
Myelodysplastic Syndromes and Metabolism

 ,
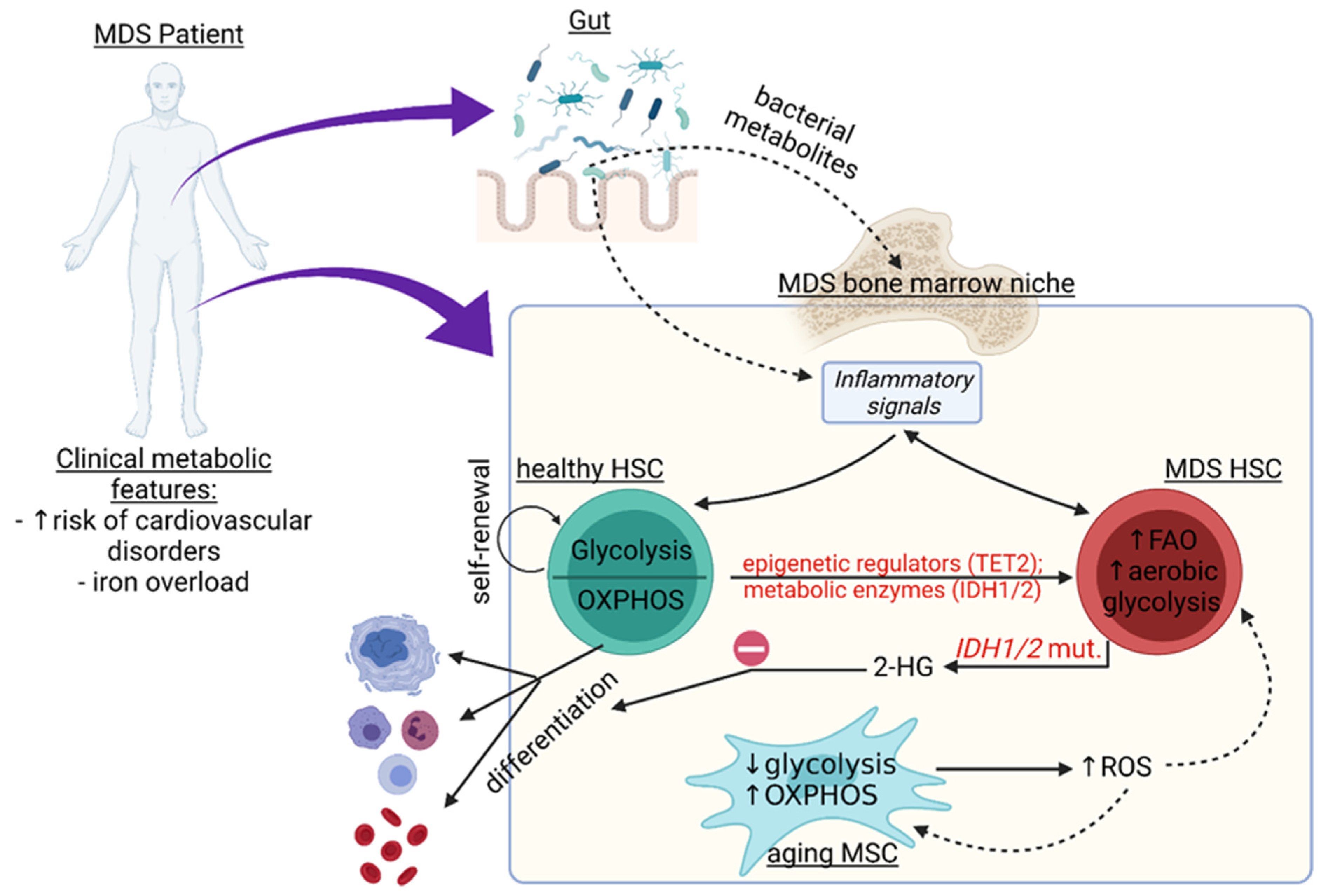
, 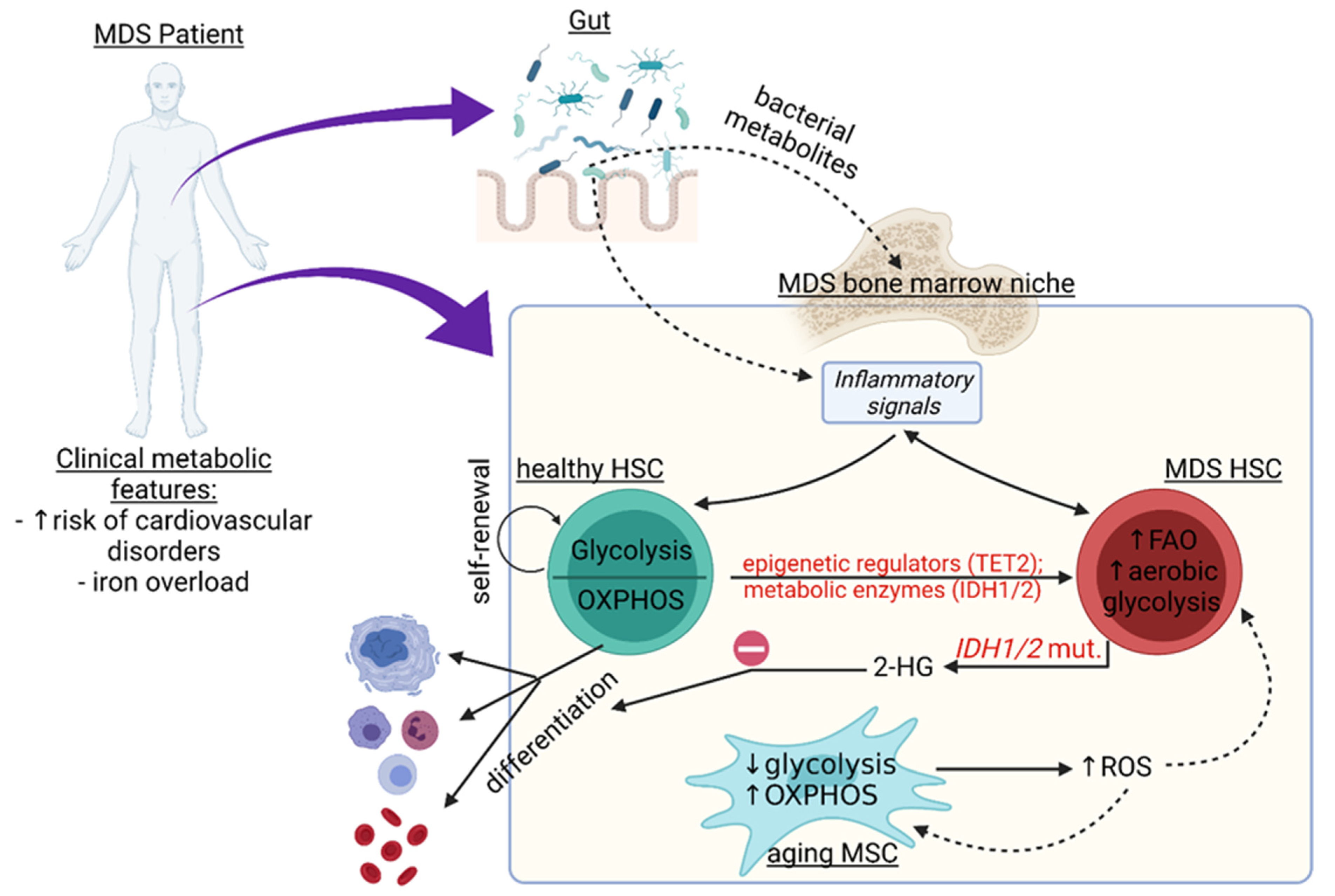{kind=link}
Abstract
:1. Introduction
2. Metabolic Changes in the HSPC Pool
Metabolic Changes and Their Association with Genetic and Epigenetic Alterations in MDS
3. Metabolic Pathways in Non-Hematopoietic Cells of the MDS Niche and Beyond
3.1. Inflammation in the Bone Marrow (BM) Niche
3.2. Iron Overload and Its Effect on Niche Metabolism in MDS
3.3. Relationship between the Microbiome and Metabolic Changes in the BM
4. Summary and Outlook: Clinical and Therapeutic Aspects of Metabolic Changes in MDS Patients
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neukirchen, J.; Schoonen, W.M.; Strupp, C.; Gattermann, N.; Aul, C.; Haas, R.; Germing, U. Incidence and prevalence of myelodysplastic syndromes: Data from the Düsseldorf MDS-registry. Leuk. Res. 2011, 35, 1591–1596. [Google Scholar] [CrossRef] [PubMed]
- De Witte, T.; Malcovati, L.; Fenaux, P.; Bowen, D.; Symeonidis, A.; Mittelman, M.; Stauder, R.; Sanz, G.; Čermák, J.; Langemeijer, S.; et al. Novel dynamic outcome indicators and clinical endpoints in myelodysplastic syndrome; the European LeukemiaNet MDS Registry and MDS-RIGHT project perspective. Haematologica 2020, 105, 2516. [Google Scholar] [CrossRef] [PubMed]
- Killick, S.B.; Ingram, W.; Culligan, D.; Enright, H.; Kell, J.; Payne, E.M.; Krishnamurthy, P.; Kulasekararaj, A.; Raghavan, M.; Stanworth, S.J.; et al. British Society for Haematology guidelines for the management of adult myelodysplastic syndromes. Br. J. Haematol. 2021, 194, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.S.; Dragoljevic, D.; Bertuzzo Veiga, C.; Wang, N.; Yvan-Charvet, L.; Murphy, A.J. Interplay between Clonal Hematopoiesis of Indeterminate Potential and Metabolism. Trends Endocrinol. Metab. 2020, 31, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P. Clinical Implications of Clonal Hematopoiesis. Mayo Clin. Proc. 2018, 93, 1122–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Vecchia, S.; Sebastián, C. Metabolic pathways regulating colorectal cancer initiation and progression. Semin. Cell Dev. Biol. 2020, 98, 63–70. [Google Scholar] [CrossRef]
- Rashkovan, M.; Ferrando, A. Metabolic dependencies and vulnerabilities in leukemia. Genes Dev. 2019, 33, 1460–1474. [Google Scholar] [CrossRef] [Green Version]
- Sellers, K.; Allen, T.D.; Bousamra, M.; Tan, J.L.; Méndez-Lucas, A.; Lin, W.; Bah, N.; Chernyavskaya, Y.; MacRae, J.I.; Higashi, R.M.; et al. Metabolic reprogramming and Notch activity distinguish between non-small cell lung cancer subtypes. Br. J. Cancer 2019, 121, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J.; Bériault, R.; Lemire, J.; Singh, R.; Chénier, D.R.; Hamel, R.D.; Appanna, V.D. The tricarboxylic acid cycle, an ancient metabolic network with a novel twist. PLoS ONE 2007, 2, e690. [Google Scholar] [CrossRef] [Green Version]
- Hajishengallis, G.; Li, X.; Chavakis, T. Immunometabolic control of hematopoiesis. Mol. Asp. Med. 2020, 77, 100923. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Chavakis, T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat. Rev. Immunol. 2021, 21, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Mitroulis, I.; Kalafati, L.; Bornhäuser, M.; Hajishengallis, G.; Chavakis, T. Regulation of the Bone Marrow Niche by Inflammation. Front. Immunol. 2020, 11, 1540. [Google Scholar] [CrossRef] [PubMed]
- Chavakis, T.; Mitroulis, I.; Hajishengallis, G. Hematopoietic progenitor cells as integrative hubs for adaptation to and fine-tuning of inflammation. Nat. Immunol. 2019, 20, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Pronk, E.; Raaijmakers, M.H.G.P. The mesenchymal niche in MDS. Blood 2019, 133, 1031–1038. [Google Scholar] [CrossRef]
- Schepers, K.; Campbell, T.B.; Passegué, E. Normal and leukemic stem cell niches: Insights and therapeutic opportunities. Cell Stem Cell 2015, 16, 254–267. [Google Scholar] [CrossRef] [Green Version]
- Hoggatt, J.; Kfoury, Y.; Scadden, D.T. Hematopoietic Stem Cell Niche in Health and Disease. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 555–581. [Google Scholar] [CrossRef]
- Olson, O.C.; Kang, Y.A.; Passegué, E. Normal hematopoiesis is a balancing act of self-renewal and regeneration. Cold Spring Harb. Perspect. Med. 2020, 10, a035519. [Google Scholar] [CrossRef]
- Caiado, F.; Pietras, E.M.; Manz, M.G. Inflammation as a regulator of hematopoietic stem cell function in disease, aging, and clonal selection. J. Exp. Med. 2021, 218, e20201541. [Google Scholar] [CrossRef]
- Papa, L.; Djedaini, M.; Hoffman, R. Mitochondrial role in stemness and differentiation of hematopoietic stem cells. Stem Cells Int. 2019, 2019, 4067162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hock, M.B.; Kralli, A. Transcriptional control of mitochondrial biogenesis and function. Annu. Rev. Physiol. 2009, 71, 177–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannini, N.; Girotra, M.; Naveiras, O.; Nikitin, G.; Campos, V.; Giger, S.; Roch, A.; Auwerx, J.; Lutolf, M.P. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat. Commun. 2016, 7, 13125. [Google Scholar] [CrossRef]
- Folmes, C.D.L.; Dzeja, P.P.; Nelson, T.J.; Terzic, A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012, 11, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Varum, S.; Rodrigues, A.S.; Moura, M.B.; Momcilovic, O.; Easley, C.A., IV; Ramalho-Santos, J.; van Houten, B.; Schatten, G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS ONE 2011, 6, e20914. [Google Scholar] [CrossRef] [Green Version]
- Ansó, E.; Weinberg, S.E.; Diebold, L.P.; Thompson, B.J.; Malinge, S.; Schumacker, P.T.; Liu, X.; Zhang, Y.; Shao, Z.; Steadman, M.; et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 2017, 19, 614–625. [Google Scholar] [CrossRef]
- Yu, W.M.; Liu, X.; Shen, J.; Jovanovic, O.; Pohl, E.E.; Gerson, S.L.; Finkel, T.; Broxmeyer, H.E.; Qu, C.K. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell 2013, 12, 62–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeffler, D.; Schroeder, T. Symmetric and asymmetric activation of hematopoietic stem cells. Curr. Opin. Hematol. 2021, 28, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.H.; et al. A PML-PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oburoglu, L.; Tardito, S.; Fritz, V.; De Barros, S.C.; Merida, P.; Craveiro, M.; Mamede, J.; Cretenet, G.; Mongellaz, C.; An, X.; et al. Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem Cell 2014, 15, 169–184. [Google Scholar] [CrossRef] [Green Version]
- Yvan-Charvet, L.; Pagler, T.; Gautier, E.L.; Avagyan, S.; Siry, R.L.; Han, S.; Welch, C.L.; Wang, N.; Randolph, G.J.; Snoeck, H.W.; et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 2010, 328, 1689–1693. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Zhang, Y.; Yokota, A.; Yan, X.; Liu, J.; Choi, K.; Li, B.; Sashida, G.; Peng, Y.; Xu, Z.; et al. Pathobiological pseudohypoxia as a putative mechanism underlying myelodysplastic syndromes. Cancer Discov. 2018, 8, 1438–1457. [Google Scholar] [CrossRef] [Green Version]
- Tong, H.; Hu, C.; Zhuang, Z.; Wang, L.; Jin, J. Hypoxia-inducible factor-1α expression indicates poor prognosis in myelodysplastic syndromes. Leuk. Lymphoma 2012, 53, 2412–2418. [Google Scholar] [CrossRef]
- Goda, N.; Kanai, M. Hypoxia-inducible factors and their roles in energy metabolism. Int. J. Hematol. 2012, 95, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, S.H.; Chen, C.T.; Wei, Y.H. Inhibitory effects of hypoxia on metabolic switch and osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2013, 31, 2779–2788. [Google Scholar] [CrossRef] [PubMed]
- Tobiasson, M.; Abdulkadir, H.; Lennartsson, A.; Katayama, S.; Marabita, F.; De Paepe, A.; Karimi, M.; Krjutskov, K.; Einarsdottir, E.; Grövdal, M.; et al. Comprehensive mapping of the effects of azacitidine on DNA methylation, repressive/permissive histone marks and gene expression in primary cells from patients with MDS and MDSrelated disease. Oncotarget 2017, 8, 28812–28825. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stergiou, I.; Kambas, K.; Giannouli, S.; Katsila, T.; Dimitrakopoulou, A.; Vidali, V.; Synolaki, E.; Papageorgiou, A.; Nezos, A.; Patrinos, G.; et al. Autophagy in Myelodysplastic Syndromes: The Role of HIF-1a/REDD1 Molecular Pathway. Blood 2018, 132, 1808. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Yang, R.; Yang, Y.; Zhao, Y.; Wakeham, A.; Li, W.Y.; Tseng, A.; Leca, J.; Berger, T.; Saunders, M.; et al. IDH1 mutation contributes to myeloid dysplasia in mice by disturbing heme biosynthesis and erythropoiesis. Blood 2020, 137, 945–958. [Google Scholar] [CrossRef]
- Poulaki, A.; Katsila, T.; Stergiou, I.E.; Giannouli, S.; Gómez-Tamayo, J.C.; Piperaki, E.T.; Kambas, K.; Dimitrakopoulou, A.; Patrinos, G.P.; Tzioufas, A.G.; et al. Bioenergetic profiling of the differentiating human mds myeloid lineage with low and high bone marrow blast counts. Cancers 2020, 12, 3520. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosc, C.; Broin, N.; Fanjul, M.; Saland, E.; Farge, T.; Courdy, C.; Batut, A.; Masoud, R.; Larrue, C.; Skuli, S.; et al. Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites. Nat. Commun. 2020, 11, 4056. [Google Scholar] [CrossRef]
- Giger, S.; Kovtonyuk, L.V.; Utz, S.G.; Ramosaj, M.; Kovacs, W.J.; Schmid, E.; Ioannidis, V.; Greter, M.; Manz, M.G.; Lutolf, M.P.; et al. A Single Metabolite which Modulates Lipid Metabolism Alters Hematopoietic Stem/Progenitor Cell Behavior and Promotes Lymphoid Reconstitution. Stem Cell Rep. 2020, 15, 566–576. [Google Scholar] [CrossRef]
- Zhou, S.; Chen, S.; Jiang, Q.; Pei, M. Determinants of stem cell lineage differentiation toward chondrogenesis versus adipogenesis. Cell. Mol. Life Sci. 2019, 76, 1653–1680. [Google Scholar] [CrossRef]
- Sbrana, F.V.; Cortini, M.; Avnet, S.; Perut, F.; Columbaro, M.; De Milito, A.; Baldini, N. The Role of Autophagy in the Maintenance of Stemness and Differentiation of Mesenchymal Stem Cells. Stem Cell Rev. Rep. 2016, 12, 621–633. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Heiden, M.G.V.; Kroemer, G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. 2013, 12, 829–846. [Google Scholar] [CrossRef]
- Pérez, L.M.; Bernal, A.; De Lucas, B.; Martin, N.S.; Mastrangelo, A.; García, A.; Barbas, C.; Gálvez, B.G. Altered metabolic and stemness capacity of adipose tissue-derived stem cells from obese mouse and human. PLoS ONE 2015, 10, e0123397. [Google Scholar] [CrossRef] [Green Version]
- Mancini, O.K.; Lora, M.; Cuillerier, A.; Shum-Tim, D.; Hamdy, R.; Burelle, Y.; Servant, M.J.; Stochaj, U.; Colmegna, I. Mitochondrial oxidative stress reduces the immunopotency of mesenchymal stromal cells in adults with coronary artery disease. Circ. Res. 2018, 122, 255–266. [Google Scholar] [CrossRef]
- Pietilä, M.; Palomäki, S.; Lehtonen, S.; Ritamo, I.; Valmu, L.; Nystedt, J.; Laitinen, S.; Leskelä, H.V.; Sormunen, R.; Pesälä, J.; et al. Mitochondrial function and energy metabolism in umbilical cord blood- and bone marrow-derived mesenchymal stem cells. Stem Cells Dev. 2012, 21, 575–588. [Google Scholar] [CrossRef]
- Plenge, U.; Belhage, B.; Guadalupe-Grau, A.; Andersen, P.R.; Lundby, C.; Dela, F.; Stride, N.; Pott, F.C.; Helge, J.W.; Boushel, R. Erythropoietin treatment enhances muscle mitochondria capacity in humans. Front. Physiol. 2012, 3, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cayla, J.L.; Maire, P.; Duvallet, A.; Wahrmann, J.P. Erythropoietin induces a shift of muscle phenotype from fast glycolytic to slow oxidative. Int. J. Sports Med. 2008, 29, 460–465. [Google Scholar] [CrossRef]
- Wang, L.; Teng, R.; Di, L.; Rogers, H.; Wu, H.; Kopp, J.B.; Noguchi, C.T. PPARa and sirt1 mediate erythropoietin action in increasing metabolic activity and browning of white adipocytes to protect against obesity and metabolic disorders. Diabetes 2013, 62, 4122–4131. [Google Scholar] [CrossRef] [Green Version]
- Santini, V.; Almeida, A.; Giagounidis, A.; Skikne, B.; Beach, C.; Weaver, J.; Tu, N.; Fenaux, P. Achievement of red blood cell transfusion independence in red blood cell transfusion-dependent patients with lower-risk non-del(5q) myelodysplastic syndromes correlates with serum erythropoietin levels. Leuk. Lymphoma 2020, 17, 1475–1483. [Google Scholar] [CrossRef]
- Weickert, M.-T.; Hecker, J.S.; Buck, M.C.; Schreck, C.; Rivière, J.; Schiemann, M.; Schallmoser, K.; Bassermann, F.; Strunk, D.; Oostendorp, R.A.J.; et al. Bone marrow stromal cells from MDS and AML patients show increased adipogenic potential with reduced Delta-like-1 expression. Sci. Rep. 2021, 11, 5944. [Google Scholar] [CrossRef]
- Ambrosi, T.H.; Scialdone, A.; Graja, A.; Gohlke, S.; Jank, A.M.; Bocian, C.; Woelk, L.; Fan, H.; Logan, D.W.; Schürmann, A.; et al. Adipocyte Accumulation in the Bone Marrow during Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell 2017, 20, 771–784.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulard, M.; Dosquet, C.; Bonnet, D. Role of the microenvironment in myeloid malignancies. Cell. Mol. Life Sci. 2018, 75, 1377. [Google Scholar] [CrossRef] [Green Version]
- Azadniv, M.; Myers, J.R.; McMurray, H.R.; Guo, N.; Rock, P.; Coppage, M.L.; Ashton, J.; Becker, M.W.; Calvi, L.M.; Liesveld, J.L. Bone marrow mesenchymal stromal cells from acute myelogenous leukemia patients demonstrate adipogenic differentiation propensity with implications for leukemia cell support. Leukemia 2020, 34, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Nuschke, A.; Rodrigues, M.; Wells, A.W.; Sylakowski, K.; Wells, A. Mesenchymal stem cells/multipotent stromal cells (MSCs) are glycolytic and thus glucose is a limiting factor of in vitro models of MSC starvation. Stem Cell Res. Ther. 2016, 7, 179. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Muñoz, N.; Bunnell, B.A.; Logan, T.M.; Ma, T. Density-dependent metabolic heterogeneity in human mesenchymal stem cells. Stem Cells 2015, 33, 3368–3381. [Google Scholar] [CrossRef] [Green Version]
- Pattappa, G.; Heywood, H.K.; de Bruijn, J.D.; Lee, D.A. The metabolism of human mesenchymal stem cells during proliferation and differentiation. J. Cell. Physiol. 2011, 226, 2562–2570. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Grayson, W.L.; Fröhlich, M.; Vunjak-Novakovic, G. Hypoxia and stem cell-based engineering of mesenchymal tissues. Biotechnol. Prog. 2009, 25, 32–42. [Google Scholar] [CrossRef]
- Yin, J.Q.; Zhu, J.; Ankrum, J.A. Manufacturing of primed mesenchymal stromal cells for therapy. Nat. Biomed. Eng. 2019, 3, 90–104. [Google Scholar] [CrossRef]
- Najafi, R.; Sharifi, A.M. Deferoxamine preconditioning potentiates mesenchymal stem cell homing in vitro and in streptozotocin-diabetic rats. Expert Opin. Biol. Ther. 2013, 13, 959–972. [Google Scholar] [CrossRef]
- Teofili, L.; Martini, M.; Nuzzolo, E.R.; Capodimonti, S.; Iachininoto, M.G.; Cocomazzi, A.; Fabiani, E.; Voso, M.T.; Larocca, L.M. Endothelial Progenitor Cell Dysfunction in Myelodysplastic Syndromes: Possible Contribution of a Defective Vascular Niche to Myelodysplasia. Neoplasia 2015, 17, 401. [Google Scholar] [CrossRef]
- Lu, L.; Payvandi, F.; Wu, L.; Zhang, L.H.; Hariri, R.J.; Man, H.W.; Chen, R.S.; Muller, G.W.; Hughes, C.C.W.; Stirling, D.I.; et al. The anti-cancer drug lenalidomide inhibits angiogenesis and metastasis via multiple inhibitory effects on endothelial cell function in normoxic and hypoxic conditions. Microvasc. Res. 2009, 77, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Cain, D.W.; Snowden, P.B.; Sempowski, G.D.; Kelsoe, G. Inflammation triggers emergency granulopoiesis through a density-dependent feedback mechanism. PLoS ONE 2011, 6, e19957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Long, L.; Zhou, P.; Chapman, N.M.; Chi, H. mTOR signaling at the crossroads of environmental signals and T-cell fate decisions. Immunol. Rev. 2020, 295, 15–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, D.T.; Liu, X.G. mTOR signaling in T cell immunity and autoimmunity. Int. Rev. Immunol. 2015, 34, 50–66. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The metabolic signature of macrophage responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; O’Neill, L.A. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur. J. Immunol. 2016, 46, 13–21. [Google Scholar] [CrossRef]
- Kristinsson, S.Y.; Björkholm, M.; Hultcrantz, M.; Derolf, Å.R.; Landgren, O.; Goldin, L.R. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J. Clin. Oncol. 2011, 29, 2897–2903. [Google Scholar] [CrossRef] [Green Version]
- Fozza, C.; Crobu, V.; Isoni, M.A.; Dore, F. The immune landscape of myelodysplastic syndromes. Crit. Rev. Oncol. Hematol. 2016, 107, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal. Transduct. Target. Ther. 2021, 6, 1–21. [Google Scholar] [CrossRef]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef] [PubMed]
- Nagareddy, P.R.; Kraakman, M.; Masters, S.L.; Stirzaker, R.A.; Gorman, D.J.; Grant, R.W.; Dragoljevic, D.; Hong, E.S.; Abdel-Latif, A.; Smyth, S.S.; et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014, 19, 821–835. [Google Scholar] [CrossRef] [Green Version]
- Ferrone, C.K.; Blydt-Hansen, M.; Rauh, M.J. Age-associated TET2 mutations: Common drivers of myeloid dysfunction, cancer and cardiovascular disease. Int. J. Mol. Sci. 2020, 21, 626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abegunde, S.O.; Buckstein, R.; Wells, R.A.; Rauh, M.J. An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Exp. Hematol. 2018, 59, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Sano, S.; Oshima, K.; Wang, Y.; MacLauchlan, S.; Katanasaka, Y.; Sano, M.; Zuriaga, M.A.; Yoshiyama, M.; Goukassian, D.; Cooper, M.A.; et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J. Am. Coll. Cardiol. 2018, 71, 875–886. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; Hell, S.; Platzbecker, U. Controversies on the consequences of iron overload and chelation in MDS. HemaSphere 2020, 4, e409. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Ravera, S.; Calabrese, C.; Gaidano, V.; Niscola, P.; Balleari, E.; Gallo, D.; Petiti, J.; Signorino, E.; Rosso, V.; et al. Iron overload alters the energy metabolism in patients with myelodysplastic syndromes: Results from the multicenter FISM BIOFER study. Sci. Rep. 2020, 10, 9156. [Google Scholar] [CrossRef]
- Kim, C.H.; Leitch, H.A. Iron overload-induced oxidative stress in myelodysplastic syndromes and its cellular sequelae. Crit. Rev. Oncol. Hematol. 2021, 163, 103367. [Google Scholar] [CrossRef]
- Zheng, Q.Q.; Zhao, Y.S.; Guo, J.; Zhao, S.D.; Song, L.X.; Fei, C.M.; Zhang, Z.; Li, X.; Chang, C.K. Iron overload promotes erythroid apoptosis through regulating HIF-1a/ROS signaling pathway in patients with myelodysplastic syndrome. Leuk. Res. 2017, 58, 55–62. [Google Scholar] [CrossRef]
- Hu, J.; Meng, F.; Hu, X.; Huang, L.; Liu, H.; Liu, Z.; Li, L. Iron overload regulate the cytokine of mesenchymal stromal cells through ROS/HIF-1α pathway in Myelodysplastic syndromes. Leuk. Res. 2020, 93, 106354. [Google Scholar] [CrossRef]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef]
- Angelucci, E.; Li, J.; Greenberg, P.; Wu, D.; Hou, M.; Figueroa, E.H.M.; Rodriguez, M.G.; Dong, X.; Ghosh, J.; Izquierdo, M.; et al. Iron chelation in transfusion-dependent patients with low- To intermediate-1-risk myelodysplastic syndromes: A randomized trial. Ann. Intern. Med. 2020, 172, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; Porto, G.; Simmelbauer, A.; Altamura, S.; Passos, S.T.; Garbowski, M.; Silva, A.M.N.; Spaich, S.; Seide, S.E.; Sparla, R.; et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur. Heart J. 2020, 41, 2681–2695. [Google Scholar] [CrossRef] [PubMed]
- Manzo, V.E.; Bhatt, A.S. The human microbiome in hematopoiesis and hematologic disorders. Blood 2015, 126, 311–318. [Google Scholar] [CrossRef] [Green Version]
- Josefsdottir, K.S.; Baldridge, M.T.; Kadmon, C.S.; King, K.Y. Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood 2017, 129, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Meisel, M.; Hinterleitner, R.; Pacis, A.; Chen, L.; Earley, Z.M.; Mayassi, T.; Pierre, J.F.; Ernest, J.D.; Galipeau, H.J.; Thuille, N.; et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 2018, 557, 580–584. [Google Scholar] [CrossRef]
- Chan, O.; Renneville, A.; Padron, E. Chronic myelomonocytic leukemia diagnosis and management. Leukemia 2021, 35, 1552–1562. [Google Scholar] [CrossRef]
- Zeng, H.; He, H.; Guo, L.; Li, J.; Lee, M.; Han, W.; Guzman, A.G.; Zang, S.; Zhou, Y.; Zhang, X.; et al. Antibiotic treatment ameliorates Ten-eleven translocation 2 (TET2) loss-of-function associated hematological malignancies. Cancer Lett. 2019, 467, 1–8. [Google Scholar] [CrossRef]
- Galloway-Peña, J.R.; Smith, D.P.; Sahasrabhojane, P.; Ajami, N.J.; Wadsworth, W.D.; Daver, N.G.; Chemaly, R.F.; Marsh, L.; Ghantoji, S.S.; Pemmaraju, N.; et al. The role of the gastrointestinal microbiome in infectious complications during induction chemotherapy for acute myeloid leukemia. Cancer 2016, 122, 2186–2196. [Google Scholar] [CrossRef] [Green Version]
- Hueso, T.; Ekpe, K.; Mayeur, C.; Gatse, A.; Joncquel-Chevallier Curt, M.; Gricourt, G.; Rodriguez, C.; Burdet, C.; Ulmann, G.; Neut, C.; et al. Impact and consequences of intensive chemotherapy on intestinal barrier and microbiota in acute myeloid leukemia: The role of mucosal strengthening. Gut Microbes 2020, 12, 1800897. [Google Scholar] [CrossRef] [PubMed]
- Galloway-Peña, J.R.; Smith, D.P.; Sahasrabhojane, P.; Wadsworth, W.D.; Fellman, B.M.; Ajami, N.J.; Shpall, E.J.; Daver, N.; Guindani, M.; Petrosino, J.F.; et al. Characterization of oral and gut microbiome temporal variability in hospitalized cancer patients. Genome Med. 2017, 9, e0182520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Østgård, L.S.G.; Nørgaard, M.; Pedersen, L.; Østgård, R.D.; Medeiros, B.C.; Overgaard, U.M.; Schöllkopf, C.; Severinsen, M.; Marcher, C.W.; Jensen, M.K. Autoimmune diseases, infections, use of antibiotics and the risk of acute myeloid leukaemia: A national population-based case-control study. Br. J. Haematol. 2018, 181, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.M.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef] [Green Version]
- Rattanathammethee, T.; Tuitemwong, P.; Thiennimitr, P.; Sarichai, P.; Na Pombejra, S.; Piriyakhuntorn, P.; Hantrakool, S.; Chai-Adisaksopha, C.; Rattarittamrong, E.; Tantiworawit, A.; et al. Gut microbiota profiles of treatment-naïve adult acute myeloid leukemia patients with neutropenic fever during intensive chemotherapy. PLoS ONE 2020, 15, e0236460. [Google Scholar] [CrossRef]
- Brunner, A.M.; Blonquist, T.M.; Hobbs, G.S.; Amrein, P.C.; Neuberg, D.S.; Steensma, D.P.; Abel, G.A.; Fathi, A.T. Risk and timing of cardiovascular death among patients with myelodysplastic syndromes. Blood Adv. 2017, 1, 2032–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Kreitz, J.; Schönfeld, C.; Seibert, M.; Stolp, V.; Alshamleh, I.; Oellerich, T.; Steffen, B.; Schwalbe, H.; Schnütgen, F.; Kurrle, N.; et al. Metabolic Plasticity of Acute Myeloid Leukemia. Cells 2019, 8, 805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillebeen, C.; Lam, N.H.; Chow, S.; Botta, A.; Sweeney, G.; Pantopoulos, K. Regulatory connections between iron and glucose metabolism. Int. J. Mol. Sci. 2020, 21, 7773. [Google Scholar] [CrossRef]
- Wong, C.A.C.; Leitch, H.A. Delayed time from RBC transfusion dependence to first cardiac event in lower IPSS risk MDS patients receiving iron chelation therapy. Leuk. Res. 2019, 83, 106170. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.D.; Marteyn, B.; Farnoud, N.R.; de Botton, S.; Bernard, O.A.; et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Valdemarin, F.; Caffa, I.; Persia, A.; Cremonini, A.L.; Ferrando, L.; Tagliafico, L.; Tagliafico, A.; Guijarro, A.; Carbone, F.; Ministrini, S.; et al. Safety and feasibility of fasting-mimicking diet and effects on nutritional status and circulating metabolic and inflammatory factors in cancer patients undergoing active treatment. Cancers 2021, 13, 4013. [Google Scholar] [CrossRef]
- Meyer, A.; Montastier, E.; Hager, J.; Saris, W.H.M.; Astrup, A.; Viguerie, N.; Valsesia, A. Plasma metabolites and lipids predict insulin sensitivity improvement in obese, nondiabetic individuals after a 2-phase dietary intervention. Am. J. Clin. Nutr. 2018, 108, 13–23. [Google Scholar] [CrossRef]
- Bouronikou, E.; Georgoulias, P.; Giannakoulas, N.; Valotassiou, V.; Palassopoulou, M.; Vassilopoulos, G.; Papadoulis, N.; Matsouka, P. Metabolism-related cytokine and hormone levels in the serum of patients with myelodysplastic syndromes. Acta Haematol. 2013, 130, 27–33. [Google Scholar] [CrossRef]
- Dalamaga, M.; Karmaniolas, K.; Chamberland, J.; Nikolaidou, A.; Lekka, A.; Dionyssiou-Asteriou, A.; Mantzoros, C.S. Higher fetuin-A, lower adiponectin and free leptin levels mediate effects of excess body weight on insulin resistance and risk for myelodysplastic syndrome. Metabolism 2013, 62, 1830–1839. [Google Scholar] [CrossRef]
- Rangan, P.; Choi, I.; Wei, M.; Navarrete, G.; Guen, E.; Brandhorst, S.; Enyati, N.; Pasia, G.; Maesincee, D.; Ocon, V.; et al. Fasting-Mimicking Diet Modulates Microbiota and Promotes Intestinal Regeneration to Reduce Inflammatory Bowel Disease Pathology. Cell Rep. 2019, 26, 2704–2719. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Chen, S.L.; Dai, Y.J.; Wang, Y.; Qin, Z.Y.; Li, H.; Shu, L.L.; Li, J.Y.; Huang, H.Y.; Liang, Y. Identification of a metabolic gene panel to predict the prognosis of myelodysplastic syndrome. J. Cell. Mol. Med. 2020, 24, 6373–6384. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balaian, E.; Wobus, M.; Bornhäuser, M.; Chavakis, T.; Sockel, K. Myelodysplastic Syndromes and Metabolism. Int. J. Mol. Sci. 2021, 22, 11250. https://doi.org/10.3390/ijms222011250
Balaian E, Wobus M, Bornhäuser M, Chavakis T, Sockel K. Myelodysplastic Syndromes and Metabolism. International Journal of Molecular Sciences. 2021; 22(20):11250. https://doi.org/10.3390/ijms222011250
Chicago/Turabian StyleBalaian, Ekaterina, Manja Wobus, Martin Bornhäuser, Triantafyllos Chavakis, and Katja Sockel. 2021. "Myelodysplastic Syndromes and Metabolism" International Journal of Molecular Sciences 22, no. 20: 11250. https://doi.org/10.3390/ijms222011250
APA StyleBalaian, E., Wobus, M., Bornhäuser, M., Chavakis, T., & Sockel, K. (2021). Myelodysplastic Syndromes and Metabolism. International Journal of Molecular Sciences, 22(20), 11250. https://doi.org/10.3390/ijms222011250