Abstract
Starting from isosteviol, a series of diterpenoid 1,3-aminoalcohol derivatives were stereoselectively synthesised. The acid-catalysed hydrolysis and rearrangement of natural stevioside gave isosteviol, which was transformed to the key intermediate methyl ester. In the next step, Mannich condensation of diterpenoid ketone, paraformaldehyde, and secondary amines resulted in the formation of 1,3-aminoketones with different stereoselectivities. During the Mannich condensation with dibenzylamine, an interesting N-benzyl → N-methyl substituent exchange was observed. Reduction of 1,3-aminoketones produced diastereoisomeric 1,3-aminoalcohols. Alternatively, aminoalcohols were obtained via stereoselective hydroxy-formylation, followed by oxime preparation, reduction, and finally, reductive alkylation of the obtained primary aminoalcohols. An alternative 1,3-aminoalcohol library was prepared by reductive amination of the intermediate 3-hydroxyaldehyde obtained from isosteviol in two-step synthesis. Cytotoxic activity of compounds against human tumour cell lines (A2780, SiHa, HeLa, MCF-7 and MDA-MB-231) was investigated. In our preliminary study, the 1,3-aminoalcohol function and N-benzyl substitution seemed to be essential for the reliable antiproliferative activity. To extend their application, a diterpenoid condensed with 2-phenylimino-1,3-thiazine and -1,3-oxazine was also attempted to prepare, but only formation of thioether intermediate was observed.
1. Introduction
Terpenoids, also known as isoprenoids, are the most numerous and structurally diverse group of natural products present in most plants []. Several studies have confirmed that this class of compounds displays a wide array of very important pharmacological properties []. Between terpenoids, the diterpenoid stevioside with a complex ent-kaurane skeleton and three glucose moieties has been the focus of attention in recent decades [,]. Stevioside is extracted from the plant Stevia rebaudiana, which is a perennial herbal shrub of the Asteraceae family that originated from Brazil and Paraguay in South America, while cultivated for its sweet leaves []. It is applied in food chemistry as a commercial sweetener considered to be a non-caloric sugar substitute. In recent years, stevioside and steviol, its aglycon, have attracted scientific attention because of their broad spectrum of biological activities, including antihyperglycemic, [] antihypertensive, [,] antitumour, [,] and immunomodulatory actions [] beside several other biological activities [,].
Isosteviol, a structural isomer of steviol, is a tetracyclic diterpenoid with an ent-beyerane skeleton obtained by acidic hydrolysis of stevioside []. In recent years, isosteviol derivatives have drawn high interest because of their biological activities, including anti-inflammatory, [] glucocorticoid agonist, [] antibacterial [], anticancer [,], or even cardioprotective properties [].
Cytotoxic activities of isosteviol derivatives, obtained by microbial and chemical transformations, have also induced much attention in recent years [,]. Several of the novel isosteviol derivatives have been successfully synthesised by chemical modification of isosteviol, and some of these derivatives exhibited good cytotoxic activity as potential drug molecules. Li and co-workers reported compounds with an α-methylenecyclopentanone moiety in the D-ring of isosteviol displaying remarkable anticancer activity against MDA-MB-231 cell line with an IC50 value of 1.58 μM []. Jayachandra and co-workers synthesised isosteviol analogues showing a potential protective effect against DOX-induced cardiotoxicity in zebrafish embryos in vivo []. Tao and co-workers reported that Esophageal carcinoma cells were more sensitive to 1,3-aminoalcohols, exhibiting anticancer activities superior to Cisplatin with an IC50 value 4.01 μM [].
1,3-Aminoalcohols may also serve as a building blocks of many natural and synthetic products, and they exhibit wide-ranging biological and catalytic activities [,,]. In a previous work, Tao and co-workers prepared a series of compounds by modifying a crucial aminoalcohol fragment of the D-ring of isosteviol affording significantly improved anticancer activities []. In a similar study, a hydroxythiourea derivative has been described as a useful candidate for the treatment of tumours on different cell lines [].
In the present contribution, we report the preparation of a new library of isosteviol-based chiral bifunctional synthons, such as β-aminoketones, 1,3-aminoalcohols, and 1,3-heterocycles fused with ent-beyerane, starting from commercially available natural stevioside. We also planned to investigate the preliminary study of the effect of keto-amine and 1,3-aminoalcohol functions and the stereochemistry and substitution level of amine function on antiproliferative activity on multiple human cancer cell lines.
2. Results and Discussion
2.1. Synthesis of Isosteviol-Based 1,3-Aminoalcohols
Starting from commercially available stevioside or mixtures of steviol glycosides, key intermediates 3-hydroxyaldehyde 4 and primary 1,3-aminoalcohol 6 were prepared in a four- and six-step synthesis (Scheme 1). Isosteviol 1 was obtained from commercially available natural stevioside by acid-catalysed hydrolysis and rearrangement [].
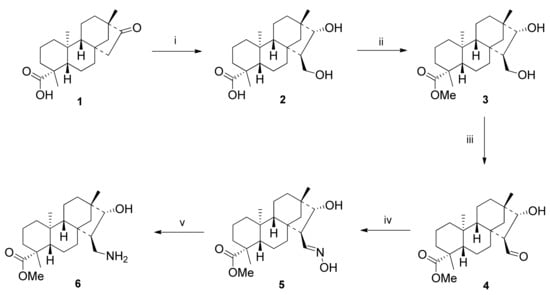
Scheme 1.
Stereoselective synthesis of isosteviol-based 1,3-aminoalcohol 6. (i) HCHO, NaOH (2 eq.), EtOH,1 h, 60 °C, 70%; (ii) CH2N2, Et2O, 5 min, 25 °C, 72%; (iii) 10 mol% TEMPO, NCS (2 eq.), TBAB (1 eq.), DCM/H2O, 12 h, reflux, 90%; (iv) H2NOH-HCl (2 eq.), EtOH, 12 h, reflux, 76%; (v) Raney-Ni, H2 (10 atm), THF, 12 h, 25 °C, 83%.
Diol 2 was synthesised in a stereoselective manner with good yield in a one-pot Aldol-Cannizzaro process in two steps according to literature methods [,]. Esterification of 2 was carried out with diazomethane in Et2O resulting in methyl ester 3 []. The TBAB-catalysed oxidisation of 3 with TEMPO and NCS gave regioselectively 4 in excellent yield. In the next step, compound 5 was obtained via oximation of 4 with hydroxylamine hydrochloride in the presence of NaHCO3 in ethanol and then it was converted to 1,3-aminoalcohol 6 with hydrogenation catalysed by Raney-Ni in THF in good yield (Scheme 1).
2.2. Synthesis of Isosteviol-Based 1,3-Aminoalcohols via Schiff Bases
The results of our previous study on N-substituted steviol-based aminodiols and related literature data on the antiproliferative activity of primary 1,3-aminoalcohols based on isosteviol predicted the interest of N-substituted 1,3-aminoalcohols. Therefore, a small library of 1,3-aminoalcohols was prepared to study structure–bioactivity relationship of N-substitution and antiproliferative activity [,]. Syntheses were accomplished via two pathways: reductive amination of hydroxyaldehyde 4 with primary amines or reductive alkylation of primary 1,3-aminoalcohol 6 with different aldehydes via formation of Schiff bases, followed by reduction with NaBH4 under mild conditions. The desired N-substituted 1,3-aminoalcohols (7–12) were isolated in acceptable yields. Reaction conditions and yields are presented in Scheme 2 and Table 1.
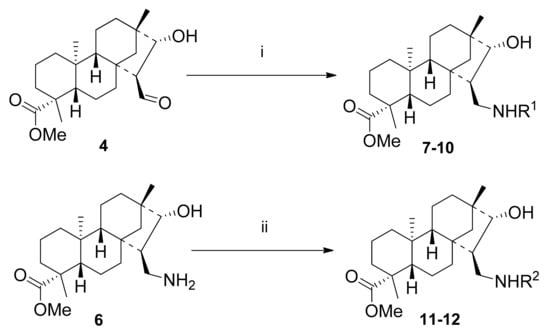
Scheme 2.
Synthesis of isosteviol-based 1,3-aminoalcohols. (i) 1) R1NH2 (1 eq.), dry EtOH, 3 h, 25 °C; 2) dry MeOH, NaBH4 (2 eq.), 3–4 h, 25 °C, 64–83%; (ii) 1) aldehydes (1 eq.), dry EtOH, 3 h, 25 °C; 2) dry MeOH, NaBH4 (2 eq.), 3–4 h, 25 °C, 64–65%.

Table 1.
Synthesis of aminoalcohols 7–12 via Schiff products.
2.3. Syntheses and Reduction of Isosteviol-Based 1,3-Aminoketones Obtained via Mannich Condensation
Isosteviol methyl ester 13 was prepared from 1 with diazomethane in excellent yield []. The Mannich condensation of 13 was accomplished with paraformaldehyde and different secondary amine HCl salts in glacial acetic acid, resulting in a library of aminoketones with good to moderate yields (Scheme 3, Table 2) []. The condensation reaction took place in an exclusive stereoselective manner, forming a single diastereoisomer with (7R) configuration of the new stereocenter at C15. Results are collected in Table 2.
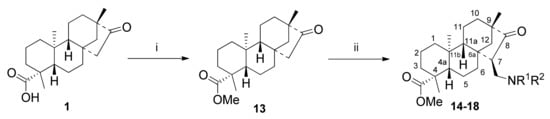
Scheme 3.
Synthesis of aminoketones 14–18 via Mannich condensation. (i) CH2N2, Et2O, 5 min, 25 °C, 79%; (ii) NHR1R2*HCl (1 eq.), (CH2O)n (2 eq.), AcOH, 24 h, reflux, 13–68%.
Table 2.
Synthesis of aminoketones 14–18 via Mannich condensation.
As entry 6 of Table 2 shows, the condensation of 13 with dibenzylamine hydrochloride surprisingly led to N-methyl-N-benzyl derivative 15 instead of the expected N,N-dibenzyl-substituted product, although with a low yield (13%). When the reaction was repeated with both N-benzyl-N-(S)-α-methylbenzylamine and the corresponding (7R) enantiomer, 15 as a single product could be isolated again (Scheme 4).
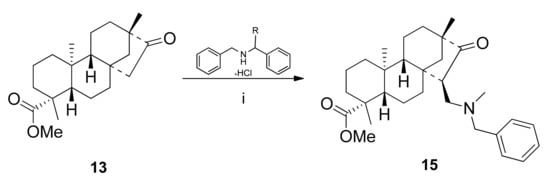
Scheme 4.
Substituent exchange under Mannich condensation. R = H, Me(S), Me(R). (i) (CH2O)n (2 eq.), AcOH, 24 h, reflux, 13%.
This interesting N-benzyl → N-methyl substituent exchange can be explained with the special steric hindrance of the diterpenoid skeleton with N-methyl-N-benzylamine representing the limit of the Mannich condensation in the case of this special ring system (Scheme 5). According to the classical mechanism of Mannich condensation, the first step is the formation of an iminium ion in the reaction of dibenzylamine and formaldehyde (Scheme 5). Because of steric hindrance, iminium species C cannot react with the enolate of the ketone. Rather, under the applied conditions, isomeric iminium salt D is formed. This step is followed by water addition and benzaldehyde elimination, resulting in N-methyl-N-benzylamine (F), ready for the condensation to give 15.
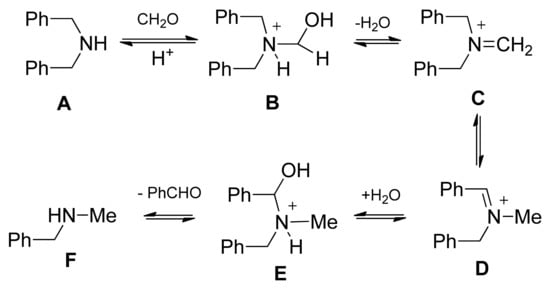
Scheme 5.
Proposed substituent exchange under Mannich condensation of dibenzylamine.
The reduction of aminoketones 14–18 with NaBH4 under mild conditions provided diastereomeric mixtures of 1,3-aminoalcohols. Reaction routes are outlined in Scheme 6. When pyrrolidinoaminoketone (16) or dimethylaminoketone (17) derivatives were applied, the reaction proceeded in a highly stereoselective way, resulting in the formation of 21 and 22 as single diastereoisomers. In other cases, diastereomeric mixtures were formed. Data are presented in Table 3.
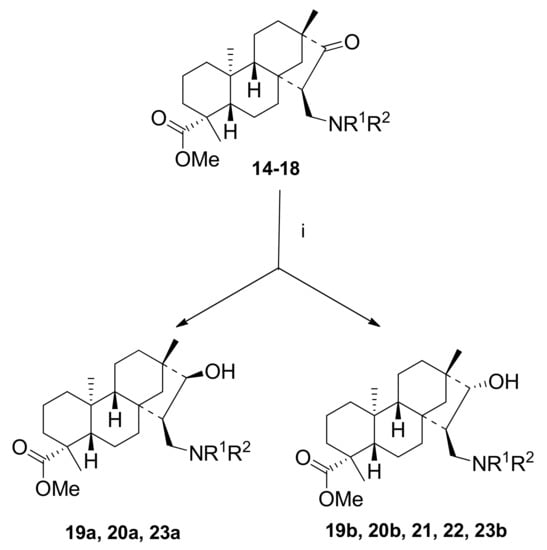
Scheme 6.
Synthesis of aminoalcohols 19a–23b via reduction of 14–18 with NaBH4. (i) NaBH4 (2 eq.), dry MeOH, 2–3 h, 25 °C, 10–75%.
Table 3.
Synthesis of aminoketones 14–18 via Mannich condensation.
The different steric hindrances of N-substituents can explain the different stereoselectivity of reduction of amino ketones. Probably in the case of less hindrance aminomethyl substituents (15-17), a cyclic complex can be formed with the protic solvent, and the hydride can attack from only the less sterically hindrance side, while this complex cannot be formed in the case of bulky N-substitution (14 and 18) and therefore the attack of the hydride can take place both side of the carbonyl function, resulting in a mixture of diastereoisomers [].
The relative, therefore absolute configurations of the new stereocenters of aminoalcohols 19-23 at position 7 and 8 were determined by NMR with NOESY spectral analysis, based on the observation of NOE effects between H-C(12) and H-C(8), H-C(8) and H-C(15), H-C(12) and Me-C(17), as well as between H-C(12) and H-C(15). Thus, the structure of 19b, 20b, 21, 22, and 23b was determined as outlined in Figure 1. Similarly, NOE effects were observed in the case of 19a, 20a, and 23a.
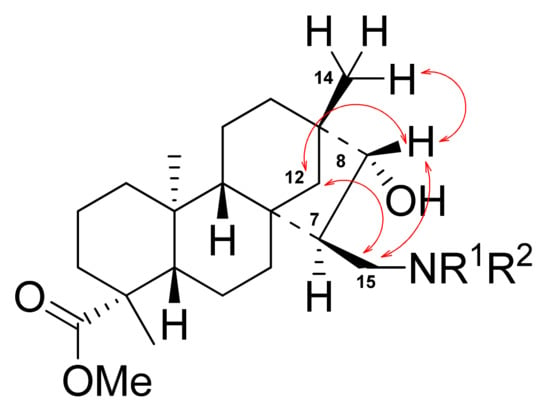
Figure 1.
Determination of the structure of 1,3-aminoalcohols by NOESY.
Beside the NOESY experiments, the configurations of the newly formed stereocenters of 1,3-aminoalcohols were determined via two alternative synthetic pathways (Scheme 7). Reductive amination of 4 (obtained from 3 with known stereochemistry) with benzylamine followed by methylation of 8 with iodomethane yielded a product that was identical with 20b obtained as a major product of the reduction of aminoketone 15. Alternatively, debenzylation of 20b over 5% Pd/C catalyst in methanol resulted in N-methyl aminoalcohol identical with 7 obtained by reductive amination of 4 with methylamine. Diastereoisomer 24 was also prepared by debenzylation of 20a over 5% Pd/C catalyst (Scheme 7).
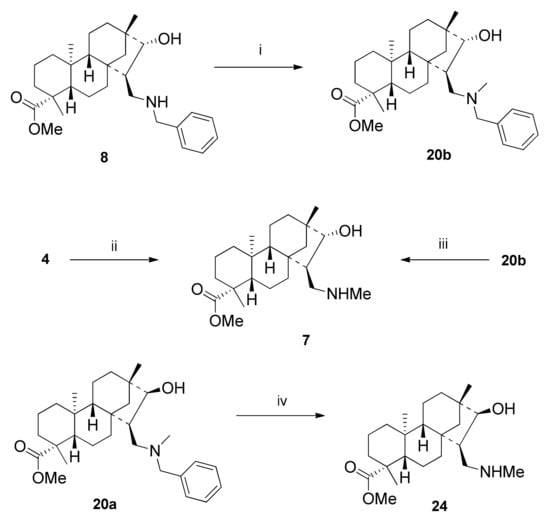
Scheme 7.
Alternative determination of the structure of 1,3-aminoalcohols via synthesis. (i) DCM, Et3N (1 eq.), MeI (1 eq.), 4 h, 25 °C, 77%; (ii) 1) MeNH2 (1 eq.), EtOH, 3 h, 25 °C; 2) MeOH, NaBH4 (2 eq.), 3–4 h, 25 °C, 77%; (iii) MeOH, 5% Pd/C, H2 (1 atm), 12 h, 25 °C, 24%; (iv) MeOH, 5% Pd/C, H2 (1 atm), 12 h, 25 °C, 28%.
2.4. Attempted Ring Closure of Isosteviol-Based Thiourea Derivatives
In our earlier studies we observed expressed cytotoxic activity of monoterpene-fused 2-phenylimino-1,3-oxazines and -1,3-thiazines on human cancer cell lines [,]. Consequently, we decided to convert primary 1,3-aminoalcohol 6 into its 1,3-oxazine and 1,3-thiazine derivatives. Reaction of 6 with phenyl isothiocyanate in DCM at room temperature provided the corresponding thiourea 25 with excellent yield (Scheme 8) []. The ring closure of thiourea 25 was attempted in a two-step procedure, which involved the treatment of 25 with methyl iodide, followed by alkaline-induced elimination of methyl mercaptol. Unfortunately, only the formation of thiomethyl ether intermediate 26 was observed. Alternatively, acid-promoted dehydrative cyclisation of 25 by treatment with EtOH containing 18% hydrochloric acid gave surprisingly only thioethyl ether 28 instead of the expected 2-phenylimino-1,3-thiazine 29 in moderate yield []. This reaction can be explained by the steric hindrance of the diterpene skeleton which inhibits the attack of sulphur on H-C(16) carbon, meanwhile the reaction of HCl with EtOH under the applied conditions can generate EtCl in situ, which reacts with thiourea 25 similarly to MeI, resulting in thioether 28 (Scheme 8).
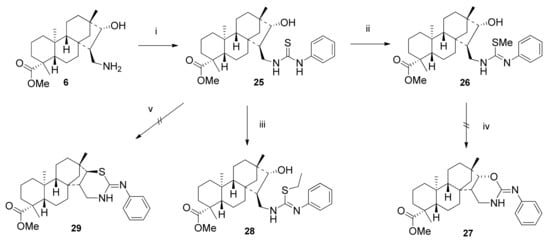
Scheme 8.
Attempted synthesis of 2-phenylimino-1,3-oxazine and -1,3-thiazine. (i) PhNCS (1 eq.), DCM, 2 h, 25 °C, 91%; (ii) MeI (5 eq.), EtOH, 2 h, 25 °C, 69%; (iii) 18% HCl, dry EtOH, 12 h, reflux, 46%; (iv) 10% KOH, EtOH, 4 h, reflux; (v) 18% HCl, EtOH, 12 h, reflux.
2.5. Antiproliferative Properties of the Prepared Diterpenes
The antiproliferative activities of the prepared diterpene analogues were determined by means of MTT assay on a panel of human adherent cancer lines, including cells from cervical (HeLa, SiHa), breast (MDA-MB-231, MCF-7), and ovary cancers (A2780) as given in Table 4. Based on the obtained activities, some conclusions could be arrived at with respect to structure–activity relationships. Since the original diol (3), its aldehyde analogue (4), and the corresponding oxime (5) elicited no relevant effect on the growth of cancer cells, an amino function seems to be essential for antiproliferative activity (Table 4). Primary amine 6 as well as secondary amine derivatives 7–12 exerted similarly pronounced antiproliferative action, and the calculated IC50 values of these compounds are comparable to or lower than those of reference agent cisplatin. The cell line-independent IC50 values may be interpreted as a marker of general cytotoxic property of molecules 6–12. From the results presented in Table 4, it seems to be clear that both the aminoalcohol function and the N-benzyl substitution (8–12), but not the aliphatic substitution (7 and 24), are essential for the remarkable antiproliferative activity. Cervical cell lines are especially sensitive to these agents. Aminoketones 14–18 are much less effective and most of them exert only negligible activity. Reduced analogues (19–23/24), bearing tertiary amino function, proved to elicit more pronounced action compared only with aminoketones, and the orientation of the newly formed alcohol function has no substantial impact on the efficacy of the product. Phenylthioureido analogue 25 exerted some modest activities with IC50 values between 10 and 23 μM, while the thioether type compounds 26 and 28 do not favour the antiproliferative action of the diterpene skeleton.
Table 4.
Antiproliferative properties of the tested diterpene analogues.
3. Conclusions
In summary, a series of novel isosteviol derivatives containing 1,3-aminoalcohol and thiourea moieties have been synthesised with moderated to good yields, and their cytotoxic activities against five human cancer cell lines (HeLa, Siha, MCF7, MDA-MB-231, A2780) have been investigated. Starting from commercially available stevioside, a new family of isosteviol-based chiral 1,3-aminoalcohols and thioureid derivatives were prepared through hydroxyaldehyde and isosteviol methyl ester as key intermediates via stereoselective transformations. The resulting 1,3-aminoalcohols exert remarkable antiproliferative action of human cancer cell lines. The in vitro pharmacological studies have clearly shown that the N-benzyl substituent at the amino function is essential and some of the prepared molecules proved to be more potent than anticancer agent cisplatin used clinically.
4. Materials and Methods
General methods: Commercially available reagents were used as obtained from suppliers (Molar Chemicals Ltd., Halásztelek, Hungary; Merck Ltd., Budapest, Hungary and VWR International Ltd., Debrecen, Hungary), while solvents were dried according to standard procedures. Optical rotations were measured in MeOH at 20 °C with a Perkin-Elmer 341 polarimeter (PerkinElmer Inc., Shelton, CT, USA). Chromatographic separations and monitoring of reactions were carried out on Merck Kieselgel 60 (Merck Ltd., Budapest, Hungary). Melting points were determined on a Kofler apparatus (Nagema, Dresden, Germany). 1H- and 13C-NMR spectra were recorded on Brucker Avance DRX 500 spectrometer (Bruker Biospin, Karlsruhe, Baden Württemberg, Germany) [500 MHz (1H) and 125 MHz (13C), δ = 0 (TMS)]. Chemical shifts are expressed in ppm (δ) relative to TMS as internal reference. J values are given by Hz. All 1H/13C NMR, NOESY, 2D-HMBC, and 2D-HMQC spectra are available in Supporting Information file. HRMS flow injection analysis was performed with a Thermo Scientific Q Exactive Plus hybrid quadrupole-Orbitrap (Thermo Fisher Scientific, Waltham, MA, USA) mass spectrometer coupled to a Waters Acquity I-Class UPLC™ (Waters, Manchester, UK).
Starting materials: Stevioside was obtained from Molar Chemicals Ltd., Halásztelek, Hungary. Isosteviol 1 was prepared from commercially available stevioside or a mixture of steviol glycosides in a one-step synthesis according to the literature method, and all its spectroscopic data were the same as described in the literature [].
Compounds 2, 3, and 13 were prepared by literature methods. Their spectroscopic data and physical and chemical properties were similar to those reported therein [,]. 1H, 13C, COSY, HSQC, HMBC, and NOESY NMR spectra of new compounds are available in Supplementary Materials.
(4R,6aS,7R,8R,9S,11bS)-Methyl 7-formyl-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (4): To a solution of 3 (4.70 mmol, 1.73 g) in DCM/H2O (50 / 50 mL), TEMPO (10 mol%, 73 mg), NCS (9.40 mmol, 1.26 g), and TBAB (4.70 mmol, 1.52 g) was added. After 12 h reflux the reaction was found to be completed (indicated by TLC), and the mixture was extracted with DCM (3 × 50 mL). The combined organic phase was extracted with water (1 × 50 mL), dried (Na2SO4), filtered, and concentrated. The purification of the crude product was accomplished by column chromatography on silica gel with an appropriate solvent mixture (n-hexane/EtOAc = 4:1). Yield: 1.53 g (90%); white crystals; m.p. 151–152 °C; = –117 (c 0.24 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.86 (s, 3H), 0.89–0.93 (m, 1H), 0.97 (s, 3H), 1.01–1.06 (m, 3H), 1.12–1.14 (m, 1H), 1.17 (s, 3H), 1.20–1.27 (m, 2H), 1.37–1.40 (m, 1H), 1.42–1.45 (m, 1H), 1.53–1.85 (m, 8H), 2.18 (d, 2H, J = 13.3 Hz), 2.92 (s, 1H), 3.64 (s, 3H), 4.26 (d, 1H, J = 4.9 Hz), 9.93 (d, 1H, J = 2.5 Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm) 13.0 (CH3), 18.8 (CH2), 19.7 (CH2), 21.6 (CH2), 24.5 (CH3), 28.8 (CH3), 33.0 (CH2), 35.9 (CH2), 37.9 (Cq), 38.2 (Cq), 39.6 (CH2), 41.2 (Cq), 43.7 (Cq), 46.5 (Cq), 51.2 (CH3), 53.9 (CH2), 56.8 (CH), 57.4 (CH), 61.7 (CH), 78.3 (CH), 177.8 (C=O), 204.4 (CH). C22H34O4 (362.50): 363.26. HRMS (ESI+): m/z calcd. for C22H35O4 [M + H]+ 363.2535; found 363.25230.
(4R,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-7-((hydroxyimino)methyl)-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (5): A mixture of compound 4 (4.20 mmol, 1.53 g) and hydroxylamine hydrochloride (8.40 mmol, 0.58 g) in 50 mL EtOH was stirred in presence of NaHCO3 (4.20 mmol, 0.35 g) at 60 °C for 2 h, then the reaction mixture was concentrated under vacuum and extracted with 50 mL DCM and 50 mL water. The water phase was extracted further with DCM (3 × 50 mL) and the combined organic phase was washed with saturated NaCl aqueous solution (1 × 50 mL), dried (Na2SO4), and concentrated under vacuum. The product obtained was purified by column chromatography (n-hexane/EtOAc = 2:1). Yield: 1.21 g (76%); white crystals; m.p. 113–114 °C; = –90 (c 0.40 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.75 (s, 3H), 0.84–0.90 (m, 1H), 0.94 (s, 3H), 0.97–1.06 (m, 4H), 1.11–1.17 (m, 4H), 1.19–1.26 (m, 1H), 1.40 (dd, 2H, J = 2.6 Hz, 12.0 Hz), 1.56–1.72 (m, 6H), 1.76–1.85 (m, 2H), 2.16 (d, 1H, J = 13.3 Hz), 2.71–2.73 (m, 1H), 3.17 (s, 1H), 3.63 (s, 3H), 3.76 (d, 1H, J = 4.8 Hz), 7.47 (d, 1H, J = 8.5 Hz), 8.90 (s, 1H,); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.1 (CH3), 18.8 (CH2), 19.5 (CH2), 20.8 (CH2), 24.9 (CH3), 28.8 (CH3), 33.0 (CH2), 35.8 (CH2), 38.0 (CH2), 38.2 (Cq), 39.6 (CH2), 41.4 (Cq), 43.7 (Cq), 44.8 (Cq), 49.9 (CH), 51.3 (CH3), 53.9 (CH2), 56.9 (CH), 57.3 (CH), 83.7 (CH), 153.9 (CH), 178.1 (C=O). HRMS (ESI+): m/z calcd. for C22H36NO4 [M + H]+ 378.2644; found 378.2639.
(4R,6aS,7R,8R,9S,11bS)-Methyl 7-aminomethyl-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (6): To a suspension of Raney nickel (0.20 g) in THF (20 mL) solution of oxime 5 (1.53 g, 4.10 mmol) in THF (30 mL) was added and the mixture was stirred under H2 atmosphere (10 atm) at room temperature for 12 h. The mixture was then filtered and evaporated, and the crude product was purified by crystallisation (n-hexane/DCM). Compound 6: 1.24 g (83%); white crystals; m.p. 165–166 °C; = −42 (c 0.33 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.73 (s, 3H), 0.84–0.90 (m, 1H), 0.92 (s, 3H), 0.94–1.07 (m, 5H), 1.16–1.20 (m, 4H), 1.34–1.43 (m, 2H), 1.60–1.85 (m, 9H), 2.15–2.17 (m, 3H), 2.44 (t, 1H, J = 11.8 Hz), 3.13 (dd, 1H, J = 3.4 Hz, 11.3 Hz), 3.50 (d, 1H, J = 4.4 Hz), 3.63 (s, 3H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.1 (CH3), 19.0 (CH2), 19.5 (CH2), 22.1 (CH2), 25.1 (CH3), 28.9 (CH3), 33.2 (CH2), 35.0 (CH2), 38.0 (CH2), 38.2 (Cq), 39.7 (CH2), 40.9 (Cq), 42.6 (Cq), 43.8 (Cq), 44.1 (CH2), 50.7 (CH), 51.2 (CH3), 54.2 (CH2), 57.2 (CH), 57.8 (CH), 87.7 (CH), 177.9 (C=O). HRMS (ESI+): m/z calcd. for C22H38NO3 [M + H]+ 364.2852; found 364.2846.
General procedure for preparation of aminoalcohol with primary amines and aldehydes: Method A: To a solution of 4 (0.10 g, 0.28 mmol) in dry EtOH (10 mL), primary amines (0.28 mmol) were added in one portion and the solution was stirred at room temperature for 3 h and then evaporated to dryness. The residue was dissolved in dry EtOH (10 mL), stirred for a further 1 h, and evaporated to dryness again. The product was dissolved in dry MeOH (10 mL) and NaBH4 (0.56 mmol, 0.02 g) was added in small portions to the mixture under ice cooling. After stirring for 4 h at room temperature, the mixture was evaporated to dryness, and the residue was dissolved in H2O (20 mL) and extracted with DCM (3 × 20 mL). The combined organic layer was dried (Na2SO4), filtered and evaporated to dryness. The crude product obtained was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1).
Method B: To a solution of 6 (0.10 g, 0.28 mmol) in dry EtOH (10 mL), aldehydes (0.28 mmol) were added in one portion, and the solution was stirred at room temperature for 3 h and then evaporated to dryness. The product was dissolved in dry EtOH (10 mL) and stirred for a further 1 h and evaporated to dryness again. The crude product was dissolved in dry MeOH (10 mL) and NaBH4 (0.56 mmol, 0.02 g) was added in small portions to the mixture under ice cooling. After stirring for 4 h at room temperature, the mixture was evaporated to dryness, and the residue was dissolved in H2O (20 mL) and extracted with DCM (3 × 20 mL). The combined organic layer was dried (Na2SO4), filtered and evaporated to dryness. The crude product obtained was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1).
(4R,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-4,9,11b-trimethyl-7-((methylamino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (7): The reaction was accomplished starting from compound 4 with 33 wt% methylamine (0.28 mmol, 0.02 mL) according to the general procedure Method A. Yield: 0.09 g (80%). An alternative synthesis of 7 was accomplished from 26 with a yield of 0.03 g (24%). The product (0.15 g, 0.33 mmol) in MeOH (25 mL) was added to a suspension of palladium-on-carbon (5% Pd/C, 0.10 g), and the mixture was stirred under a H2 atmosphere (1 atm) at room temperature. After completion of the reaction (monitored by TLC, 24 h), the mixture was filtered through a Celite pad, and the solution was evaporated to dryness. The crude product was purified by column chromatography on silica gel (CHCl3/MeOH = 9:1). Compound 7: white crystals; m.p. 135–136 °C; = –66 (c 0.37 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.73 (s, 3H), 0.85–0.88 (m, 1H), 0.92 (s, 3H), 0.94–1.07 (m, 5H), 1.16–1.21 (m, 4H), 1.36–1.43 (m, 2H), 1.59–1.82 (m, 8H), 1.87–1.89 (m, 1H), 2.01 (s, 2H), 2.16 (d, 1H, J = 13.3 Hz), 2.33 (t, 1H, J = 11.3 Hz), 2.48 (s, 3H), 2.92 (d, 1H, J = 11.0 Hz), 3.46–3.47 (m, 1H), 3.63 (s, 3H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.1 (CH3), 19.0 (CH2), 19.6 (CH2), 22.1 (CH2), 25.0 (CH3), 28.9 (CH3), 33.1 (CH2), 35.1 (CH2), 36.8 (CH3), 38.0 (CH2), 38.1 (Cq), 39.6 (CH2), 40.8 (Cq), 42.4 (Cq), 43.8 (Cq), 47.9 (CH), 51.1 (CH3), 54.3 (CH2), 54.7 (CH2), 57.2 (CH), 57.9 (CH), 88.3 (CH), 177.8 (C=O). HRMS (ESI+): m/z calcd. for C23H40NO3 [M + H]+ 378.3008; found 378.3003.
(4R,6aS,7R,8R,9S,11bS)-Methyl 7-((benzylamino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (8): The reaction was accomplished starting from compound 4 with benzylamine (0.28 mmol, 0.02 mL) according to the general procedure Method A. Yield: 0.11 g (83%); white crystals; m.p. 114–115 °C; = –75 (c 0.33 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.70 (s, 3H), 0.83–0.90 (m, 2H), 0.92 (s, 3H), 0.94–1.06 (m, 4H), 1.15 (s, 3H), 1.17–1.21 (m, 1H), 1.34 (dd, 1H, J = 2.5 Hz, 11.3 Hz), 1.39–1.42 (m, 1H), 1.58–1.62 (m, 4H), 1.70–1.81 (m, 4H), 1.84–1.88 (m, 1H), 2.15 (d, 1H, J = 13.3 Hz), 2.35 (t, 1H, J = 11.9 Hz), 3.02 (dd, 1H, J = 4.0 Hz, 11.0 Hz), 3.47 (d, 1H, J = 4.9 Hz), 3.60 (s, 3H), 3.76 (d, 1H, J = 13.4 Hz), 3.88 (d, 1H, J = 13.4 Hz), 7.22–7.26 (m, 1H), 7.30–7.34 (m, 4H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.1 (CH3), 19.0 (CH2), 19.6 (CH2), 22.1 (CH2), 25.1 (CH3), 29.0 (CH3), 33.0 (CH2), 35.0 (CH2), 38.0 (CH2), 38.1 (Cq), 39.6 (CH2), 40.6 (Cq), 42.3 (Cq), 43.7 (Cq), 48.5 (CH), 51.2 (CH3), 51.8 (CH2), 54.2 (CH2), 54.3 (CH2), 57.1 (CH), 57.8 (CH), 88.7 (CH), 126.9 (CH), 128.0 (2xCH), 128.4 (2xCH), 140.6 (Cq), 177.9 (C=O). HRMS (ESI+): m/z calcd. for C29H44NO3 [M + H]+ 454.3321; found 454.3223.
(4R,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-4,9,11b-trimethyl-7-((((S)-1-phenylethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (9): The reaction was accomplished starting from compound 4 with (S)-(–)-α-methylbenzylamine (0.28 mmol, 0.04 mL) according to the general procedure Method A. Yield: 0.09 g (70%); white crystals; m.p. 108–110 °C; = +63 (c 0.29 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.59 (s, 3H), 0.78–0.84 (m, 1H), 0.89 (s, 3H), 0.92–1.03 (m, 5H), 1.09 (s, 3H), 1.11–1.23 (m, 2H), 1.36–1.41 (m, 2H), 1.56–1.69 (m, 4H), 1.75–1.83 (m, 2H), 1.94 (d, 3H, J = 6.7 Hz), 2.12 (d, 1H, J = 13.1 Hz), 2.34 (d, 1H, J = 13.0 Hz), 2.81 (t, 1H, J = 13.0 Hz), 3.00 (dd, 1H, J = 3.6 Hz, 12.5 Hz), 3.59 (s, 3H), 3.75 (d, 1H, J = 4.6 Hz), 4.54–4.58 (m, 1H), 7.36–7.39 (m, 1H), 7.44 (t, 2H, J = 7.3 Hz), 7.66 (d, 2H, J = 7.4 Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.4 (CH3), 18.8 (CH2), 19.4 (CH2), 21.8 (CH2), 21.9 (CH3), 24.6 (CH3), 28.8 (CH3), 33.1 (CH2), 34.7 (CH2), 37.9 (CH2), 38.0 (Cq), 39.6 (CH2), 41.4 (Cq), 43.0 (CH), 43.0 (Cq), 43.5 (Cq), 48.5 (CH2), 51.4 (CH3), 53.5 (CH2), 57.1 (CH), 58.1 (CH), 58.5 (CH), 84.3 (CH), 127.7 (2xCH), 129.2 (CH), 129.5 (2xCH), 136.4 (Cq), 177.5 (C=O). HRMS (ESI+): m/z calcd. for C30H46NO3 [M + H]+ 468.3478; found 468.3472.
(4R,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-4,9,11b-trimethyl-7-((((R)-1-phenylethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (10): The reaction was accomplished starting from compound 4 with (R)-(–)-α-methylbenzylamine (0.28 mmol, 0.04 mL) according to the general procedure Method A. Yield: 0.08 g (64%); white crystals; m.p. 104–105 °C; = –18 (c 1.01 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.61 (s, 3H), 0.81–0.91 (m, 3H), 0.93 (s, 3H), 0.96–1.02 (m, 3H), 1.11 (s, 3H), 1.17–1.21 (m, 1H), 1.36 (d, 3H, J = 6.6 Hz), 1.38–1.42 (m, 1H), 1.44–1.47 (m, 1H), 1.55–1.60 (m, 3H), 1.64–1.70 (m, 3H), 1.76–1.80 (m, 3H), 2.13 (d, 1H, J = 13.3 Hz), 2.34 (t, 1H, J = 11.2 Hz), 2.84 (dd, 1H, J = 4.2 Hz, 11.1 Hz), 3.48 (d, 1H, J = 5.0 Hz), 3.55 (s, 3H), 3.77 (dd, 1H, J = 6.6 Hz, 6.6 Hz), 7.20–7.24 (m, 1H), 7.29–7.32 (m, 4H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 12.8 (CH3), 18.9 (CH2), 19.5 (CH2), 22.1 (CH2), 24.1 (CH3), 25.1 (CH3), 28.8 (CH3), 33.0 (CH2), 35.0 (CH2), 38.1 (CH2) 38.1 (Cq), 39.6 (CH2), 40.6 (Cq), 42.3 (Cq), 43.7 (Cq), 48.9 (CH), 50.2 (CH2), 51.0 (CH), 54.4 (CH2), 57.1 (CH), 57.9 (CH), 58.8 (CH), 88.7 (CH), 126.4 (2xCH), 126.9 (CH), 128.4 (2xCH), 146.2 (Cq), 177.9 (C=O). HRMS (ESI+): m/z calcd. for C30H46NO3 [M + H]+ 468.3478; found 468.3472.
(4R,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-7-(((4-methoxybenzyl)amino)methyl)-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (11): The reaction was accomplished starting from compound 6 with 4-anisaldehyde (0.28 mmol, 0.03 mL) according to the general procedure Method B. Yield: 0.09 g (64%); white crystals; m.p. 123–125 °C; = –34 (c 0.17 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.65 (s, 3H), 0.78–0.84 (m, 1H), 0.89 (s, 3H), 0.91–1.06 (m, 5H), 1.10–1.16 (m, 4H), 1.26–1.44 (m, 5H), 1.58–1.63 (m, 2H), 1.68–1.69 (m, 2H), 1.77–1.82 (m, 2H), 2.13 (d, 1H, J = 13.7 Hz), 2.31 (d, 1H, J = 12.6 Hz), 2.79 (t, 1H, J = 12.9 Hz), 3.03 (dd, 1H, J = 3.5 Hz, 12.1 Hz), 3.59 (s, 3H), 3.66 (d, 1H, J = 4.8 Hz), 3.75 (s, 3H), 4.00 (d, 1H, J = 13.1 Hz), 4.20 (d, 1H, J = 13.1 Hz), 6.91 (d, 2H, J = 8.5 Hz), 7.54 (d, 2H, J = 8.5 Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.2 (CH3), 18.8 (CH2), 19.4 (CH2), 21.8 (CH2), 24.7 (CH3), 28.9 (CH3), 33.0 (CH2), 34.8 (CH2), 37.9 (CH2), 38.0 (Cq), 39.5 (CH2), 41.4 (Cq), 42.9 (Cq), 43.6 (Cq), 43.7 (CH), 48.7 (CH2), 49.9 (CH2), 51.3 (CH3), 53.4 (CH2), 55.2 (CH3), 56.9 (CH), 57.8 (CH), 84.9 (CH), 114.4 (2xCH), 122.3 (Cq), 131.8 (2xCH), 160.2 (Cq), 177.6 (C=O). HRMS (ESI+): m/z calcd. for C30H46NO4 [M + H]+ 484.3427; found 484.3421.
(4R,6aS,7R,8R,9S,11bS)-Methyl 7-(((4-fluorobenzyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (12): The reaction was accomplished starting from compound 6 with 4-fluorobenzaldehyde (0.28 mmol, 0.03 mL), according to the general procedure Method B. Yield: 0.09 g (65%); white crystals; m.p. 118–119 °C; = –27 (c 0.15 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.70 (s, 3H), 0.82–0.87 (m, 1H), 0.91–0.92 (m, 4H), 0.96–1.06 (m, 4H), 1.15–1.21 (m, 4H), 1.34 (dd, 1H, J = 2.2 Hz, 11.8 Hz), 1.40 (d, 1H, J = 14.1 Hz), 1.58–1.88 (m, 10H), 2.16 (d, 1H, J = 13.1 Hz), 2.34 (d, 1H, J = 11.8 Hz), 3.00 (dd, 1H, J = 3.9 Hz, 10.7 Hz), 3.45 (d, 1H, J = 4.7 Hz), 3.60 (s, 3H), 3.73 (d, 1H, J = 13.0 Hz), 3.84 (d, 1H, J = 13.0 Hz), 7.00 (t, 2H, J = 8.5 Hz), 7.26–7.31 (m, 2H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.1 (CH3), 19.0 (CH2), 19.6 (CH2), 22.1 (CH2), 25.0 (CH3), 28.9 (CH3), 33.0 (CH2), 35.0 (CH2), 38.0 (CH2), 38.1 (Cq), 39.6 (CH2), 40.7 (Cq), 42.3 (Cq), 43.8 (Cq), 48.4 (CH), 51.1 (CH3), 51.7 (CH2), 53.4 (CH2), 54.3 (CH2), 57.2 (CH), 57.8 (CH), 88.6 (CH), 115.0 (CH), 115.2 (CH), 129.5 (CH), 129.6 (CH), 136.3 (Cq-F), 136.4 (Cq-F) 161.0 (Cq-F), 162.9 (Cq-F), 177.8 (C=O); 19F-NMR (470 MHz, CDCl3) δ (ppm): -116.2 (Cq-F). C29H42FNO3 (471.65): 472.32. HRMS (ESI+): m/z calcd. for C29H43FNO3 [M + H]+ 472.3227; found 472.3222.
General procedure for the preparation ofamino ketones: To a solution of isosteviol methyl ester 13 (1.80 mmol, 0.60 g) in glacial acetic acid (4 mL), paraformaldehyde (3.60 mmol, 0.10 g) and then secondary amine hydrochlorides (1.80 mmol) was added, and the mixture was treated under reflux conditions for 1.5 h. The solvent was evaporated, and the residue was dissolved in DCM (100 mL). The solution was washed with 5% aqueous KOH (100 mL) and the aqueous phase was extracted with DCM (2 × 100 mL). The combined organic layer was dried with Na2SO4, filtered, and evaporated. The crude product was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1).
(4R,6aS,7R,9S,11bS)-Methyl 4,9,11b-trimethyl-7-(morpholinomethyl)-8-oxotetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (14): The reaction was accomplished starting from compound 13 with morpholine hydrochloride (1.80 mmol, 0.22 g) according to the general procedure. Yield: 0.53 g (68%); white crystals; m.p. 148–149 °C; = –33 (c 0.56 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.69 (s, 3H), 0.87–0.90 (m, 1H), 0.93 (s, 3H), 1.17–1.24 (m, 7H), 1.26–1.27 (m, 1H), 1.32–1.38 (m, 1H), 1.57–1.60 (m, 1H), 1.68–1.72 (m, 3H), 1.78–1.88 (m, 2H), 1.97–2.02 (m, 1H), 2.10–2.14 (m, 1H), 2.18 (d, 1H, J = 13.3 Hz), 2.31 (d, 1H, J = 10.1 Hz), 2.39–2.41 (m, 2H), 2.48–2.55 (m, 4H), 3.63 (s, 3H), 3.68–3.75 (m, 4H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.0 (CH3), 19.0 (CH2), 19.5 (CH2), 20.2 (CH3), 22.1 (CH2), 28.9 (CH3), 36.2 (CH2), 37.3 (CH2), 38.2 (CH2), 38.2 (Cq), 39.6 (CH2), 41.6 (Cq), 43.8 (Cq), 48.0 (Cq), 50.9 (CH), 51.4 (CH3), 53.0 (CH2), 54.1 (2xCH2), 57.1 (CH), 57.3 (CH), 57.7 (CH2), 67.1 (2xCH2), 177.8 (C=O), 224.1 (C=O). HRMS (ESI+): m/z calcd. for C26H44NO4 [M + H]+ 432.3114; found 432.3108.
(4R,6aS,7R,9S,11bS)-Methyl 7-((benzyl(methyl)amino)methyl)-4,9,11b-trimethyl-8-oxotetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (15): The reaction was accomplished starting from compound 13 with N-methyl-N-benzylamine hydrochloride (1.80 mmol, 0.28 g) according to the general procedure. Yield: 0.40 g (47%); white crystals; m.p. 154–155 °C; = –40 (c 0.73 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.71 (s, 3H), 0.87–0.90 (m, 1H), 0.93 (s, 3H), 0.99–1.05 (m, 1H), 1.16–1.17 (m, 1H), 1.21 (m, 4H), 1.23–1.29 (m, 2H), 1.32–1.38 (m, 1H), 1.43 (d, 1H, J = 14.1 Hz), 1.60 (d, 1H, J = 12.3 Hz), 1.67–1.70 (m, 3H), 1.79–1.86 (m, 2H), 2.02–2.10 (m, 2H), 2.15–2.19 (m, 4H), 2.35 (d, 1H, J = 4.0 Hz, 12.6 Hz), 2.26–2.60 (m, 1H), 2.66–2.70 (m, 1H), 3.43 (d, 1H, J = 13.1 Hz), 3.51 (s, 3H), 3.59 (d, 1H, J = 13.1 Hz), 7.22–7.25 (m, 1H), 7.30–7.36 (m, 4H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.1 (CH3), 18.9 (CH2), 19.6 (CH2), 20.2 (CH3), 22.5 (CH2), 28.8 (CH3), 35.9 (CH2), 37.5 (CH2), 38.0 (CH2), 38.2 (Cq), 39.7 (CH2), 41.8 (CH3), 41.8 (Cq), 43.8 (Cq), 47.9 (Cq), 50.9 (CH3), 51.3 (CH), 52.8 (CH2), 57.1 (CH), 57.3 (Cq), 57.3 (CH), 63.5 (CH2), 126.9 (CH), 128.1 (2xCH), 129.1 (2xCH), 139.1 (Cq), 177.9 (C=O), 224.2 (C=O). HRMS (ESI+): m/z calcd. for C30H44NO3 [M + H]+ 466.3321; found 466.3316.
(4R,6aS,7R,9S,11bS)-Methyl 4,9,11b-trimethyl-8-oxo-7-(pyrrolidin-1-ylmethyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (16): The reaction was accomplished starting from compound 13 with pyrrolidine hydrochloride (1.80 mmol, 0.19 g) according to the general procedure. Yield: 0.44 g (59%); white crystals; m.p. 138–139 °C; = –61 (c 0.77 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.69 (s, 3H), 0.86–0.92 (m, 1H), 0.95 (s, 3H), 0.99–1.05 (m, 1H), 1.12–1.24 (m, 7H), 1.24–1.26 (m, 1H), 1.31–1.37 (m, 1H), 1.40–1.44 (m, 1H), 1.57–1.60 (m, 1H), 1.66–1.70 (m, 2H), 1.74–1.85 (m, 7H), 1.87–1.93 (m, 1H), 2.10–2.18 (m, 2H), 2.45–2.52 (m, 6H), 2.72–2.76 (m, 1H), 3.62 (s, 3H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.0 (CH3), 18.9 (CH2), 19.6 (CH2), 20.3 (CH3), 21.9 (CH2), 23.9 (2xCH2), 28.7 (CH3), 35.8 (CH2), 37.6 (CH2), 38.1 (CH2), 38.2 (Cq), 39.7 (CH2), 41.7 (Cq), 43.8 (Cq), 47.9 (Cq), 51.0 (CH3), 52.3 (CH), 52.8 (CH2), 54.5 (2xCH2), 54.7 (CH2), 57.2 (CH), 57.3 (CH), 180.0 (C=O), 224.3 (C=O). HRMS (ESI+): m/z calcd. for C26H42NO3 [M + H]+ 416.3165; found 416.3159.
(4R,6aS,7R,9S,11bS)-Methyl 7-((dimethylamino)methyl)-4,9,11b-trimethyl-8-oxotetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (17): The reaction was accomplished starting from compound 13 with dimethylamine hydrochloride (1.80 mmol, 0.15 g) according to the general procedure. Yield: 0.44 g (62%); white crystals; m.p. 129–130 °C; = –66 (c 0.16 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.71 (s, 3H), 0.86–0.92 (m, 1H), 0.95 (s, 3H), 0.98–1.05 (m, 1H), 1.13–1.16 (m, 2H), 1.18 (m, 4H), 1.22–1.28 (m, 2H), 1.31–1.37 (m, 1H), 1.41–1.45 (m, 1H), 1.58–1.62 (m, 1H), 1.65–1.70 (m, 2H), 1.73 (d, 1H, J = 2.7 Hz, 11.9 Hz),1.77–1.86 (m, 2H), 1.88–1.97 (m, 1H), 2.00–2.04 (m, 1H), 2.17 (d, 1H, J = 13.3 Hz), 2.23 (s, 6H), 2.24–2.28 (m, 1H), 2.44–2.50 (m, 2H), 3.62 (s, 3H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.1 (CH3), 18.9 (CH2), 19.5 (CH2), 20.3 (CH3), 21.9 (CH2), 28.7 (CH3), 35.9 (CH2), 37.6 (CH2), 38.1 (CH2), 38.2 (Cq), 39.7 (CH2), 41.7 (Cq), 43.8 (Cq), 45.9 (2xCH3), 48.0 (Cq), 50.9 (CH3), 51.0 (CH), 52.7 (CH2), 57.1 (CH), 57.3 (CH), 58.4(CH2), 177.9 (C=O), 224.0 (C=O). HRMS (ESI+): m/z calcd. for C24H40NO3 [M + H]+ 390.3008; found 390.3004.
(4R,6aS,7R,9S,11bS)-Methyl 7-((diethylamino)methyl)-4,9,11b-trimethyl-8-oxotetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (18): The reaction was accomplished starting from compound 13 with diethylamine hydrochloride (1.80 mmol, 0.20 g) according to the general procedure. Yield: 0.42 g (56%); white crystals; m.p. 110–111 °C; = –18 (c 0.41 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.71 (s, 3H), 0.86–0.91 (m, 1H), 0.93 (s, 3H), 0.98–1.06 (m, 7H), 1.13–1.17 (m, 2H), 1.18 (m, 4H), 1.22–1.27 (m, 2H), 1.30–1.36 (m, 1H), 1.57–1.59 (m, 1H), 1.65–1.70 (m, 3H), 1.74–1.89 (m, 2H), 1.97–2.03 (m, 1H), 2.06–2.10 (m, 1H), 2.17 (d, 1H, J = 13.3 Hz), 2.33 (dd, 1H, J = 3.7 Hz, 12.9 Hz), 2.42–2.52 (m, 3H), 2.59–2.68 (m, 3H), 3.61 (s, 3H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 11.1 (2xCH3), 13.0 (CH3), 18.9 (CH2), 19.5 (CH2), 20.2 (CH3), 22.1 (CH2), 28.8 (CH3), 36.1 (CH2), 37.4 (CH2), 38.2 (CH2), 38.3 (Cq), 39.7 (CH2), 41.7 (Cq), 43.8 (Cq), 46.3 (2xCH2), 47.9 (Cq), 51.2 (CH3), 51.7 (CH), 53.0 (CH2), 53.1 (CH2), 57.3 (CH), 57.5 (CH), 178.0 (C=O), 224.6 (C=O). HRMS (ESI+): m/z calcd. for C26H44NO3 [M + H]+ 418.3297; found 418.3302.
General procedure for preparation of aminoalcohols with Sodium borohydride: To a solution of amino ketones 14–18 (0.92 mmol) in dry MeOH (10 mL) NaBH4 (1.84 mmol, 0.07 g) was added in small portions under ice cooling. After stirring for 2–3 h, the mixture was evaporated to dryness, and the residue was dissolved in H2O (20 mL) and extracted with DCM (3 × 20 mL). The combined organic layer was dried (Na2SO4), filtered and evaporated to dryness. The crude product obtained was purified by column chromatography on silica gel (CHCl3/MeOH = 19:1).
(4R,6aS,7R,8S,9S,11bS)-Methyl 8-hydroxy-4,9,11b-trimethyl-7-(morpholinomethyl)tetradecahydro-6a,9 methanocyclohepta[a]naphthalene-4-carboxylate (19a): The reaction was accomplished starting from compound 14 according to the general procedure. Yield: 0.12 g (28%); white crystals; m.p. 123–124 °C; = –46 (c 0.38 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.69 (s, 3H), 0.75 (d, 1H, J = 11.3 Hz), 1.87–0.92 (m, 2H), 0.95 (s, 3H), 1.00–1.03 (m, 1H), 1.06–1.09 (m, 1H), 1.14–1.18 (m, 4H), 1.19–1.24 (m, 2H), 1.40–1.47 (m, 2H), 1.58–1.60 (m, 2H), 1.64–1.71 (m, 2H), 1.75–1.82 (m, 3H), 2.17 (d, 1H, J = 13.5 Hz), 2.31–2.46 (m, 4H), 2.67–2.75 (m, 3H), 3.64 (s, 3H), 3.68–3.70 (m, 4H), 3.79 (d, 1H, J = 6.2 Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.1 (CH3), 19.0 (CH2), 19.4 (CH2), 21.1 (CH3), 22.0 (CH2), 28.8 (CH3), 34.0 (CH2), 38.0 (CH2), 38.1 (Cq), 38.2 (CH2), 39.2 (CH), 39.7 (CH2), 42.1 (Cq), 43.8 (Cq), 44.8 (Cq), 51.0 (CH3), 52.7 (CH2), 53.0 (2xCH2), 56.7 (CH), 57.2 (2xCH), 66.9 (2xCH2), 88.1 (CH), 177.8 (C=O). HRMS (ESI+): m/z calcd. for C26H44NO4 [M + H]+ 434.3270; found 434.3265.
(4R,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-4,9,11b-trimethyl-7-(morpholinomethyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (19b): The reaction was accomplished starting from compound 14 according to the general procedure. Yield: 0.10 g (24%); white crystals; m.p. 147–148 °C; = –58 (c 0.41 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.70 (s, 3H), 0.85–0.91 (m, 2H), 0.93 (s, 3H), 0.96–1.08 (m, 4H), 1.16–1.22 (m, 4H), 1.35 (dd, 1H, J = 2.5 Hz, 11.5 Hz), 1.40–1.43 (m, 1H), 1.58–1.65 (m, 4H), 1.68–1.72 (m, 2H), 1.76–1.82 (m, 3H), 2.00–2.02 (m, 1H), 2.15–2.23 (m, 2H), 2.34 (s, 1H), 2.53 (dd, 1H, J = 3.9 Hz, 11.7 Hz), 2.68 (s, 2H), 3.44 (d, 1H, J = 5.1 Hz), 3.63 (s, 3H), 3.66–3.68 (m, 2H), 3.72–3.76 (m, 2H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 12.9 (CH3), 18.9 (CH2), 19.5 (CH2), 22.2 (CH2), 25.1 (CH3), 28.9 (CH3), 32.9 (CH2), 34.9 (CH2), 38.0 (CH2), 38.0 (Cq), 39.5 (CH2), 40.5 (Cq), 41.8 (Cq), 43.7 (Cq), 44.1 (CH), 51.1 (CH3), 54.2 (3xCH2), 57.0 (CH), 57.8 (CH), 61.9 (CH2), 67.3 (2xCH2), 88.7 (CH), 177.9 (C=O). HRMS (ESI+): m/z calcd. for C26H44NO4 [M + H]+ 434.3270; found 434.3265.
(4R,6aS,7R,8S,9S,11bS)-Methyl 7-((benzyl(methyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (20a): The reaction was accomplished starting from compound 15 according to the general procedure. Yield: 0.02 g (10%); white crystals; m.p. 125–127 °C; = –37 (c 0.38 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.70 (s, 3H), 0.76 (d, 1H, J = 11.5 Hz), 0.87–0.93 (m, 2H), 0.98 (s, 3H), 1.01–1.04 (m, 1H), 1.08 (d, 1H, J = 12.1 Hz), 1.16–1.25 (m, 6H), 1.40–1.47 (m, 2H), 1.57–1.62 (m, 1H), 1.65–1.71 (m, 3H), 1.78–1.82 (m, 3H), 2.15–2.21 (m, 4H), 2.41–2.50 (m, 2H), 2.90 (t, 1H, J = 11.8 Hz), 3.45–3.51 (m, 1H), 3.63–3.67 (m, 4H), 3.85 (d, 1H, J = 3.1 Hz), 7.24–7.33 (m, 5H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.2 (CH3), 19.0 (CH2), 19.4 (CH2), 21.2 (CH3), 22.1 (CH2), 29.0 (CH3), 34.0 (CH2), 37.9 (CH2), 38.1 (CH2), 38.2 (CH2), 39.6 (CH2), 40.5 (CH), 40.8 (CH3), 42.1 (Cq), 43.8 (Cq), 44.9 (Cq), 51.2 (CH3), 52.8 (CH2), 56.8 (CH2), 57.2 (CH), 57.2 (CH), 61.8 (CH2), 80.4 (CH), 127.3 (CH), 128.4 (2xCH), 128.9 (2xCH), 138.1 (Cq), 177.9 (C=O). HRMS (ESI+): m/z calcd. for C30H46NO3 [M + H]+ 468.3478; found 468.3472.
(4R,6aS,7R,8R,9S,11bS)-Methyl 7-((benzyl(methyl)amino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (20b): The reaction was accomplished starting from compound 15 according to the general procedure. Yield: 0.18 g (42%). Alternative procedure for the synthesis of 24b: To a solution of 8 (0.12 mmol, 0.05 g) in DCM (5 mL), Et3N (0.12 mmol, 15 µL) and iodomethane (0.12 mmol, 7 µL) were added. The solution was stirred for 4 h at room temperature. Then water (20 mL) was added, and the mixture was extracted with DCM (3 × 15 mL). The organic phase was dried (Na2SO4) and evaporated to dryness. The crude product was purified by column chromatography on silica gel (CHCl3/MeOH 9:1). Yield: 0.04 g (77%); white crystals; m.p. 113–114 °C; = –21 (c 0.24 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.73 (s, 3H), 0.84–0.90 (m, 5H), 0.94–1.07 (m, 4H), 1.13–1.20 (m, 4H), 1.26–1.28 (m, 1H), 1.38–1.42 (m, 1H), 1.55–1.60 (m, 2H), 1.62–1.73 (m, 3H), 1.75–1.83 (m, 3H), 2.03–2.07 (m, 1H), 2.16 (d, 1H, J = 12.6 Hz), 2.27 (s, 3H), 2.36 (t, 1H, J = 11.9 Hz), 2.49–2.52 (m, 1H), 3.37–3.42 (m, 2H), 3.75 (d, 1H, J = 13.3 Hz), 7.23–7.28 (m, 1H), 7.30–7.32 (m, 4H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.0 (CH3), 19.0 (CH2), 19.6 (CH2), 22.2 (CH2), 25.1 (CH3), 28.9 (CH3), 33.1 (CH2), 34.9 (CH2), 38.0 (CH2), 38.1 (CHq), 39.6 (CH2), 40.5 (Cq), 42.2 (Cq), 42.6 (CH3), 43.8 (Cq), 45.2 (CH), 51.0 (CH3), 54.3 (CH2), 57.1 (CH), 58.0 (CH), 60.5 (CH2), 63.2 (CH2), 88.4 (CH), 127.2 (CH), 128.4 (2xCH), 129.2 (2xCH), 139.2 (Cq), 177.8 (C=O). HRMS (ESI+): m/z calcd. for C30H46NO3 [M + H]+ 468.3478; found 468.3472.
(4R,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-4,9,11b-trimethyl-7-(pyrrolidin-1-ylmethyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (21): The reaction was accomplished starting from compound 16 according to the general procedure. Yield: 0.30 g (70%); white crystals; m.p. 143–144 °C; = –20 (c 0.35 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.68 (s, 3H), 0.81 (d, 1H, J = 11.7 Hz), 0.85–0.92 (m, 2H), 0.94 (s, 3H), 0.99–1.10 (m, 1H), 1.09 (dd, 1H, J = 2.1 Hz, 12.1 Hz), 1.13–1.21 (m, 4H), 1.22–1.26 (m, 1H), 1.27–1.32 (m, 1H), 1.42–1.54 (m, 3H), 1.59–1.72 (m, 5H), 1.82–1.86 (m, 1H), 2.08 (s, 2H), 2.17–2.19 (m, 1H), 2.35–2.39 (m, 3H), 2.71–2.73 (m, 1H), 2.88 (s, 1H), 3.58–3.63 (m, 1H), 3.64 (s, 3H), 3.86 (s, 1H), 3.96 (s, 1 H), 4.07 (d, 1H, J = 6.7 Hz), 4.76 (s, 1H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.6 (CH3), 19.0 (CH2), 19.3 (CH2), 21.5 (CH2), 21.7 (CH3), 23.2 (CH2), 23.6 (CH2), 29.1 (CH3), 34.2 (CH2), 37.3 (CH2), 37.6 (CH2), 37.9 (Cq), 39.4 (CH2), 41.4 (CH), 42.6 (Cq), 43.8 (Cq), 44.9 (Cq), 51.4 (CH3), 53.0 (CH2), 53.3 (CH2), 55.3 (CH2), 56.4 (CH2), 56.8 (CH), 56.9 (CH), 76.4 (CH), 177.7 (C=O). HRMS (ESI+): m/z calcd. for C26H44NO3 [M + H]+ 418.3321; found 418.3316.
(4R,6aS,7R,8R,9S,11bS)-Methyl 7-((dimethylamino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (22): The reaction was accomplished starting from compound 17 according to the general procedure. Yield: 0.32 g (75%); white crystals; m.p. 149–150 °C; = –70 (c 0.33 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.70 (s, 3H), 0.82 (d, 1H, J = 11.7 Hz), 0.85–0.91 (m, 1H), 0.92–0.96 (m, 4H), 0.99–1.05 (m, 1H), 1.09 (dd, 1H, J = 2.1 Hz, 12.1 Hz), 1.17 (s, 3H), 1.19–1.32 (m, 3H), 1.42–1.53 (m, 3H), 1.60–1.64 (m, 3H), 1.70–1.77 (m, 2H), 1.83–1.86 (m, 1H), 2.18 (d, 1H, J = 13.4 Hz),1.42–1.47 (m, 2H), 2.73 (dd, 1H, J = 2.0 Hz, 11.6 Hz), 2.84 (s, 6H), 3.40 (t, 1H, J = 12.4 Hz), 3.63 (s, 3H), 4.07 (d, 1H, J = 6.8 Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.6 (CH3), 18.9 (CH2), 19.3 (CH2), 21.5 (CH3), 21.5 (CH2), 29.0 (CH3), 34.2 (CH2), 37.3 (CH2), 37.6 (CH2), 38.0 (Cq), 39.4 (CH2), 40.3 (CH), 41.7 (CH3), 42.6 (Cq), 43.8 (Cq), 44.9 (Cq), 45.9 (CH3), 51.3 (CH3), 52.9 (CH2), 56.9 (CH), 57.0 (CH), 57.7 (CH2), 76.7 (CH), 177.6 (C=O), 224.0 (C=O). HRMS (ESI+): m/z calcd. for C24H42NO3 [M + H]+ 392.3165; found 392.3159.
(4R,6aS,7R,8S,9S,11bS)-Methyl 7-((diethylamino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (23a): The reaction was accomplished starting from compound 18 according to the general procedure. Yield: 0.04 g (10%); white crystals; m.p. 111–112 °C; = –51 (c 0.24 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.69 (s, 3H), 0.82 (d, 1H, J = 11.6 Hz), 0.85–0.88 (m, 1H), 0.92–0.95 (m, 4H), 1.00–1.05 (m, 1H), 1.10 (dd, 1H, J = 1.7 Hz, 11.9 Hz), 1.14–1.20 (m, 4H), 1.26–1.32 (m, 2H), 1.40–1.54 (m, 9H), 1.60–1.63 (m, 3H), 1.70–1.78 (m, 2H), 1.86 (m, 1 H, J = 13.9 Hz), 2.19 (d, 1H, J = 13.4 Hz), 2.42–2.45 (m, 1H), 2.75 (d, 1H, J = 10.5 Hz), 3.16 (m, 3H), 3.35 (t, 1H, J = 12.6 Hz), 3.61 (s, 3H), 4.07 (d, 1H, J = 6.1 Hz), 4.79 (s, 1H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 7.4 (CH3), 9.0 (CH3), 13.5 (CH3), 18.9 (CH2), 19.3 (CH2), 21.6 (CH3), 21.7 (CH2), 28.9 (CH3), 34.3 (CH2), 37.3 (CH2), 37.7 (CH2), 38.0 (Cq), 39.4 (CH2), 39.8 (CH), 42.6 (Cq), 43.8 (Cq), 44.7 (CH2), 45.1 (Cq), 49.2 (CH2), 50.8 (CH2), 51.2 (CH3), 53.0 (CH2), 56.9 (CH), 57.0 (CH), 76.4 (CH), 177.6 (C=O). HRMS (ESI+): m/z calcd. for C26H46NO3 [M + H]+ 420.3478; found 420.3472.
(4R,6aS,8R,9S,11bS)-Methyl 7-((diethylamino)methyl)-8-hydroxy-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (23b): The reaction was accomplished starting from compound 18 according to the general procedure. Yield: 0.04 g (10%); white crystals; m.p. 120–121 °C; = –41 (c 0.11 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.72 (s, 3H), 0.87–0.90 (m, 1H), 0.94 (s, 3H), 0.99–1.05 (m, 3H), 1.08–1.12 (m, 1H), 1.15–1.22 (m, 5H), 1.33–1.36 (m, 4H), 1.43–1.46 (m, 1H), 1.51–1.54 (m, 5H), 1.61–1.64 (m, 2H), 1.73–1.80 (m, 2H), 1.84–1.90 (m, 2 H), 2.19 (d, 1H, J = 13.0 Hz), 2.26 (d, 1H, J = 13.0 Hz), 2.94 (t, 1H, J = 12.7 Hz), 3.01–3.03 (m, 1H), 3.17–3.27 (m, 3H), 3.46–3.48 (m, 1H), 3.61 (s, 3H), 3.87 (m, 1H), 4.95 (s, 1H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 6.8 (CH3), 9.2 (CH3), 13.2 (CH3), 18.9 (CH2), 19.5 (CH2), 22.0 (CH2), 24.6 (CH3), 28.9 (CH3), 33.0 (CH2), 34.9 (CH2), 37.7 (CH2), 38.1 (Cq), 39.4 (CH2), 41.6 (Cq), 42.4 (CH), 43.2 (Cq), 43.8 (Cq), 44.3 (CH2), 49.5 (CH2), 51.2 (CH3), 53.6 (CH2), 55.7 (CH2), 56.9 (CH), 57.8 (CH), 84.3 (CH), 177.6 (C=O). HRMS (ESI+): m/z calcd. for C26H46NO3 [M + H]+ 420.3478; found 420.3472.
(4R,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-4,9,11b-trimethyl-7-((methylamino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (24): To a suspension of palladium-on-carbon (5% Pd/C, 0.10 g) in MeOH (15 mL), 20a was added (0.24 mmol, 0.10 g) in MeOH (15 mL), and the mixture was stirred under a H2 atmosphere (1 atm) at room temperature. After completion of the reaction (as monitored by TLC, 24 h), the mixture was filtered through a Celite pad, and the solution was evaporated to dryness. The crude product was purified by column chromatography on silica gel (CHCl3/MeOH = 9:1). Yield: 0.02 g (28%); white crystals; m.p. 122–123 °C; = –65 (c 0.19 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.70 (s, 3H), 0.79 (d, 1H, J = 11.6 Hz), 0.85–0.92 (m, 2H), 0.94 (s, 3H), 0.98–1.04 (m, 1H), 1.08 (d, 1H, J = 11.8 Hz), 1.17 (m, 3H), 1.21–1.26 (s, 3H), 1.40–1.44 (m, 1H), 1.48–1.54 (m, 2H), 1.58–1.83 (m, 6H), 2.18 (d, 1H, J = 13.3 Hz), 2.41–2.44 (m, 1H), 2.55–2.57 (m, 1H), 2.68 (s, 3H), 3.20 (m, 1H), 3.63 (s, 3H), 3.97 (d, 1H, J = 6.6 Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.4 (CH3), 18.9 (CH2), 19.3 (CH2), 21.3 (CH3), 21.7 (CH2), 29.0 (CH3), 34.1 (CH2), 37.5 (CH2), 37.7 (CH2), 38.0 (CH2), 39.5 (CH2), 40.3 (CH), 42.4 (Cq), 43.8 (Cq), 44.1 (CH3), 44.9 (Cq), 51.2 (CH3), 52.8 (CH2), 57.0 (CH), 57.0 (CH), 57.8 (CH2), 77.8 (CH), 177.6 (C=O). HRMS (ESI+): m/z calcd. for C23H40NO3 [M + H]+ 378.3008; found 478.3003.
(4R,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-4,9,11b-trimethyl-7-((3-phenylthioureido)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (25): Compound 6 (0.20 g, 0.55 mmol) was dissolved in DCM (10 mL) and phenyl isothiocyanate (0.55 mmol, 0.06 mL) was added. The reaction mixture was stirred for 2 h at room temperature. After completion of the reaction (monitored by TLC), the mixture was extracted with water (3 × 10 mL). The water phase was extracted with DCM (3 × 10 mL) and the combined organic phase was dried with Na2SO4 and concentrated under vacuum. The crude product was purified by column chromatography on silica gel with CHCl3/MeOH 9:1. Yield: 0.25 g (91%); white crystals; m.p. 153–154 °C; = +9 (c 0.48 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.72 (s, 3H), 0.84–0.86 (m, 1H), 0.87 (s, 3H), 0.95–1.01 (m, 3H), 1.04–1.12 (m, 2H), 1.16 (s, 3H), 1.19–1.24 (m, 1H), 1.38–1.43 (m, 2H), 1.46–1.50 (m, 1H), 1.57–1.59 (m, 1H), 1.64–1.77 (m, 4H), 1.79–1.83 (m, 2H), 1.99–2.02 (m, 1H), 2.16 (d, 1H, J = 13.3 Hz), 3.40 (d, 1H, J = 4.9 Hz), 1.43–1.46 (m, 1H), 3.63 (s, 3H), 3.92–3.97 (m, 1H), 6.52 (s, 1H), 7.25–7.29 (m, 3H), 7.41 (t, 2H, J = 8.3 Hz); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.0 (CH3), 18.9 (CH2), 19.4 (CH2), 22.2 (CH2), 24.7 (CH3), 28.7 (CH3), 32.9 (CH2), 34.9 (CH2), 38.1 (CH2), 38.1 (Cq), 39.7 (CH2), 41.1 (Cq), 42.5 (Cq), 43.7 (Cq), 46.9 (CH), 48.8 (CH2), 51.3 (CH3), 53.8 (CH2), 57.1 (CH), 57.6 (CH), 87.6 (CH), 125.0 (2xCH), 127.0 (CH), 130.0 (2xCH), 136.3 (Cq), 177.9 (C=O), 181.0 (Cq). HRMS (ESI+): m/z calcd. for C29H43N2O3S [M + H]+ 498.2994; found 498.2989.
(4R,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-4,9,11b-trimethyl-7-((((methylthio)(phenylimino)methyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (26): To a solution of 25 (0.30 mmol, 0.15 g), EtOH (10 mL) and iodomethane (1.50 mmol, 0.14 mL) were added. The solution was stirred for 2 h at room temperature then water (20 mL) was added, and the mixture was extracted with DCM (3 × 15 mL). The organic phase was dried with Na2SO4 and evaporated to dryness. The crude product was purified by column chromatography on silica gel (CHCl3/MeOH 9:1). Yield: 0.11 g (69%); white crystals; m.p. 164–165 °C; = –92 (c 0.10 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.71 (s, 3H), 0.82–0.88 (m, 4H), 0.92–1.07 (m, 5H), 1.12–1.17 (m, 4H), 1.29–1.32 (m, 2H), 1.38–1.43 (m, 1H), 1.51–1.70 (m, 4H), 1.67–1.70 (m, 2H), 1.76–1.82 (m, 2H), 1.87–1.89 (m, 1H), 2.16 (d, 1H, J = 13.3 Hz), 2.83 (t, 1H, J = 10.8 Hz), 3.30 (s, 1H), 3.47 (d, 1H, J = 12.0 Hz), 3.64 (s, 3H), 3.86 (s, 3H), 4.28 (s, 1H), 6.92 (d, 2H, J = 7.6 Hz), 6.98 (t, 1H, J = 7.4 Hz), 7.26–7.30 (m, 3H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 12.8 (CH3), 18.9 (CH2), 19.4 (CH2), 22.1 (CH2), 24.9 (CH3), 28.9 (CH3), 33.0 (CH2), 34.9 (CH2), 38.0 (CH2), 38.1 (Cq), 39.6 (CH2), 41.0 (Cq), 42.4 (Cq), 43.8 (Cq), 44.4 (CH2), 47.5 (CH), 51.2 (CH3), 53.5 (CH3), 53.9 (CH2), 57.2 (CH), 57.6 (CH), 87.5 (CH), 122.4 (CH), 122.9 (2xCH), 129.4 (2xCH), 148.8 (Cq), 154.0 (Cq), 177.8 (C=O). HRMS (ESI+): m/z calcd. for C30H45N2O3S [M + H]+ 513.3151; found 513.3145.
(4R,6aS,7R,8R,9S,11bS)-Methyl 8-hydroxy-4,9,11b-trimethyl-7-((((ethylthio)(phenylimino)methyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (28): A solution of 25 (0.60 mmol, 0.30 g) in dry EtOH (10 mL) was added EtOH containing 18% hydrochloric acid (10 mL) and mixture was stirred at reflux temperature for 12 h. The reaction mixture was then concentrated under vacuum, dissolved in water (20 mL) and extracted with DCM (3 × 15 mL). The combined organic layer was washed with saturated NaCl aqueous solution (15 mL), dried (Na2SO4) and concentrated under vacuum. The crude product was purified by column chromatography on silica gel with CHCl3/MeOH 9:1.Yield: 0.15 g (46%); white crystals; m.p. 114–115 °C; = –37 (c 0.28 MeOH); 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.76 (s, 3H), 0.85–0.89 (m, 1H), 0.92 (s, 3H), 0.96–1.08 (m, 5H), 1.15 (s, 3H), 1.20–1.36 (m, 5H), 1.39–1.41 (m, 2H), 1.61–1.80 (m, 8H), 2.06–2.08 m, 1H), 2.16 (d, 1H, J = 13.3 Hz), 2.68–2.84 (m, 2H), 3.15 (t, 1H, J = 12.1 Hz), 3.49–3.50 (m, 1H), 3.56 (s, 3H), 3.68 (d, 1H, J = 9.1 Hz), 6.90 (d, 2H, J = 7.4 Hz), 6.99 (t, 1H, J = 7.4 Hz), 7.25–7.26 (m, 2H); 13C-NMR (125 MHz, CDCl3) δ (ppm): 13.0 (CH3), 14.6 (CH3), 18.9 (CH2), 19.5 (CH2), 22.2 (CH2), 24.8 (CH3), 25.3 (CH2), 28.8 (CH3), 33.0 (CH2), 34.9 (CH2), 38.1 (CH2), 38.2 (Cq), 39.7 (CH2), 41.1 (Cq), 42.5 (Cq), 43.8 (Cq), 46.1 (CH2), 47.3 (CH), 51.1 (CH3), 54.0 (CH2), 57.2 (CH), 57.7 (CH), 88.2 (CH), 122.3 (2xCH), 122.5 (CH), 128.7 (2xCH), 149.8 (Cq), 177.9 (C=O). HRMS (ESI+): m/z calcd. for C29H41N2O2S [M + H]+ 527.3307; found 527.3301.
Determination of antiproliferative effect: The growth-inhibitory effects of the isosteviol-based 1,3-aminoalcohols were determined by a standard MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay on a panel containing five cell lines, including Hela and Siha (cervical cancers), MDA-MB-231 and MCF-7 (breast cancers), and A2780 ovarian cancer cells []. All cell lines were purchased from European Collection of Cell Cultures (Salisbury, UK) except the SiHa, which was obtained from American Tissue Culture Collection (Manassas, VA, USA). The cells were maintained in minimal essential medium supplemented with 10% foetal bovine serum, 1% non-essential amino acids, and 1% penicillin-streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. All media and supplements were obtained from Lonza Group Ltd., (Basel, Switzerland). Cancer cells were seeded into 96-well plates (5000 cells/well), after an overnight incubation the test compound was added in two different concentrations (10 µM and 30 µM) and incubated for other 72 h under cell-culturing condition. In the next step, 20 μL of 5 mg/mL MTT solution was added to each well and incubated for a further 4 h. The medium was removed, and the precipitated formazan crystals were dissolved in DMSO during 60 min of shaking at 37 °C. As the final step, the absorbance was measured at 545 nm by using a microplate reader. Untreated cells were included as controls. In the case of the most effective compounds (i.e., compounds eliciting higher than 50 or 85% at 10 or 30 μM, respectively), the assays were repeated with a set of dilutions (0.1–30 μM) in order to determine the IC50 values. Two independent experiments were performed with five wells for each condition. Cisplatin (Ebewe GmbH, Unterach, Austria), a clinically used anticancer agent, was used as a positive control. Calculations were performed by means of the GraphPad Prism 5.01 software (GraphPad Software Inc., San Diego, CA, USA).
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/ijms222011232/s1, 1H, 13C, COSY, HSQC, HMBC, and NOESY NMR spectra of new compounds are available.
Author Contributions
Z.S. and I.Z. conceived and designed the experiments; D.O. and V.N. performed the experiments, analysed the data and wrote the experimental part; Z.S. and I.Z. discussed the results and contributed to write the paper. All authors have read and agreed to the published version of the manuscript.
Funding
We are grateful for financial support from the Hungarian Research Foundation (NKFI K138871), UNKP-21-3-SZTE-272 and Ministry of Human Capacities, Hungary grant 20391-3/2018/FEKUSTRAT.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gupta, P.; Phulara, S. Terpenoids: Types and their application. In Biotechnology of Terpenoid Production from Microbial Cell Factories; Elsevier: Amsterdam, The Netherlands, 2021; pp. 47–78. ISBN 978-0-12-819917-6. [Google Scholar]
- Ludwiczuk, A.; Skalicka-Woźniak, K.; Georgiev, M.I. Terpenoids. In Pharmacognosy; Elsevier: Amsterdam, The Netherlands, 2017; pp. 233–266. ISBN 978-0-12-802104-0. [Google Scholar]
- Chatsudthipong, V.; Muanprasat, C. Stevioside and Related Compounds: Therapeutic Benefits beyond Sweetness. Pharmacol. Ther. 2009, 121, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Moons, N.; De Borggraeve, W.; Dehaen, W. Stevioside and Steviol as Starting Materials in Organic Synthesis. Curr. Org. Chem. 2012, 16, 1986–1995. [Google Scholar] [CrossRef]
- Geuns, J.M. Stevioside. Phytochemistry 2003, 64, 913–921. [Google Scholar] [CrossRef]
- Bhasker, S.; Madhav, H.; Chinnamma, M. Molecular Evidence of Insulinomimetic Property Exhibited by Steviol and Stevioside in Diabetes Induced L6 and 3T3L1 Cells. Phytomedicine 2015, 22, 1037–1044. [Google Scholar] [CrossRef]
- Lee, C.-N.; Wong, K.-L.; Liu, J.-C.; Chen, Y.-J.; Cheng, J.-T.; Chan, P. Inhibitory Effect of Stevioside on Calcium Influx to Produce Antihypertension. Planta Med. 2001, 67, 796–799. [Google Scholar] [CrossRef]
- Liu, J.-C.; Kao, P.-K.; Chan, P.; Hsu, Y.-H.; Hou, C.-C.; Lien, G.-S.; Hsieh, M.-H.; Chen, Y.-J.; Cheng, J.-T. Mechanism of the Antihypertensive Effect of Stevioside in Anesthetized Dogs. Pharmacology 2003, 67, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, M.; Konoshima, T.; Kozuka, M.; Tokuda, H.; Takayasu, J.; Nishino, H.; Miyakoshi, M.; Mizutani, K.; Lee, K.-H. Cancer Preventive Agents. Part 8: Chemopreventive Effects of Stevioside and Related Compounds. Bioorg. Med. Chem. 2009, 17, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.-P.; Yin, X.-Y.; Yu, H.-Y.; Xiao, H.-F. Stevioside Induced Cytotoxicity in Colon Cancer Cells via Reactive Oxygen Species and Mitogen-Activated Protein Kinase Signaling Pathways-Mediated Apoptosis. Oncol. Lett. 2017, 13, 2337–2343. [Google Scholar] [CrossRef]
- Boonkaewwan, C.; Ao, M.; Toskulkao, C.; Rao, M.C. Specific Immunomodulatory and Secretory Activities of Stevioside and Steviol in Intestinal Cells. J. Agric. Food Chem. 2008, 56, 3777–3784. [Google Scholar] [CrossRef]
- Singh, N.; Jaggi, A.; Sharma, D.; Puri, M.; Tiwary, A. Antiamnesic Effect of Stevioside in Scopolamine-Treated Rats. Indian J. Pharmacol. 2010, 42, 164. [Google Scholar] [CrossRef]
- Holvoet, P.; Rull, A.; García-Heredia, A.; López-Sanromà, S.; Geeraert, B.; Joven, J.; Camps, J. Stevia-Derived Compounds Attenuate the Toxic Effects of Ectopic Lipid Accumulation in the Liver of Obese Mice: A Transcriptomic and Metabolomic Study. Food Chem. Toxicol. 2015, 77, 22–33. [Google Scholar] [CrossRef]
- Kinghorn, A.D.; Soejarto, D.D.; Nanayakkara, N.P.D.; Compadre, C.M.; Makapugay, H.C.; Hovanec-Brown, J.M.; Medon, P.J.; Kamath, S.K. A Phytochemical Screening Procedure for Sweet Ent-Kaurene Glycosides in the Genus Stevia. J. Nat. Prod. 1984, 47, 439–444. [Google Scholar] [CrossRef]
- Yasukawa, K.; Kitanaka, S.; Seo, S. Inhibitory Effect of Stevioside on Tumor Promotion by 12-O-Tetradecanoylphorbol-13-Acetate in Two-Stage Carcinogenesis in Mouse Skin. Biol. Pharm. Bull. 2002, 25, 1488–1490. [Google Scholar] [CrossRef]
- Chang, S.-F.; Yang, L.-M.; Lo, C.-H.; Liaw, J.-H.; Wang, L.-H.; Lin, S.-J. Microbial Transformation of Isosteviol and Bioactivities against the Glucocorticoid/Androgen Response Elements. J. Nat. Prod. 2008, 71, 87–92. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, C.-J.; Liu, X.; Dai, G.-F.; Du, J.-Y.; Tao, J.-C. Stereoselective Synthesis, Characterization, and Antibacterial Activities of Novel Isosteviol Derivatives with D-Ring Modification. Helv. Chim. Acta 2010, 93, 2052–2069. [Google Scholar] [CrossRef]
- Liu, C.-J.; Zhang, T.; Yu, S.-L.; Dai, X.-J.; Wu, Y.; Tao, J.-C. Synthesis, Cytotoxic Activity, and 2D- and 3D-QSAR Studies of 19-Carboxyl-Modified Novel Isosteviol Derivatives as Potential Anticancer Agents. Chem. Biol. Drug Des. 2017, 89, 870–887. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Munir, S.; Mabkhot, Y.; Badshah, S. Bioactivity Profile of the Diterpene Isosteviol and Its Derivatives. Molecules 2019, 24, 678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, B.; Xu, G.; Xu, C.; Ou, E.; Liu, J.; Sun, X.; Zhao, Y. Synthesis and in Vivo Screening of Isosteviol Derivatives as New Cardioprotective Agents. Eur. J. Med. Chem. 2021, 219, 113396. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Wu, J.; Shi, L.; Wang, K.; Zhou, B.; Tang, Y.; Zhang, D.; Wu, Y.; Hua, W.; Wu, X. Synthesis and Evaluation of Cytotoxic Effects of Novel α-Methylenelactone Tetracyclic Diterpenoids. Bioorg. Med. Chem. Lett. 2012, 22, 1922–1925. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.-J.; Yang, C.-L.; Kuo, Y.-C.; Chang, Y.-C.; Yang, L.-M.; Chou, B.-H.; Lin, S.-J. Synthesis and Anti-Hepatitis B Virus Activity of C4 Amide-Substituted Isosteviol Derivatives. Bioorg. Med. Chem. 2015, 23, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, D.; Wu, X. Synthesis and Biological Evaluation of Novel Exo-Methylene Cyclopentanone Tetracyclic Diterpenoids as Antitumor Agents. Bioorg. Med. Chem. Lett. 2011, 21, 130–132. [Google Scholar] [CrossRef] [PubMed]
- Jayachandra, R.; Zhao, H.; Cheng, Z.; Luo, L.; Sun, T.; Tan, W. Synthesis of Isosteviol Analogues as Potential Protective Agents against Doxorubicin-Induced Cardiomyopathy in Zebrafish Embryos. Bioorg. Med. Chem. Lett. 2019, 29, 1705–1709. [Google Scholar] [CrossRef]
- Zhang, T.; Lu, L.-H.; Liu, H.; Wang, J.-W.; Wang, R.-X.; Zhang, Y.-X.; Tao, J.-C. D-Ring Modified Novel Isosteviol Derivatives: Design, Synthesis and Cytotoxic Activity Evaluation. Bioorg. Med. Chem. Lett. 2012, 22, 5827–5832. [Google Scholar] [CrossRef] [PubMed]
- Szakonyi, Z.; Balázs, Á.; Martinek, T.A.; Fülöp, F. Enantioselective Addition of Diethylzinc to Aldehydes Catalyzed by γ-Amino Alcohols Derived from (+)- and (−)-α-Pinene. Tetrahedron Asymmetry 2006, 17, 199–204. [Google Scholar] [CrossRef]
- Shah, B.A.; Kumar, A.; Gupta, P.; Sharma, M.; Sethi, V.K.; Saxena, A.K.; Singh, J.; Qazi, G.N.; Taneja, S.C. Cytotoxic and Apoptotic Activities of Novel Amino Analogues of Boswellic Acids. Bioorg. Med. Chem. Lett. 2007, 17, 6411–6416. [Google Scholar] [CrossRef] [PubMed]
- Szakonyi, Z.; Gonda, T.; Ötvös, S.B.; Fülöp, F. Stereoselective Syntheses and Transformations of Chiral 1,3-Aminoalcohols and 1,3-Diols Derived from Nopinone. Tetrahedron Asymmetry 2014, 25, 1138–1145. [Google Scholar] [CrossRef]
- Liu, C.-J.; Yu, S.-L.; Liu, Y.-P.; Dai, X.-J.; Wu, Y.; Li, R.-J.; Tao, J.-C. Synthesis, Cytotoxic Activity Evaluation and HQSAR Study of Novel Isosteviol Derivatives as Potential Anticancer Agents. Eur. J. Med. Chem. 2016, 115, 26–40. [Google Scholar] [CrossRef]
- Melis, M.S. Renal Excretion of Stevioside in Rats. J. Nat. Prod. 1992, 55, 688–690. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, J.-H.; Dai, G.-F.; Liu, C.-J.; Tian, G.-Q.; Ma, W.-Y.; Tao, J.-C. Stereoselective Synthesis of Bioactive Isosteviol Derivatives as α-Glucosidase Inhibitors. Bioorg. Med. Chem. 2009, 17, 1464–1473. [Google Scholar] [CrossRef]
- Tao, J.-C.; Tian, G.-Q.; Zhang, Y.-B.; Fu, Y.-Q.; Dai, G.-F.; Wu, Y. Synthesis and Bioactivity of Isosteviol Derivatives: A Facile Method for Preparation of Ent-16α-Hydroxy-15β-Hydroxymethylbeyeran-19-Oic Acid. Chin. Chem. Lett. 2005, 16, 1441–1444. [Google Scholar]
- Ozsvár, D.; Nagy, V.; Zupkó, I.; Szakonyi, Z. Stereoselective Synthesis and Antiproliferative Activity of Steviol-Based Diterpen Aminodiols. Int. J. Mol. Sci. 2020, 21, 184. [Google Scholar] [CrossRef]
- Coates, R.M.; Kang, H.Y. Synthesis and Evaluation of Cyclobutylcarbinyl Derivatives as Potential Intermediates in Diterpene Biosynthesis. J. Org. Chem. 1987, 52, 2065–2074. [Google Scholar] [CrossRef]
- De Carvalho, E.M.; Figueroa Villar, J.D.; Greco, S.J.; Pinheiro, S.; Carneiro, J.W.D.M. Conformational Characterization of a Camphor-Based Chiral γ-Amino Alcohol. J. Mol. Struct. 2007, 827, 121–125. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Zupkó, I.; Sillanpää, R.; Fülöp, F. Stereoselective Synthesis and Cytoselective Toxicity of Monoterpene-Fused 2-Imino-1,3-Thiazines. Molecules 2014, 19, 15918–15937. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Zupkó, I.; Fülöp, F. Stereoselective Synthesis and Antiproliferative Activity of Monoterpene-Fused 2- Imino-1,3-Oxazines. Curr. Org. Synth. 2017, 14, 612–619. [Google Scholar] [CrossRef][Green Version]
- Bernáth, G.; Szakonyi, Z.; Fülöp, F.; Sohár, P. Synthesis of Tert-Butylcyclopentane-Fused 1,3-Oxazines and 1,3-Thiazines. Heterocycles 1994, 37, 1687. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).