem. Int. 2009, 55, 581–592. [Google Scholar] [CrossRef]
- Gauron, C.; Rampon, C.; Bouzaffour, M.; Ipendey, E.; Teillon, J.; Volovitch, M.; Vriz, S. Sustained production of ROS triggers compensatory proliferation and is required for regeneration to proceed. Sci. Rep. 2013, 3, 2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, E.; Lindblad, J.L.; Bergmann, A. Tumor-promoting function of apoptotic caspases by an amplification loop involving ROS, macrophages and JNK in Drosophila. Elife 2017, 6, e26747. [Google Scholar] [CrossRef]
- Wang, A.; Jiang, H.; Liu, Y.; Chen, J.; Zhou, X.; Zhao, C.; Chen, X.; Lin, M. Rhein induces liver cancer cells apoptosis via activating ROS-dependent JNK/Jun/caspase-3 signaling pathway. J. Cancer 2020, 11, 500–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, J.J.; Hübner, A.; Zhang, C.; Flavell, R.A.; Shokat, K.M.; Davis, R.J. Chemical genetic analysis of the time course of signal transduction by JNK. Mol. Cell 2006, 21, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.J.; Abidi, S.N.F.; Skinner, A.; Tian, Y.; Smith-Bolton, R.K. The Drosophila Duox maturation factor is a key component of a positive feedback loop that sustains regeneration signaling. PLoS Genet. 2017, 13, e1006937. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.S.; Hwang, C.Y.; Shin, S.Y.; Kwon, K.S.; Cho, K.H. MLK3 is part of a feedback mechanism that regulates different cellular responses to reactive oxygen species. Sci. Signal. 2014, 7, ra52. [Google Scholar] [CrossRef]
- Hirata, Y.; Inoue, A.; Suzuki, S.; Takahashi, M.; Matsui, R.; Kono, N.; Noguchi, T.; Matsuzawa, A. trans-Fatty acids facilitate DNA damage-induced apoptosis through the mitochondrial JNK-Sab-ROS positive feedback loop. Sci. Rep. 2020, 10, 2743. [Google Scholar] [CrossRef]
- Noguchi, T.; Ishii, K.; Fukutomi, H.; Naguro, I.; Matsuzawa, A.; Takeda, K.; Ichijo, H. Requirement of reactive oxygen species-dependent activation of ASK1-p38 MAPK pathway for extracellular ATP-induced apoptosis in macrophage. J. Biol. Chem. 2008, 283, 7657–7665. [Google Scholar] [CrossRef] [Green Version]
- Sekine, Y.; Hatanaka, R.; Watanabe, T.; Sono, N.; Iemura, S.; Natsume, T.; Kuranaga, E.; Miura, M.; Takeda, K.; Ichijo, H. The Kelch repeat protein KLHDC10 regulates oxidative stress-induced ASK1 activation by suppressing PP5. Mol. Cell 2012, 48, 692–704. [Google Scholar] [CrossRef] [Green Version]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Wang, C.; Luo, T.; Lu, B.; Ma, H.; Zhou, Z.; Zhu, D.; Chi, G.; Ge, P.; Luo, Y. JNK Activation Contributes to Oxidative Stress-Induced Parthanatos in Glioma Cells via Increase of Intracellular ROS Production. Mol. Neurobiol. 2017, 54, 3492–3505. [Google Scholar] [CrossRef]
- Fogarty, C.E.; Diwanji, N.; Lindblad, J.L.; Tare, M.; Amcheslavsky, A.; Makhijani, K.; Brückner, K.; Fan, Y.; Bergmann, A. Extracellular Reactive Oxygen Species Drive Apoptosis-Induced Proliferation via Drosophila Macrophages. Curr. Biol. 2016, 26, 575–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, N.R.; Chen, Y.; Ishibashi, S.; Kritsiligkou, P.; Lea, R.; Koh, Y.; Gallop, J.L.; Dorey, K.; Amaya, E. Amputation-induced reactive oxygen species are required for successful Xenopus tadpole tail regeneration. Nat. Cell Biol. 2013, 15, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, A.S.; Adams, D.S.; Qiu, D.; Koustubhan, P.; Levin, M. Apoptosis is required during early stages of tail regeneration in Xenopus laevis. Dev. Biol. 2007, 301, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santabárbara-Ruiz, P.; López-Santillán, M.; Martínez-Rodríguez, I.; Binagui-Casas, A.; Pérez, L.; Milán, M.; Corominas, M.; Serras, F. ROS-Induced JNK and p38 Signaling Is Required for Unpaired Cytokine Activation during Drosophila Regeneration. PLoS Genet. 2015, 11, e1005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishina, T.; Komazawa-Sakon, S.; Yanaka, S.; Piao, X.; Zheng, D.M.; Piao, J.H.; Kojima, Y.; Yamashina, S.; Sano, E.; Putoczki, T.; et al. Interleukin-11 links oxidative stress and compensatory proliferation. Sci. Signal. 2012, 5, ra5. [Google Scholar] [CrossRef]
- Hansson Mild, K.; Lundström, R.; Wilén, J. Non-Ionizing Radiation in Swedish Health Care-Exposure and Safety Aspects. Int. J. Environ. Res. Public Health 2019, 16, 1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saliev, T.; Begimbetova, D.; Masoud, A.R.; Matkarimov, B. Biological effects of non-ionizing electromagnetic fields: Two sides of a coin. Prog. Biophys. Mol. Biol. 2019, 141, 25–36. [Google Scholar] [CrossRef]
- Qu, A.; Wu, X.; Li, S.; Sun, M.; Xu, L.; Kuang, H.; Xu, C. An NIR-Responsive DNA-Mediated Nanotetrahedron Enhances the Clearance of Senescent Cells. Adv. Mater. 2020, 32, e2000184. [Google Scholar] [CrossRef]
- Laubach, V.; Kaufmann, R.; Bernd, A.; Kippenberger, S.; Zöller, N. Extrinsic or Intrinsic Apoptosis by Curcumin and Light: Still a Mystery. Int. J. Mol. Sci. 2019, 20, 905. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Xu, F.; Yang, W.; Ren, J.; Ge, W.; Yang, P. Apoptosis as an underlying mechanism in lymphocytes induced by riboflavin and ultraviolet light. Transfus. Apher. Sci. 2020, 59, 102899. [Google Scholar] [CrossRef]
- Pirozzoli, M.C.; Marino, C.; Lovisolo, G.A.; Laconi, C.; Mosiello, L.; Negroni, A. Effects of 50 Hz electromagnetic field exposure on apoptosis and differentiation in a neuroblastoma cell line. Bioelectromagnetics 2003, 24, 510–516. [Google Scholar] [CrossRef]
- Yuan, L.Q.; Wang, C.; Zhu, K.; Li, H.M.; Gu, W.Z.; Zhou, D.M.; Lai, J.Q.; Zhou, D.; Lv, Y.; Tofani, S.; et al. The antitumor effect of static and extremely low frequency magnetic fields against nephroblastoma and neuroblastoma. Bioelectromagnetics 2018, 39, 375–385. [Google Scholar] [CrossRef]
- Buemi, M.; Marino, D.; Di Pasquale, G.; Floccari, F.; Senatore, M.; Aloisi, C.; Grasso, F.; Mondio, G.; Perillo, P.; Frisina, N.; et al. Cell proliferation/cell death balance in renal cell cultures after exposure to a static magnetic field. Nephron 2001, 87, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Bekhite, M.M.; Finkensieper, A.; Abou-Zaid, F.A.; El-Shourbagy, I.K.; El-Fiky, N.K.; Omar, K.M.; Sauer, H.; Wartenberg, M. Differential effects of high and low strength magnetic fields on mouse embryonic development and vasculogenesis of embryonic stem cells. Reprod. Toxicol. 2016, 65, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Bekhite, M.M.; Figulla, H.R.; Sauer, H.; Wartenberg, M. Static magnetic fields increase cardiomyocyte differentiation of Flk-1+ cells derived from mouse embryonic stem cells via Ca2+ influx and ROS production. Int. J. Cardiol. 2013, 167, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kim, S.C.; Kim, J.Y. Protective Effect of 10-Hz, 1-mT Electromagnetic Field Exposure Against Hypoxia/Reoxygenation Injury in HK-2 Cells. Biomed. Environ. Sci. 2015, 28, 231–234. [Google Scholar]
- Duong, C.N.; Kim, J.Y. Exposure to electromagnetic field attenuates oxygen-glucose deprivation-induced microglial cell death by reducing intracellular Ca(2+) and ROS. Int. J. Radiat. Biol. 2016, 92, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.; Su, L.; Lou, H.; Zhao, C.; Wang, Y.; Chen, G. The effects of 50 Hz magnetic field-exposed cell culture medium on cellular functions in FL cells. J. Radiat. Res. 2019, 60, 424–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okano, H. Effects of static magnetic fields in biology: Role of free radicals. Front. Biosci. 2008, 13, 6106–6125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, H. Exposure to Static and Extremely-Low Frequency Electromagnetic Fields and Cellular Free Radicals. Electromagn. Biol. Med. 2019, 38, 231–248. [Google Scholar] [CrossRef] [PubMed]
- McFadden, J.; Al-Khalili, J. The origins of quantum biology. Proc. Math. Phys. Eng. Sci. 2018, 474, 20180674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brocklehurst, B.; McLauchlan, K.A. Free radical mechanism for the effects of environmental electromagnetic fields on biological systems. Int. J. Radiat. Biol. 1996, 69, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ritz, T. Zeeman resonances for radical-pair reactions in weak static magnetic fields. Mol. Phys. 2006, 104, 1649–1658. [Google Scholar] [CrossRef]
- Barnes, F.; Greenebaum, B. Role of radical pairs and feedback in weak radio frequency field effects on biological systems. Environ. Res. 2018, 163, 165–170. [Google Scholar] [CrossRef]
- Barnes, F.; Greenebaum, B. Setting Guidelines for Electromagnetic Exposures and Research Needs. Bioelectromagnetics 2020, 41, 392–397. [Google Scholar] [CrossRef]
- Castello, P.R.; Hill, I.; Sivo, F.; Portelli, L.; Barnes, F.; Usselman, R.; Martino, C.F. Inhibition of cellular proliferation and enhancement of hydrogen peroxide production in fibrosarcoma cell line by weak radio frequency magnetic fields. Bioelectromagnetics 2014, 35, 598–602. [Google Scholar] [CrossRef]
- Driessen, S.; Bodewein, L.; Dechent, D.; Graefrath, D.; Schmiedchen, K.; Stunder, D.; Kraus, T.; Petri, A.K. Biological and health-related effects of weak static magnetic fields (≤ 1 mT) in humans and vertebrates: A systematic review. PLoS ONE 2020, 15, e0230038. [Google Scholar] [CrossRef]
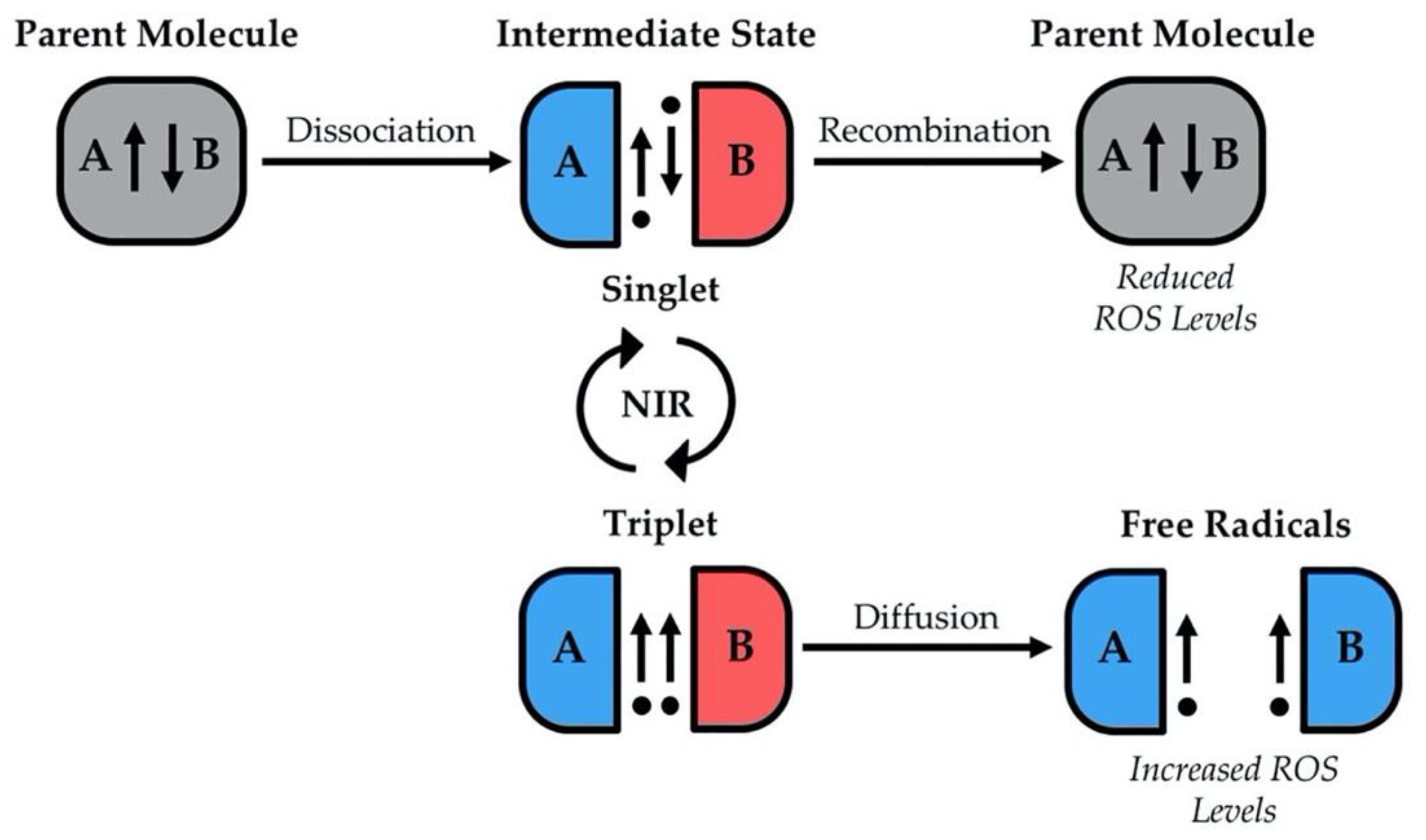

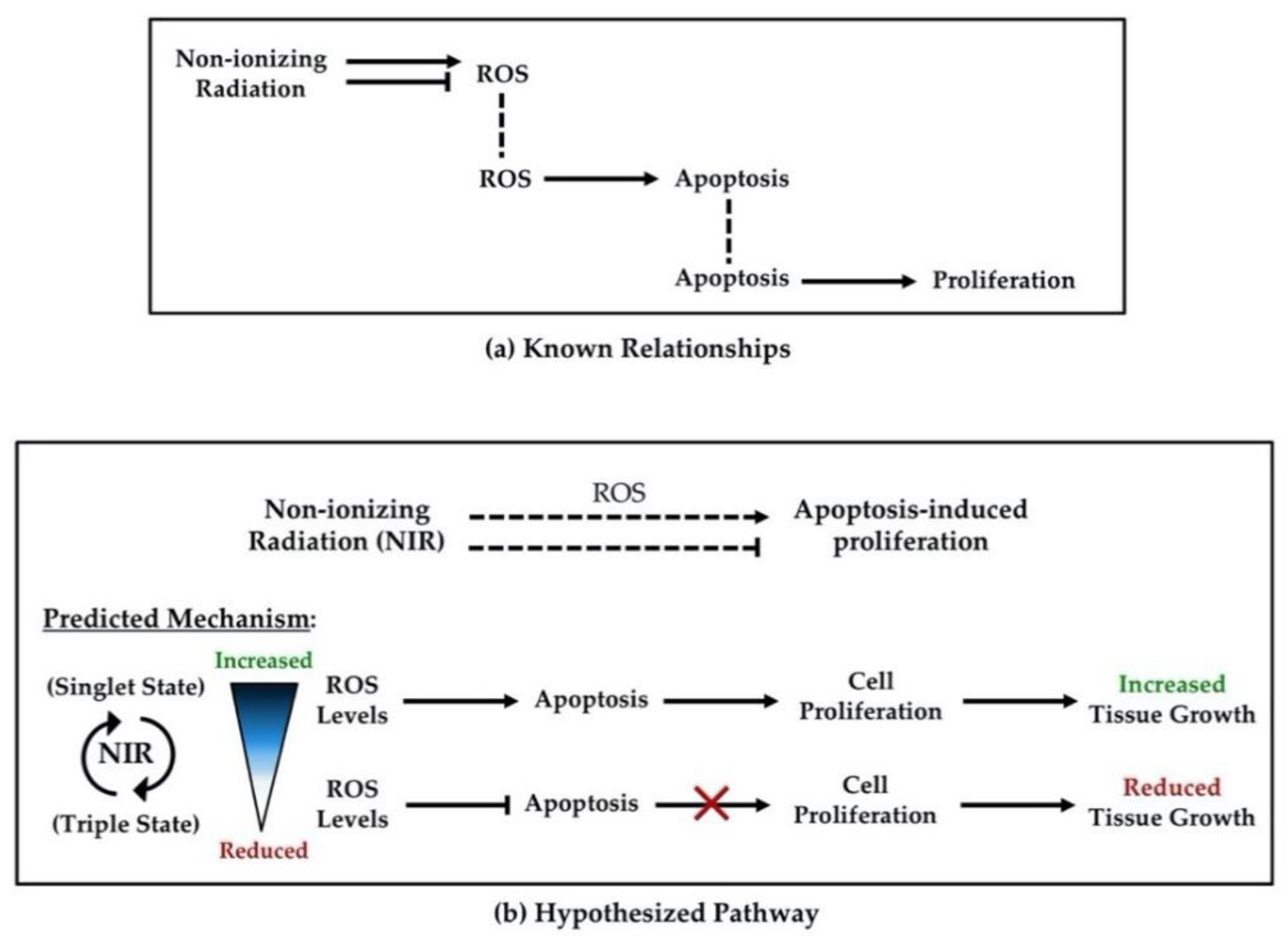

{kind=link}
{kind=link}
{kind=link}
| Terminology | Definition | Units (Symbol) |
|---|---|---|
| Reactive Oxygen Species (ROS) | A chemically reactive class of molecules containing oxygen. | - |
| Free Radicals | Highly reactive atoms or molecules characterized by a lone (unpaired) valence electron. | - |
| Spin State | A description of valence electron position, important for determining recombination or disassociation of radical pairs. | - |
| Non-ionizing Radiation (NIR) | Radiation that contains too little energy per photon to ionize an atom or molecule (remove an electron). Wavelengths greater than 280 nm. | Nanometers (nm) |
| Zeeman Effect | The splitting of spectral lines (electron decoupling) due to a static magnetic field. | - |
| Electromagnetic Fields (EMFs) | Radiation produced by the movement of charge. The interaction of electric and magnetic fields is generally described as a wave form transporting energy. | Varies |
| Magnetic Fields (MFs) | A vector field emanating from magnetic material or the result of electrical current. | Tesla (T) & Hertz (Hz) |
| Static Magnetic Fields | A magnetic field that has a constant or unchanged vector. | Tesla (T) |
| Radiofrequency | Refers to the osculation of electrical current or EMF. Magnetic fields with radiofrequency are produced by alternating electrical current. | Hertz (Hz) |
| Apoptosis-induced Proliferation | Cell division by mitosis (proliferation), induced by mitogen release from neighboring cells undergoing programed cell death (apoptosis). | - |
| Model System | NIR Type | Effects on ROS | Effects on Cellular Activities | |
|---|---|---|---|---|
| Fibrosarcoma Cells, HT-1080 Line | Static MFs [12] | Varied based on strength | Varied proliferation effects | |
| Weak RF MFs [87] | Increased ROS | Decreased proliferation | ||
| Epithelial Cells | HaCaT, A431, and A549 Cell Lines [70] | Ultraviolet light | Increased ROS | Decreased proliferation, increased apoptosis |
| A549 Cell Line [14] | Static MFs | Increased ROS | Decreased proliferation | |
| FL Cell Line [79] | Static & RF WMFs | No effects | No effects | |
| Mouse ES Cells | CCE Cell Line [75] | Static & RF MFs and WMFs | Increased ROS | Increased apoptosis |
| CGR8 Cell Line [76] | Static MFs | Decreased apoptosis | ||
| Neuroblastoma Cells | Lan-5 Line [72] | Static MFs | - | Increased proliferation, no change in apoptosis |
| CHLA-255 & N2a Lines [73] | Static & RF WMFs | Decreased proliferation, induced apoptosis | ||
| Nephroblastoma Cells, G401 Line | Static & RF WMFs [73] | - | Decreased proliferation, induced apoptosis | |
| Static MFs [14] | Increased ROS | Decreased proliferation | ||
| Renal Tubular Epithelial Cells, HK-2 Cell Line | EMFs [77] | Decreased ROS | Decreased apoptosis | |
| Leukemia Cells, THP-1 Cell Line | Static MFs [3] | Increased ROS | Decreased proliferation, increased apoptosis | |
| Lymphocytes, (whole blood, ABO/D matched) | Ultraviolet light [71] | Increased ROS | Increased apoptosis | |
| Microglial Cells, HM06 Line | EMFs [78] | Decreased ROS | Decreased apoptosis | |
| Nephroblastoma G401 Cells in Mice (in vivo) | Static & RF WMFs [73] | - | Decreased tumor mass | |
| Regenerating Adult Planarians (in vivo) | Static WMFs [13] | Decreased ROS | Decreased proliferation, inhibited regeneration | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hack, S.J.; Kinsey, L.J.; Beane, W.S. An Open Question: Is Non-Ionizing Radiation a Tool for Controlling Apoptosis-Induced Proliferation? Int. J. Mol. Sci. 2021, 22, 11159. https://doi.org/10.3390/ijms222011159
Hack SJ, Kinsey LJ, Beane WS. An Open Question: Is Non-Ionizing Radiation a Tool for Controlling Apoptosis-Induced Proliferation? International Journal of Molecular Sciences. 2021; 22(20):11159. https://doi.org/10.3390/ijms222011159
Chicago/Turabian StyleHack, Samantha J., Luke J. Kinsey, and Wendy S. Beane. 2021. "An Open Question: Is Non-Ionizing Radiation a Tool for Controlling Apoptosis-Induced Proliferation?" International Journal of Molecular Sciences 22, no. 20: 11159. https://doi.org/10.3390/ijms222011159