
Perspectives on the Role of Enzymatic Biocatalysis for the Degradation of Plastic PET




Abstract
:1. Introduction
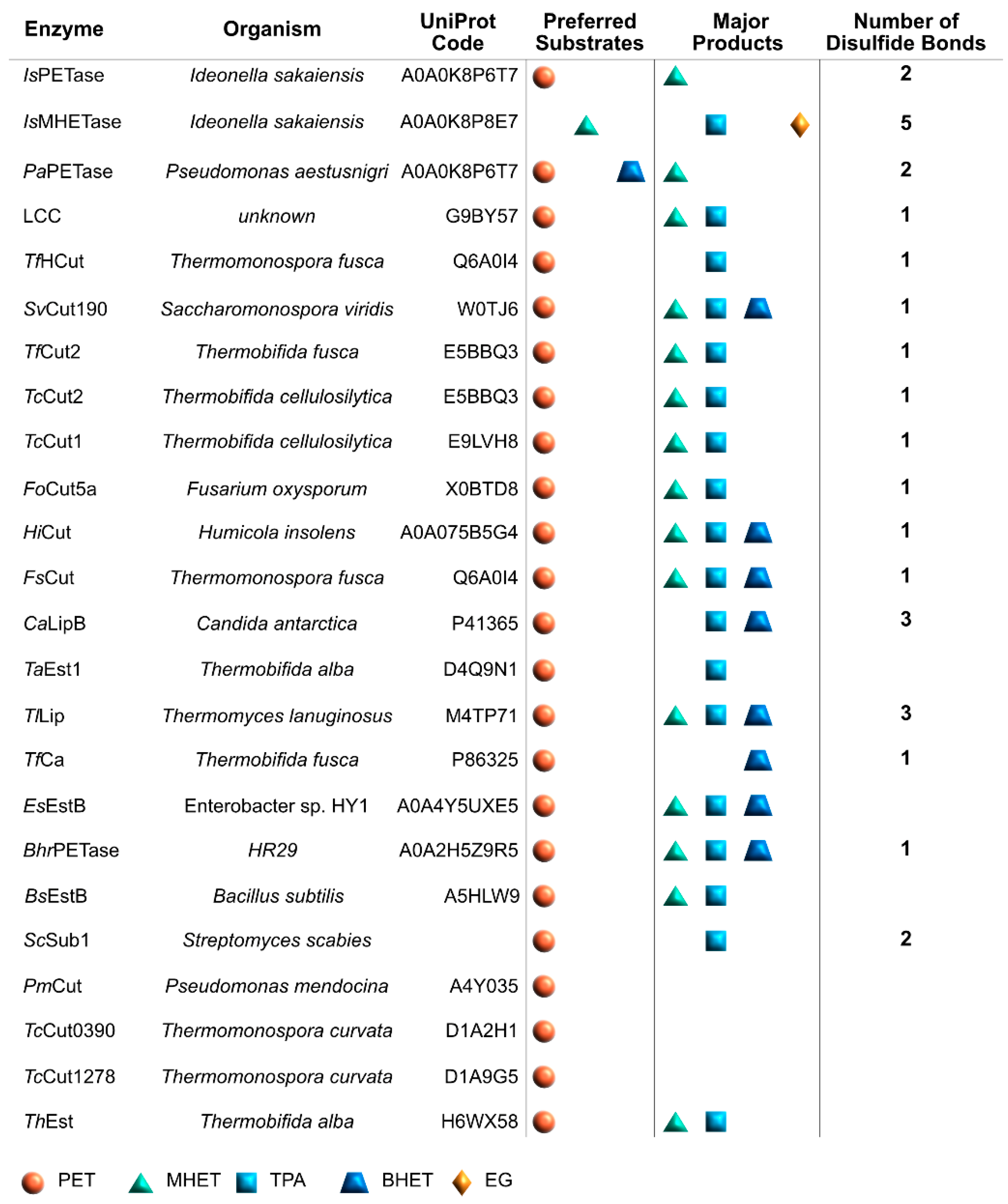
2. Enzymes Involved in PET Degradation
2.1. Ideonella sakaiensis PETase (IsPETase)
2.1.1. Discovery
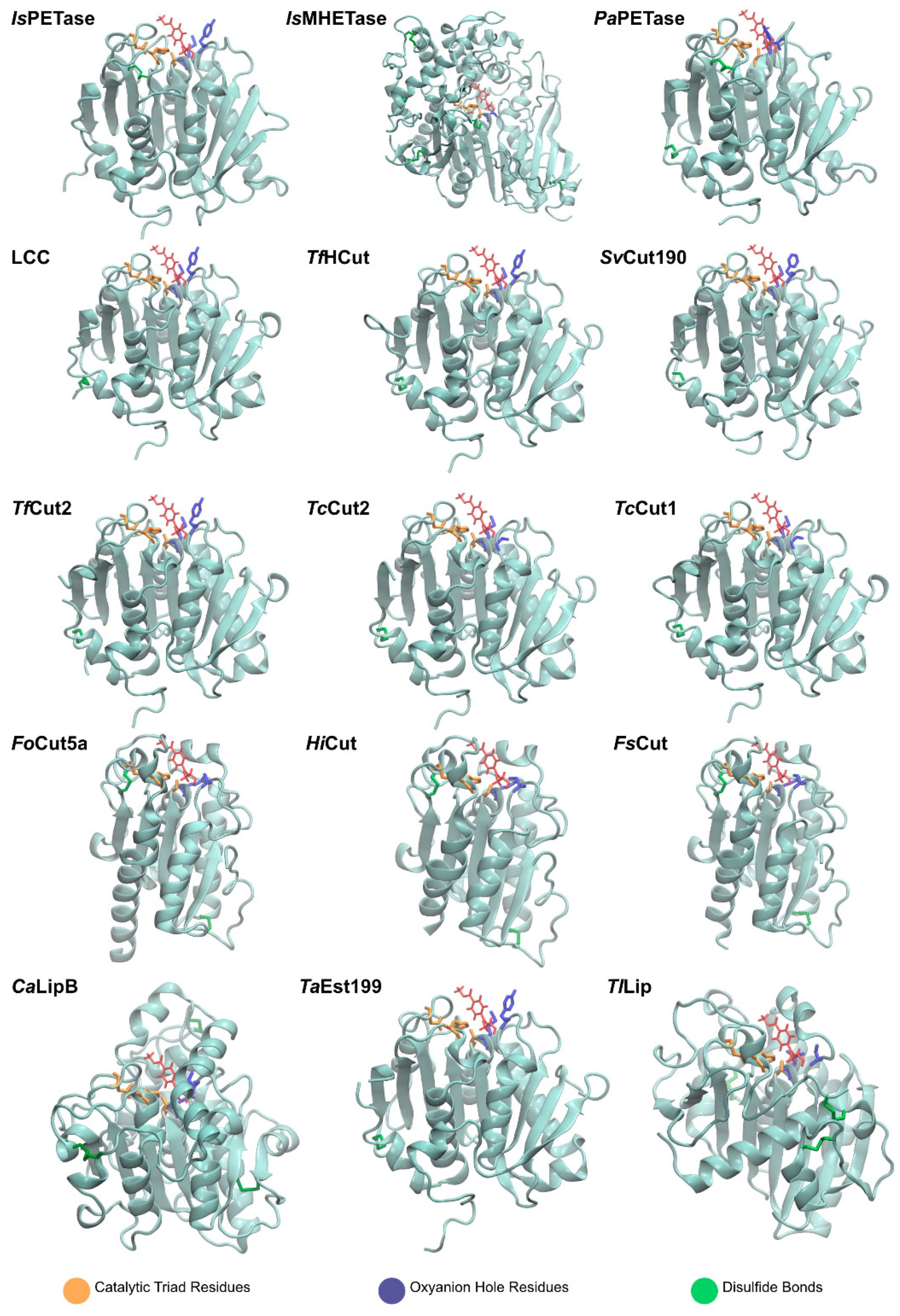
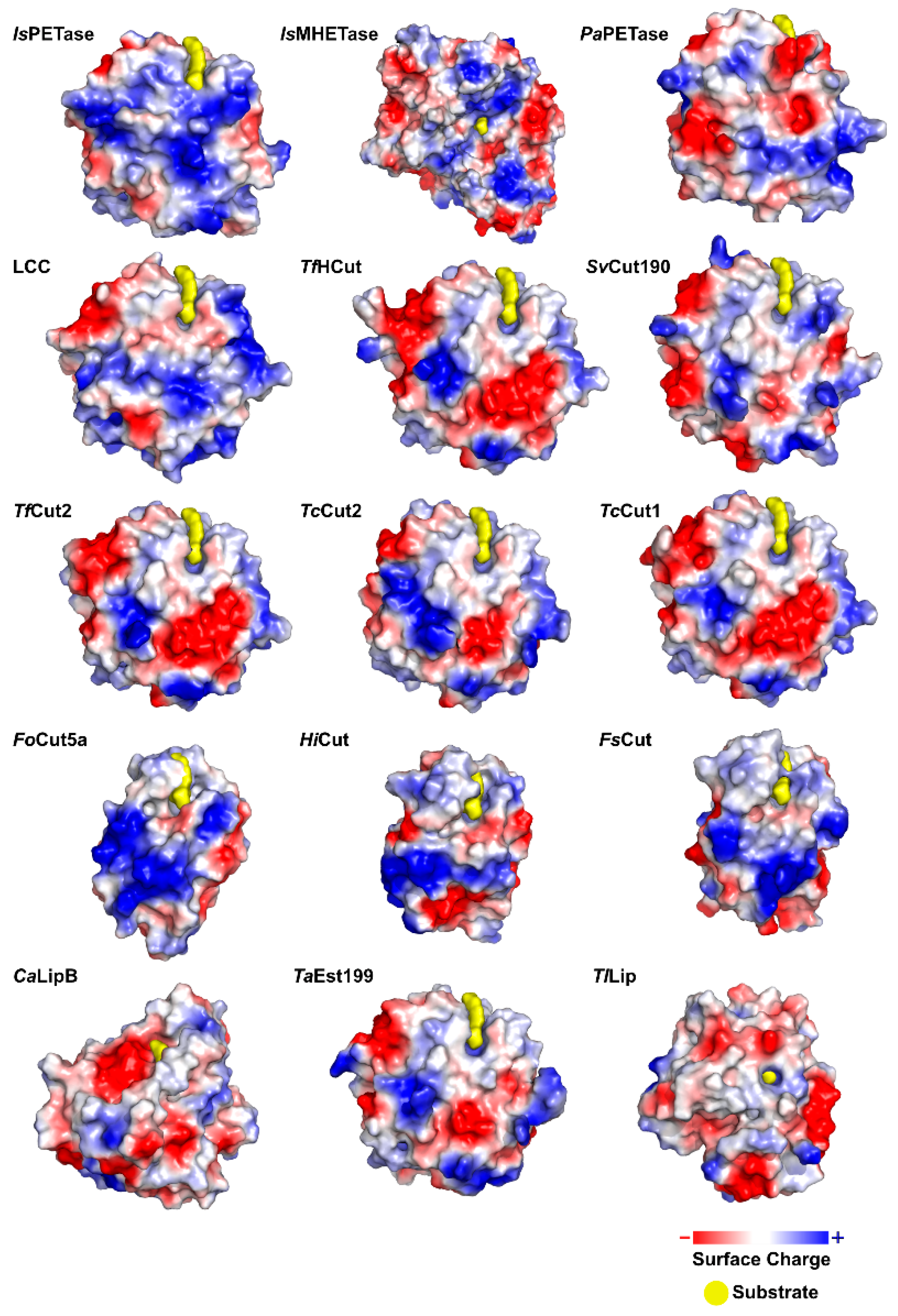
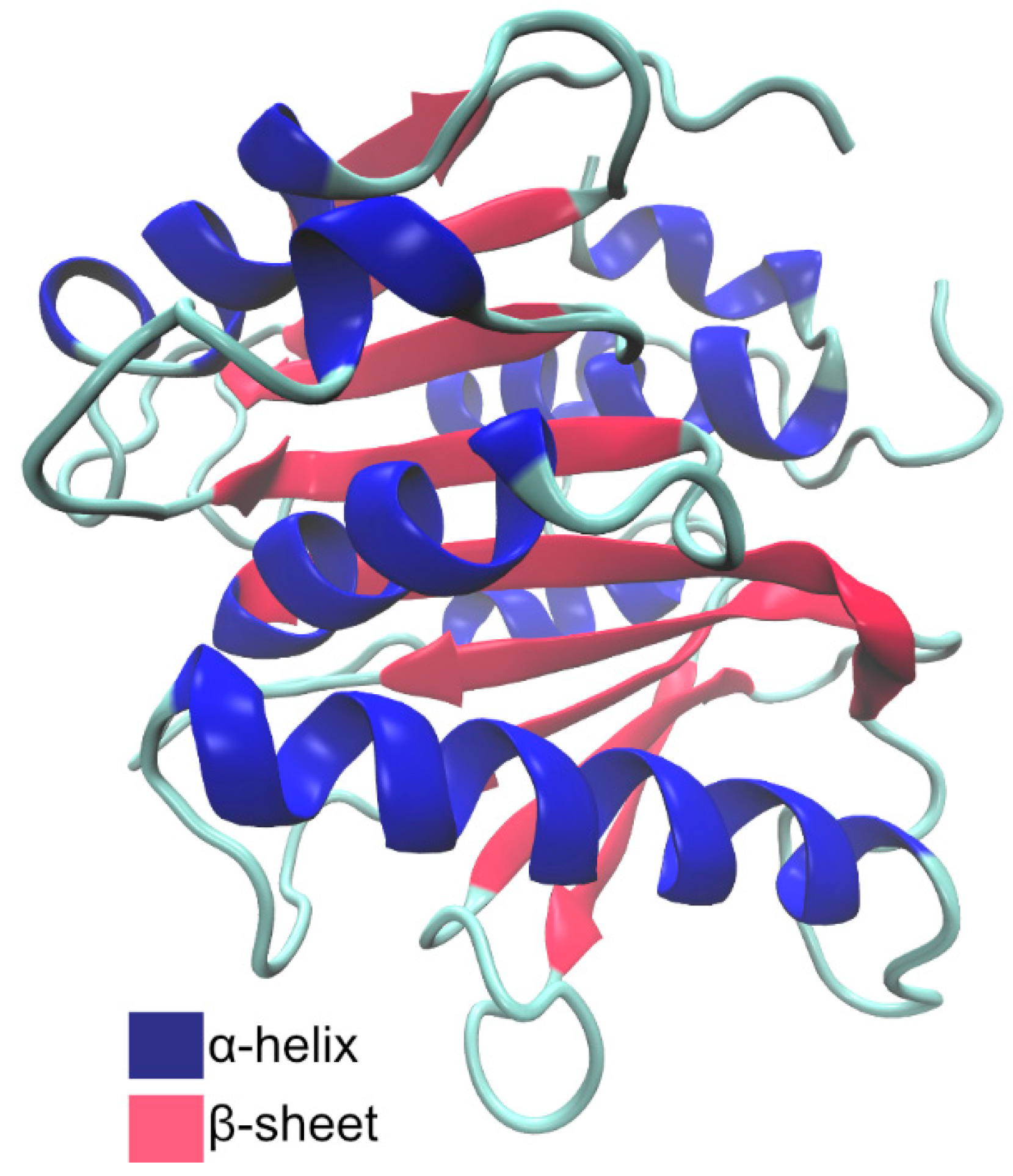
2.1.2. Structure
2.1.3. Activity
2.1.4. Proposed Mechanism
2.1.5. Future Perspectives
2.2. Ideonella sakaiensis MHETase (IsMHETase)
2.2.1. Discovery
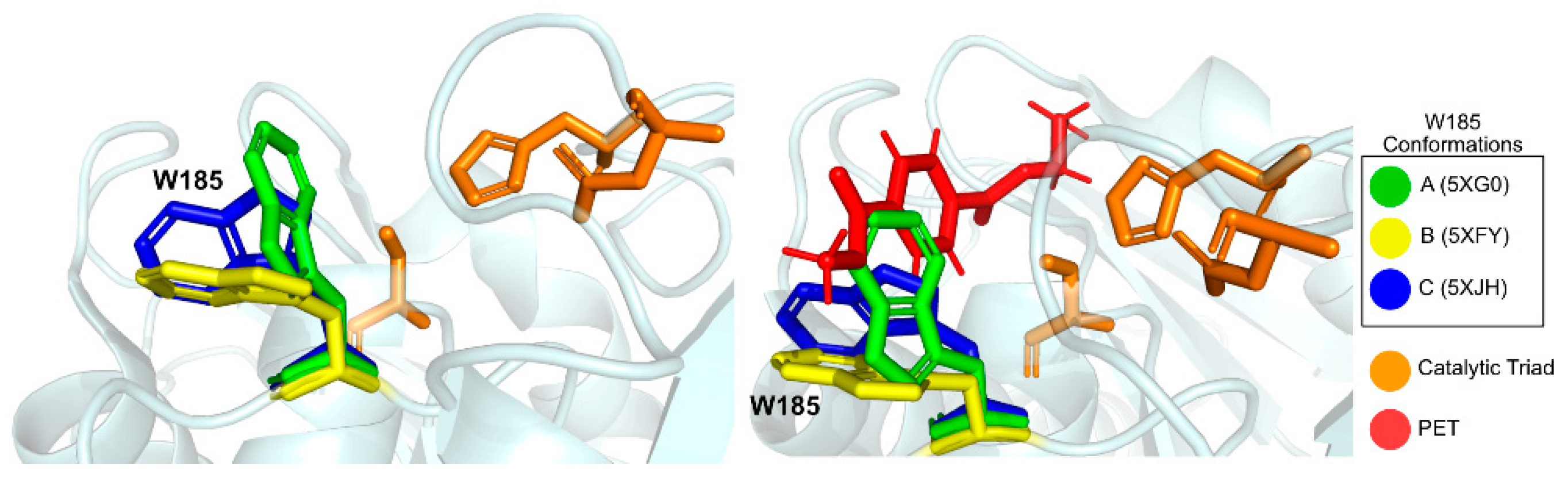
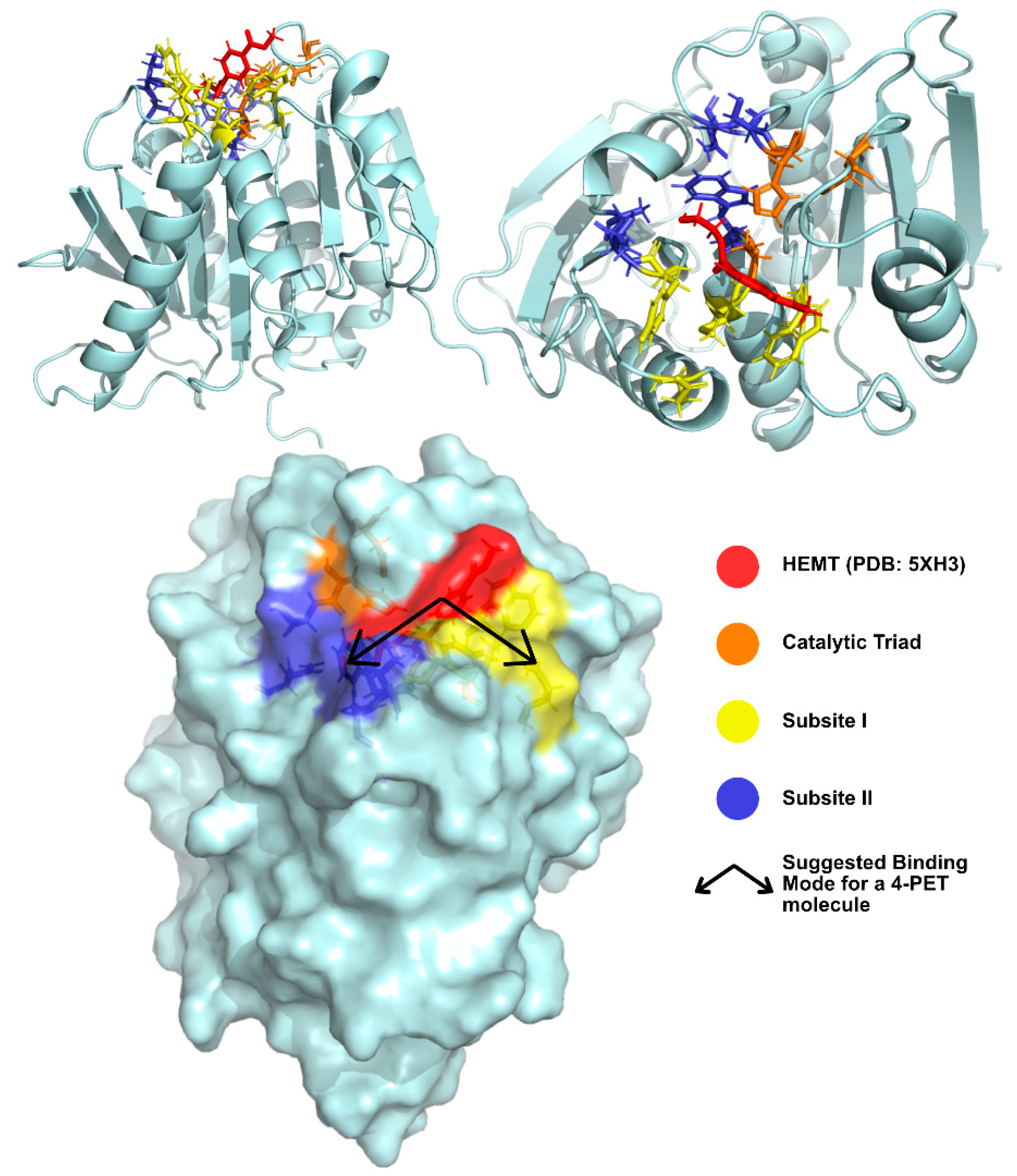
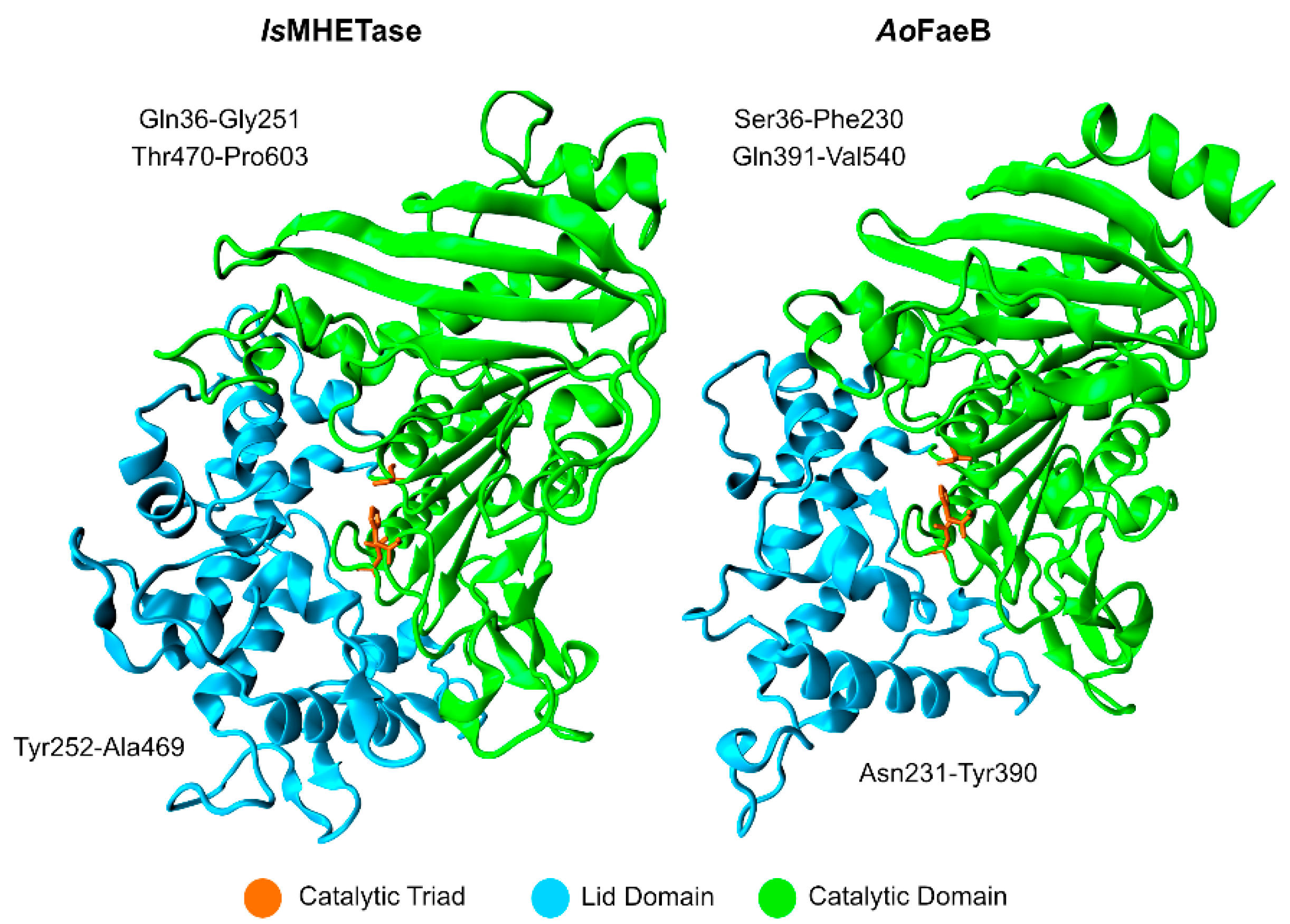
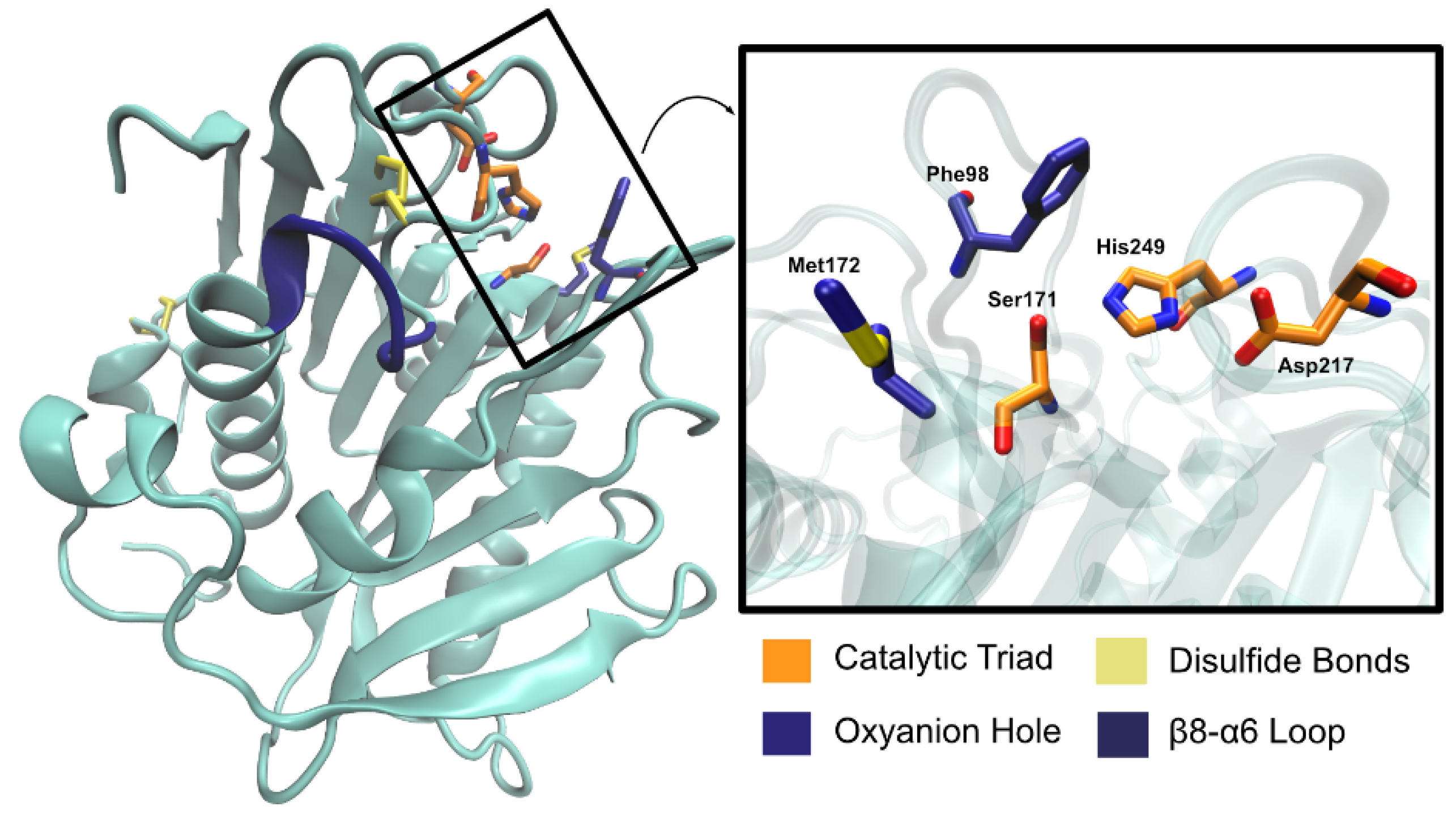
2.2.2. Structure
2.2.3. Activity
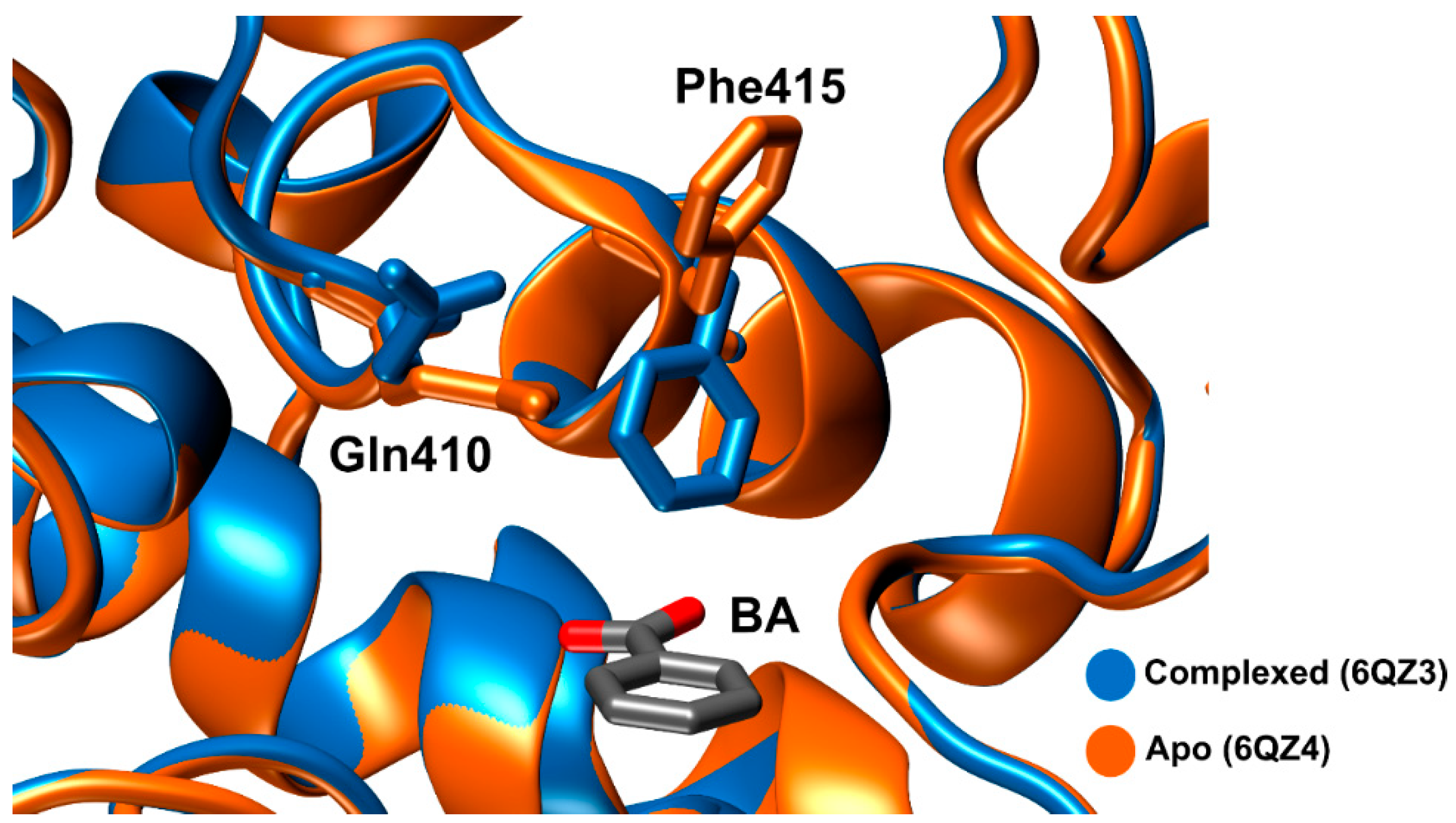
2.2.4. Proposed Mechanism
2.2.5. Future Perspectives
2.3. Pseudomonas Aestusnigri PETase (PaPETase)
2.3.1. Discovery
2.3.2. Structure
2.3.3. Activity
2.3.4. Proposed Mechanism
2.3.5. Future Perspectives
2.4. LC-Cutinase (LCC)
2.4.1. Discovery
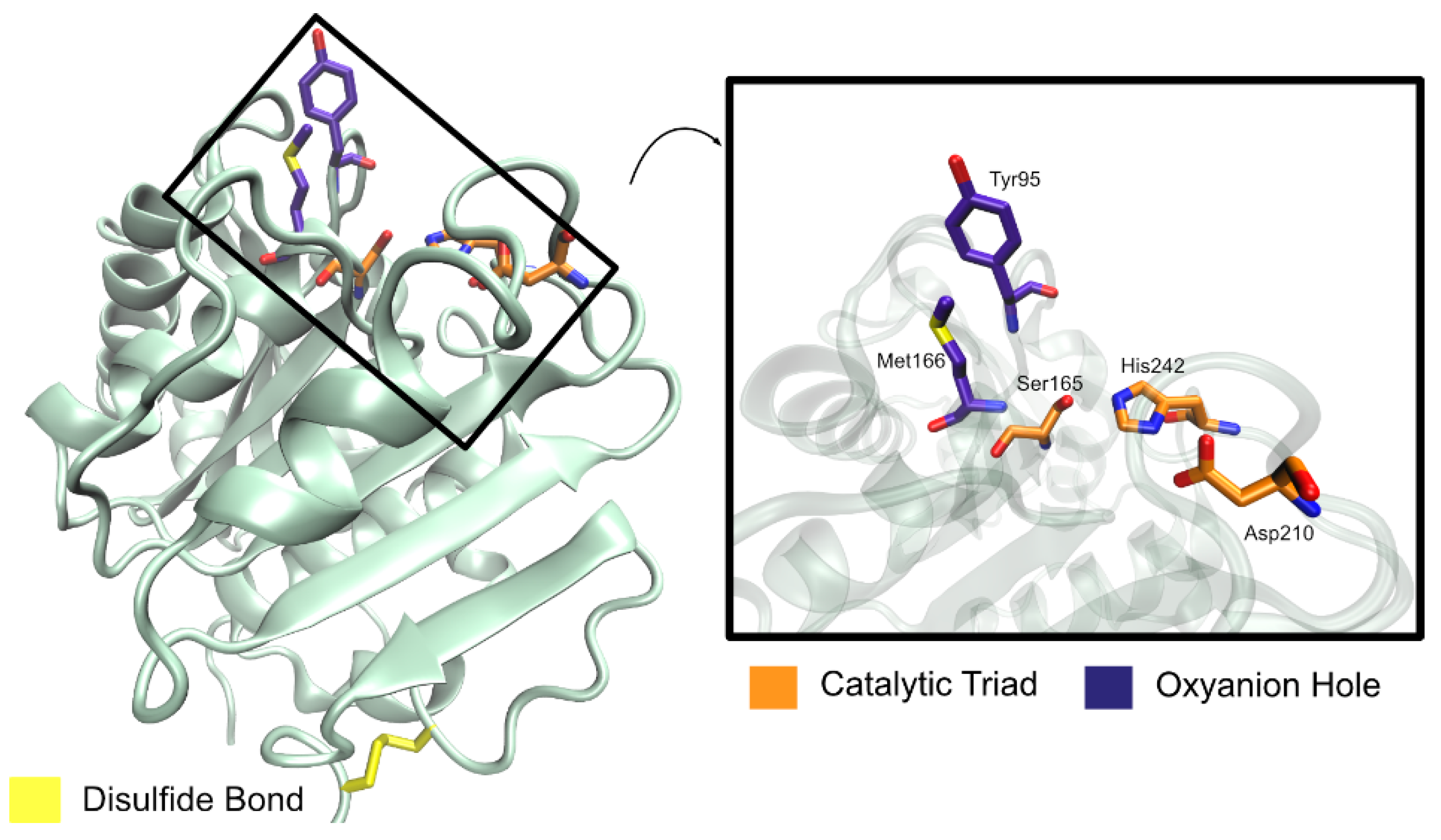
2.4.2. Structure
2.4.3. Activity
2.4.4. Future Perspectives
2.5. Thermomonospora fusca Hydrolase (TfHCut) and Thermomonospora fusca BTA Hydrolase 2 (BTA-2)
2.5.1. Discovery
2.5.2. Structure
2.5.3. Activity
2.5.4. Proposed Mechanism
2.5.5. Future Perspectives
2.6. Saccharomonospora viridis AHK190 Cutinase (SvCut190)
2.6.1. Discovery
2.6.2. Structure
2.6.3. Activity
2.6.4. Proposed Mechanism
2.6.5. Future Perspectives
2.7. Thermobifida Genus Cutinase 1 and Cutinase 2 (Cut1 and Cut2)
2.7.1. Discovery
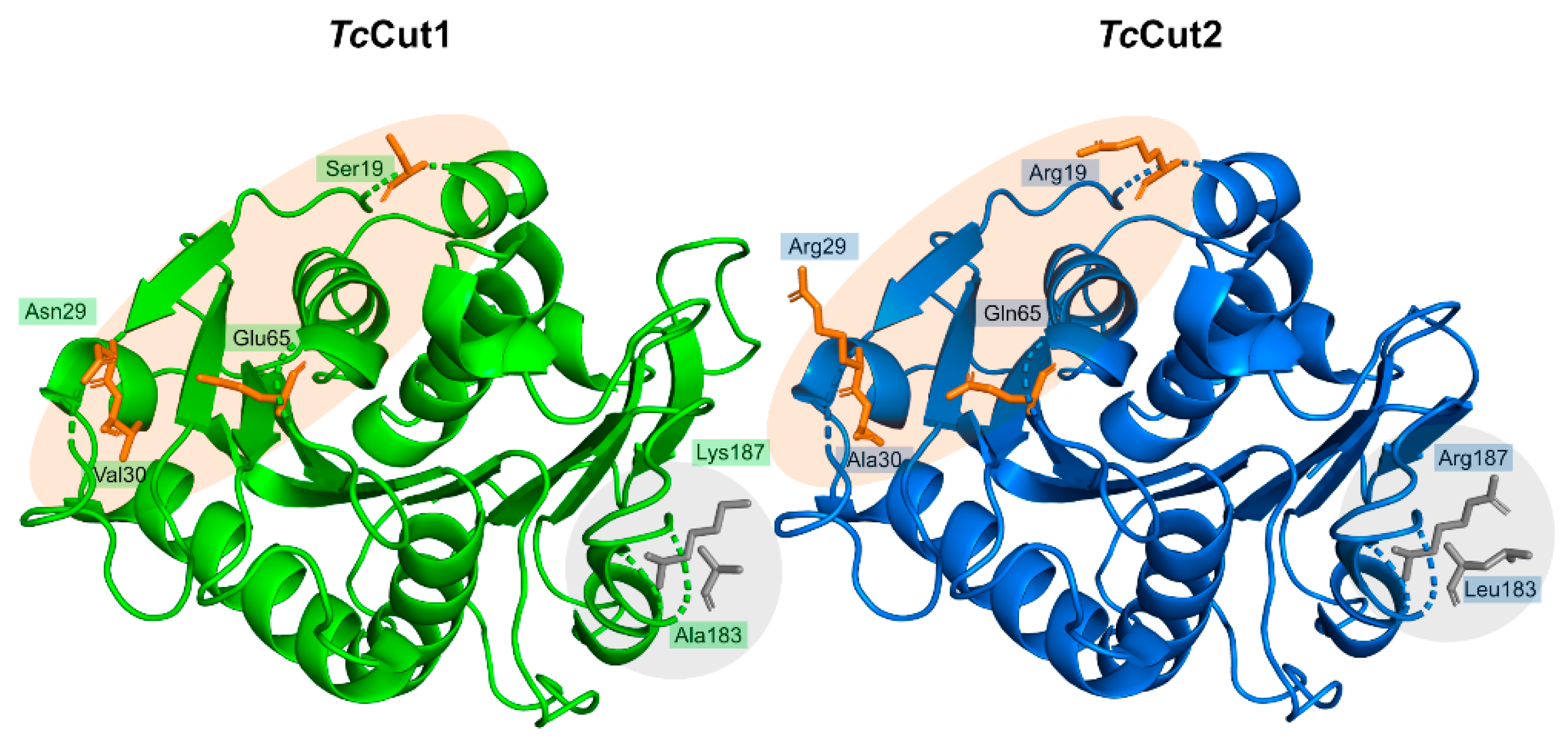
2.7.2. Structure
2.7.3. Activity
2.7.4. Future Perspectives
2.8. Fusarium oxysporum Cutinase 5 (FoCut5a)
2.8.1. Discovery
2.8.2. Structure
2.8.3. Activity
2.8.4. Future Perspectives
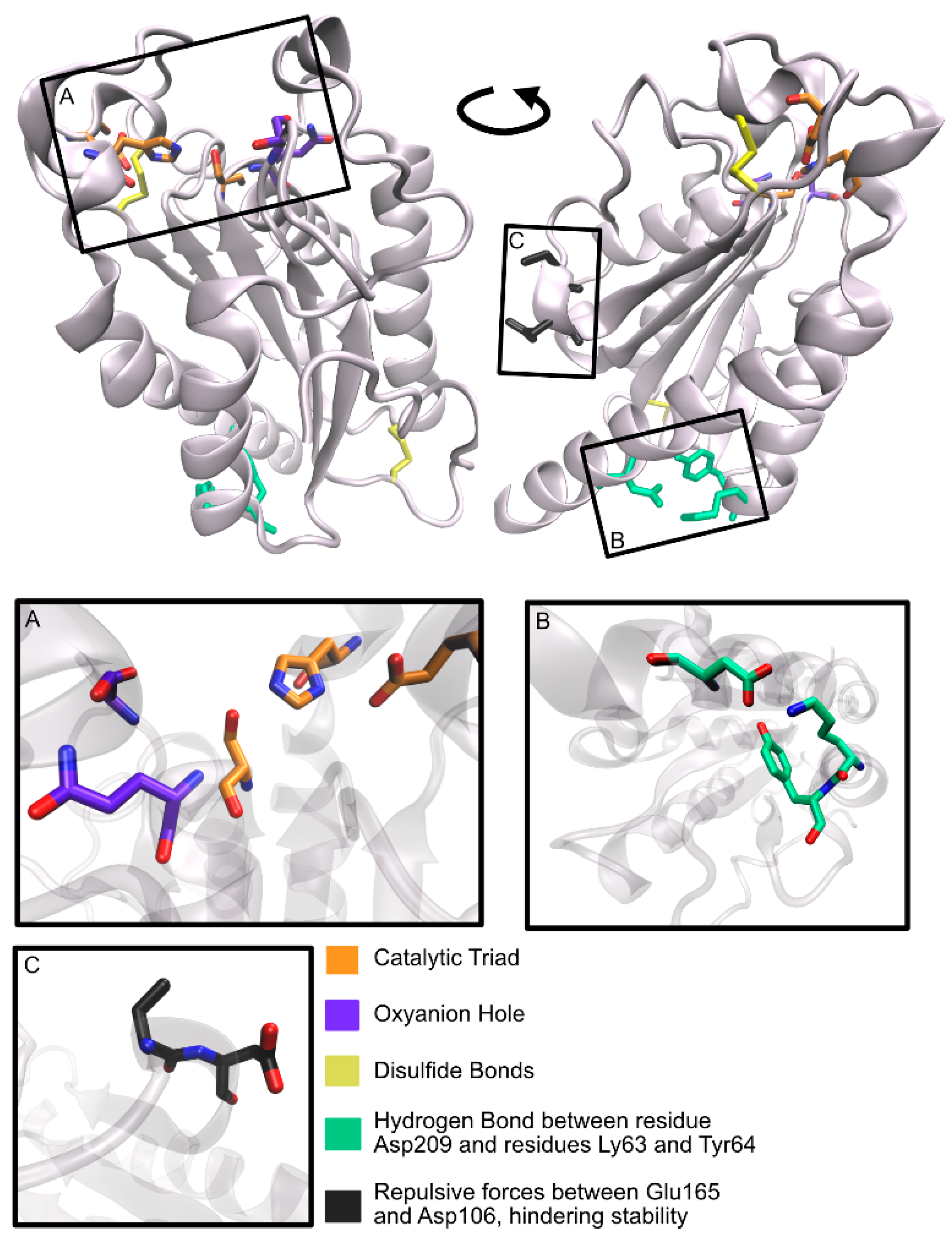
2.9. Humicola insolens Cutinase (HiCut)
2.9.1. Discovery
2.9.2. Structure
2.9.3. Activity
2.9.4. Future Perspectives
2.10. Fusarium solani Cutinase (FsCut)
2.10.1. Discovery
2.10.2. Structure
2.10.3. Activity
2.10.4. Future Perspectives
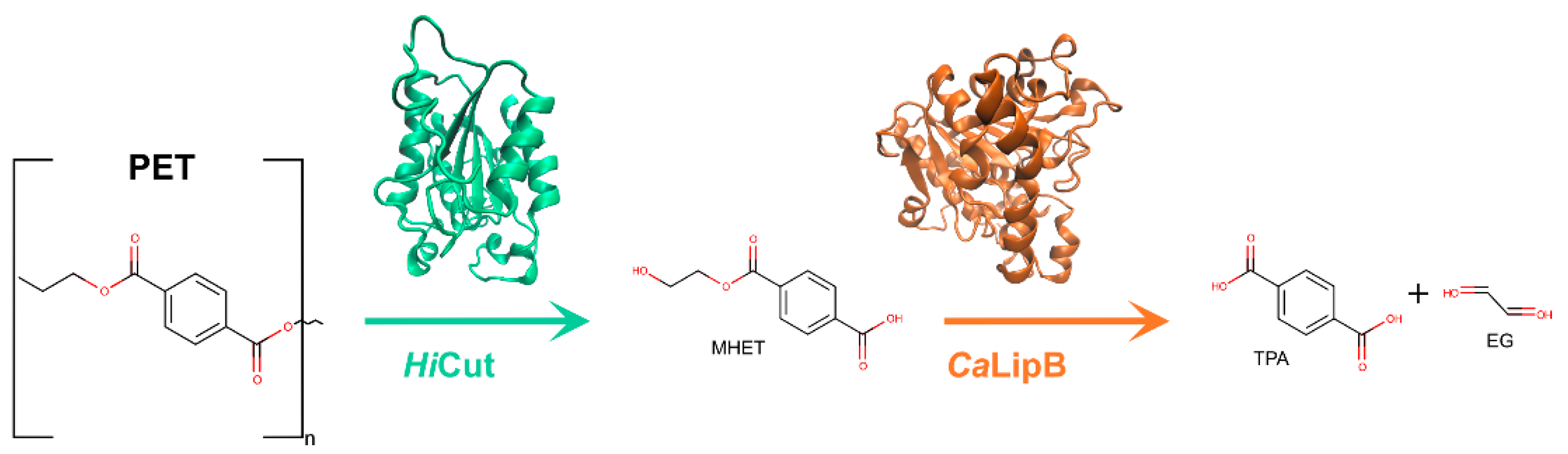
2.11. Candida antarctica Lipase B (CaLipB)
2.11.1. Discovery
2.11.2. Structure
2.11.3. Activity
2.11.4. Future Perspectives
2.12. Thermobifida alba Esterase 1 (TaEst1)
2.13. Thermomyces lanuginosus Lipase (TlLip)
2.13.1. Discovery
2.13.2. Structure
2.13.3. Activity
2.13.4. Future Perspectives
2.14. Thermobifida fusca Carboxylesterase (TfCa)
2.14.1. Discovery
2.14.2. Structure
2.14.3. Activity
2.14.4. Future Perspectives
2.15. Lesser-Known Enzymes
2.15.1. Enterobacter sp. HYI Esterase B (EsEstB)
2.15.2. HR29 Hydrolase (BhrPETase)
2.15.3. Bacillus subtilis Lipase (BsEstB)
2.15.4. Streptomyces scabies Sub1 (ScSub1)
2.15.5. Pseudomonas mendocina Cutinase (PmCut)
2.15.6. PET1–PET13
2.15.7. Thermomonospora curvata Cutinases 0390 and 1278 (TcCut0390 and TcCut1278)
2.15.8. Thermobifida halotolerans Esterase (ThEst)
3. Microorganisms with PET Degradation Activity
4. Potential PET Hydrolases with Activity to Be Confirmed
4.1. Thermobifida alba Esterase 119 (TaEst119)
4.1.1. Discovery
4.1.2. Structure
4.1.3. Activity
4.1.4. Proposed Mechanism
4.1.5. Future Perspectives
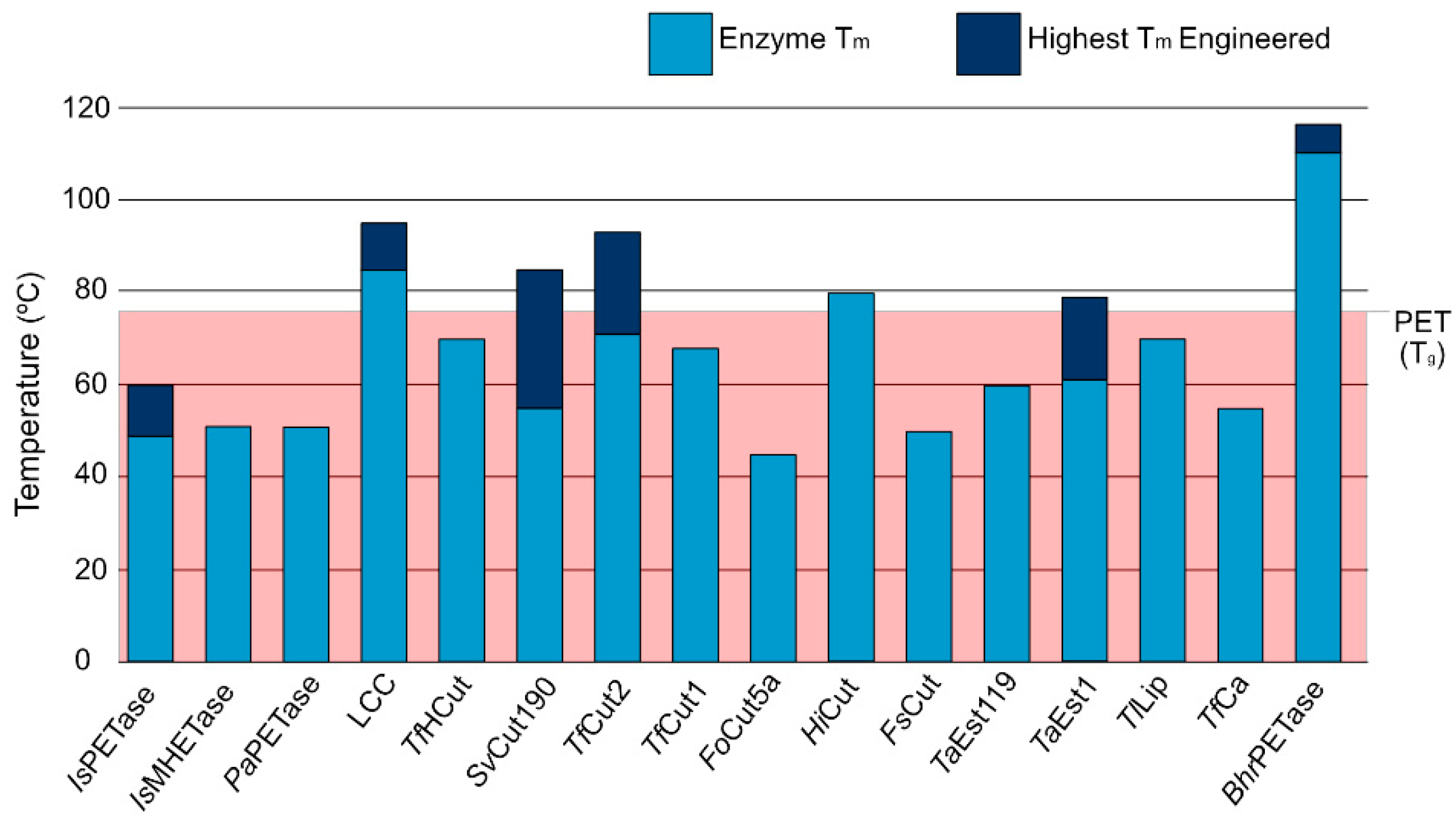
5. Summary and Outlook
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Amobonye, A.; Bhagwat, P.; Singh, S.; Pillai, S. Plastic Biodegradation: Frontline Microbes and Their Enzymes. Sci. Total Environ. 2021, 759, 143536. [Google Scholar] [CrossRef] [PubMed]
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, Use, and Fate of All Plastics Ever Made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samak, N.A.; Jia, Y.; Sharshar, M.M.; Mu, T.; Yang, M.; Peh, S.; Xing, J. Recent Advances in Biocatalysts Engineering for Polyethylene Terephthalate Plastic Waste Green Recycling. Environ. Int. 2020, 145, 106144. [Google Scholar] [CrossRef]
- Gibb, B.C. Plastics Are Forever. Nat. Chem. 2019, 11, 394–395. [Google Scholar] [CrossRef] [PubMed]
- Feil, A.; Pretz, T. Chapter 11—Mechanical Recycling of Packaging Waste; Letcher, T.M., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 283–319. [Google Scholar]
- Plastics—The Facts 2020. Available online: https://www.plasticseurope.org/en/resources/publications/4312-plastics-facts-2020 (accessed on 21 April 2021).
- Chemistry Can Help Make Plastics Sustainable—but It Isn’t the Whole Solution. Nature 2021, 590, 363–364. [CrossRef] [PubMed]
- Nielsen, T.D.; Hasselbalch, J.; Holmberg, K.; Stripple, J. Politics and the Plastic Crisis: A Review throughout the Plastic Life Cycle. WIREs Energy Environ. 2020, 9, e360. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.; Zimmermann, W. Biocatalysis as a Green Route for Recycling the Recalcitrant Plastic Polyethylene Terephthalate. Microb. Biotechnol. 2017, 10, 1302–1307. [Google Scholar] [CrossRef]
- Lahive, E.; Walton, A.; Horton, A.A.; Spurgeon, D.J.; Svendsen, C. Microplastic Particles Reduce Reproduction in the Terrestrial Worm Enchytraeus Crypticus in a Soil Exposure. Environ. Pollut. 2019, 255, 113174. [Google Scholar] [CrossRef]
- Selonen, S.; Dolar, A.; Jemec Kokalj, A.; Skalar, T.; Parramon Dolcet, L.; Hurley, R.; van Gestel, C.A.M. Exploring the Impacts of Plastics in Soil—The Effects of Polyester Textile Fibers on Soil Invertebrates. Sci. Total Environ. 2020, 700, 134451. [Google Scholar] [CrossRef]
- Jung, J.-W.; Kang, J.-S.; Choi, J.; Park, J.-W. Chronic Toxicity of Endocrine Disrupting Chemicals Used in Plastic Products in Korean Resident Species: Implications for Aquatic Ecological Risk Assessment. Ecotoxicol. Environ. Saf. 2020, 192, 110309. [Google Scholar] [CrossRef]
- Hopewell, J.; Dvorak, R.; Kosior, E. Plastics Recycling: Challenges and Opportunities. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 2115–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, R.; Vinoda, K.S.; Papireddy, M.; Gowda, A.N.S. Toxic Pollutants from Plastic Waste—A Review. Procedia Environ. Sci. 2016, 35, 701–708. [Google Scholar] [CrossRef]
- Alabi, O.A.; Ologbonjaye, K.I.; Awosolu, O.; Alalade, O.E. Public and Environmental Health Effects of Plastic Wastes Disposal: A Review. J. Toxicol. Risk Assess. 2019, 5, 1–13. [Google Scholar]
- Grigore, M.E. Methods of Recycling, Properties and Applications of Recycled Thermoplastic Polymers. Recycling 2017, 2, 24. [Google Scholar] [CrossRef] [Green Version]
- Al-Salem, S.M.; Lettieri, P.; Baeyens, J. Recycling and Recovery Routes of Plastic Solid Waste (PSW): A Review. Waste Manag. 2009, 29, 2625–2643. [Google Scholar] [CrossRef]
- Ragaert, K.; Delva, L.; Van Geem, K. Mechanical and Chemical Recycling of Solid Plastic Waste. Waste Manag. 2017, 69, 24–58. [Google Scholar] [CrossRef]
- Scalenghe, R. Resource or Waste? A Perspective of Plastics Degradation in Soil with a Focus on End-of-Life Options. Heliyon 2018, 4, e00941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schyns, Z.O.G.; Shaver, M.P. Mechanical Recycling of Packaging Plastics: A Review. Macromol. Rapid Commun. 2021, 42, 2000415. [Google Scholar] [CrossRef]
- Al-Sabagh, A.M.; Yehia, F.Z.; Eshaq, G.; Rabie, A.M.; ElMetwally, A.E. Greener Routes for Recycling of Polyethylene Terephthalate. Egypt. J. Pet. 2016, 25, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.M.; Robertson, M.L. The Future of Plastics Recycling. Science 2017, 358, 870–872. [Google Scholar] [CrossRef] [PubMed]
- Karayannidis, G.P.; Achilias, D.S. Chemical Recycling of Poly(Ethylene Terephthalate). Macromol. Mater. Eng. 2007, 292, 128–146. [Google Scholar] [CrossRef]
- Paszun, D.; Spychaj, T. Chemical Recycling of Poly(Ethylene Terephthalate). Ind. Eng. Chem. Res. 1997, 36, 1373–1383. [Google Scholar] [CrossRef]
- Focht, D.D. Biodegradation. Access Sci. 2021. [Google Scholar] [CrossRef]
- Roohi; Bano, K.; Kuddus, M.; Zaheer, M.R.; Zia, Q.; Khan, M.F.; Ashraf, G.M.; Gupta, A.; Aliev, G. Microbial Enzymatic Degradation of Biodegradable Plastics. Curr. Pharm. Biotechnol. 2017, 18, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.A.; Clemente, T.M.; Viviani, D.A.; Fong, A.A.; Thomas, K.A.; Kemp, P.; Karl, D.M.; White, A.E.; DeLong, E.F. Diversity and Activity of Communities Inhabiting Plastic Debris in the North Pacific Gyre. mSystems 2016, 1, e00024-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, R.-S.; Richardson, K.H.; Latvanen, E.J.; Hanson, C.A.; Resmini, M.; Sanders, I.A. Microbial Degradation of Plastic in Aqueous Solutions Demonstrated by CO(2) Evolution and Quantification. Int. J. Mol. Sci. 2020, 21, 1176. [Google Scholar] [CrossRef] [Green Version]
- Montazer, Z.; Habibi Najafi, M.B.; Levin, D.B. Challenges with Verifying Microbial Degradation of Polyethylene. Polymers 2020, 12, 123. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, J.; Khatri, M.; Arya, S.K. Recent Insight into Enzymatic Degradation of Plastics Prevalent in the Environment: A Mini—Review. Clean. Eng. Technol. 2021, 2, 100083. [Google Scholar] [CrossRef]
- Sousa, S.F.; Ramos, M.J.; Lim, C.; Fernandes, P.A. Relationship between Enzyme/Substrate Properties and Enzyme Efficiency in Hydrolases. ACS Catal. 2015, 5, 5877–5887. [Google Scholar] [CrossRef]
- Vedrtnam, A.; Kumar, S.; Chaturvedi, S. Experimental Study on Mechanical Behavior, Biodegradability, and Resistance to Natural Weathering and Ultraviolet Radiation of Wood-Plastic Composites. Compos. Part B Eng. 2019, 176, 107282. [Google Scholar] [CrossRef]
- Wei, R.; Tiso, T.; Bertling, J.; O’Connor, K.; Blank, L.M.; Bornscheuer, U.T. Possibilities and Limitations of Biotechnological Plastic Degradation and Recycling. Nat. Catal. 2020, 3, 867–871. [Google Scholar] [CrossRef]
- Liu, C.; Shi, C.; Zhu, S.; Wei, R.; Yin, C.-C. Structural and Functional Characterization of Polyethylene Terephthalate Hydrolase from Ideonella Sakaiensis. Biochem. Biophys. Res. Commun. 2019, 508, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Carr, C.M.; Clarke, D.J.; Dobson, A.D.W. Microbial Polyethylene Terephthalate Hydrolases: Current and Future Perspectives. Front. Microbiol. 2020, 11, 2825. [Google Scholar] [CrossRef] [PubMed]
- Webb, H.K.; Arnott, J.; Crawford, R.J.; Ivanova, E.P. Plastic Degradation and Its Environmental Implications with Special Reference to Poly(Ethylene Terephthalate). Polymers 2013, 5, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.T.H.; Qi, P.; Rostagno, M.; Feteha, A.; Miller, S.A. The Quest for High Glass Transition Temperature Bioplastics. J. Mater. Chem. A 2018, 6, 9298–9331. [Google Scholar] [CrossRef]
- Puspitasari, N.; Tsai, S.-L.; Lee, C.-K. Class I Hydrophobins Pretreatment Stimulates PETase for Monomers Recycling of Waste PETs. Int. J. Biol. Macromol. 2021, 176, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Chamas, A.; Moon, H.; Zheng, J.; Qiu, Y.; Tabassum, T.; Jang, J.H.; Abu-Omar, M.; Scott, S.L.; Suh, S. Degradation Rates of Plastics in the Environment. ACS Sustain. Chem. Eng. 2020, 8, 3494–3511. [Google Scholar] [CrossRef] [Green Version]
- Frounchi, M. Studies on Degradation of PET in Mechanical Recycling. Macromol. Symp. 1999, 144, 465–469. [Google Scholar] [CrossRef]
- Crippa, M.; Morico, B. Chapter 12—PET Depolymerization: A Novel Process for Plastic Waste Chemical Recycling. In Catalysis, Green Chemistry and Sustainable Energy; Basile, A., Centi, G., de Falco, M., Iaquaniello, G., Eds.; Elsevier Press: Amsterdam, The Netherlands, 2020; Volume 179, pp. 215–229. [Google Scholar]
- Bartolome, L.; Imran, M.; Cho, B.; Al-Masry, W.; Kim, D. Recent Developments in the Chemical Recycling of PET. In Material Recycling—Trends and Perspectives; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Kim, H.T.; Kim, J.K.; Cha, H.G.; Kang, M.J.; Lee, H.S.; Khang, T.U.; Yun, E.J.; Lee, D.-H.; Song, B.K.; Park, S.J.; et al. Biological Valorization of Poly(Ethylene Terephthalate) Monomers for Upcycling Waste PET. ACS Sustain. Chem. Eng. 2019, 7, 19396–19406. [Google Scholar] [CrossRef]
- Bano, N.; Younas, T.; Shoaib, F.; Rashid, D.; Jaffri, N. Plastic: Reduce, Recycle, and Environment. In Environmentally-Benign Energy Solutions; Springer: Cham, Switzerland, 2020; pp. 191–208. [Google Scholar]
- Kawai, F.; Thumarat, U.; Kitadokoro, K.; Waku, T.; Tada, T.; Tanaka, N.; Kawabata, T. Comparison of Polyester-Degrading Cutinases from Genus Thermobifida. In Green Polymer Chemistry: Biocatalysis and Materials II; ACS Symposium Series; American Chemical Society Press: Washington, DC, USA, 2013; Volume 1144, pp. 111–120. [Google Scholar]
- Jog, J.P. Crystallization of Polyethyleneterephthalate. J. Macromol. Sci. Part C 1995, 35, 531–553. [Google Scholar] [CrossRef]
- Biron, M. Mechanical Properties. In Material Selection for Thermoplastic Parts; William Andrew Publishing: Oxford, UK, 2016; pp. 261–337. [Google Scholar]
- Schirrer, R. Damage Mechanisms in Amorphous Glassy PolymersCrazing. In Handbook of Materials Behavior Models; Academic Press: Cambridge, MA, USA, 2001; pp. 488–499. [Google Scholar]
- Wei, R.; Breite, D.; Song, C.; Gräsing, D.; Ploss, T.; Hille, P.; Schwerdtfeger, R.; Matysik, J.; Schulze, A.; Zimmermann, W. Biocatalytic Degradation Efficiency of Postconsumer Polyethylene Terephthalate Packaging Determined by Their Polymer Microstructures. Adv. Sci. 2019, 6, 1900491. [Google Scholar] [CrossRef] [Green Version]
- Danso, D.; Chow, J.; Streit, W.R. Plastics: Environmental and Biotechnological Perspectives on Microbial Degradation. Appl. Environ. Microbiol. 2019, 85, e01095-19. [Google Scholar] [CrossRef] [Green Version]
- Müller, R.-J.; Schrader, H.; Profe, J.; Dresler, K.; Deckwer, W.-D. Enzymatic Degradation of Poly(Ethylene Terephthalate): Rapid Hydrolyse Using a Hydrolase from T. Fusca. Macromol. Rapid Commun. 2005, 26, 1400–1405. [Google Scholar] [CrossRef]
- Yoshida, S.; Hiraga, K.; Takehana, T.; Taniguchi, I.; Yamaji, H.; Maeda, Y.; Toyohara, K.; Miyamoto, K.; Kimura, Y.; Oda, K. A Bacterium That Degrades and Assimilates Poly(Ethylene Terephthalate). Science 2016, 351, 1196–1199. [Google Scholar] [CrossRef]
- Ribitsch, D.; Heumann, S.; Trotscha, E.; Herrero Acero, E.; Greimel, K.; Leber, R.; Birner-Gruenberger, R.; Deller, S.; Eiteljoerg, I.; Remler, P.; et al. Hydrolysis of Polyethyleneterephthalate by P-Nitrobenzylesterase from Bacillus Subtilis. Biotechnol. Prog. 2011, 27, 951–960. [Google Scholar] [CrossRef]
- Ribitsch, D.; Acero, E.H.; Greimel, K.; Dellacher, A.; Zitzenbacher, S.; Marold, A.; Rodriguez, R.D.; Steinkellner, G.; Gruber, K.; Schwab, H.; et al. A New Esterase from Thermobifida Halotolerans Hydrolyses Polyethylene Terephthalate (PET) and Polylactic Acid (PLA). Polymers 2012, 4, 617–629. [Google Scholar] [CrossRef]
- Silva, C.; Da, S.; Silva, N.; Matamá, T.; Araújo, R.; Martins, M.; Chen, S.; Chen, J.; Wu, J.; Casal, M.; et al. Engineered Thermobifida Fusca Cutinase with Increased Activity on Polyester Substrates. Biotechnol. J. 2011, 6, 1230–1239. [Google Scholar] [CrossRef]
- Kawai, F.; Oda, M.; Tamashiro, T.; Waku, T.; Tanaka, N.; Yamamoto, M.; Mizushima, H.; Miyakawa, T.; Tanokura, M. A Novel Ca2+-Activated, Thermostabilized Polyesterase Capable of Hydrolyzing Polyethylene Terephthalate from Saccharomonospora Viridis AHK190. Appl. Microbiol. Biotechnol. 2014, 98, 10053–10064. [Google Scholar] [CrossRef]
- Thumarat, U.; Nakamura, R.; Kawabata, T.; Suzuki, H.; Kawai, F. Biochemical and Genetic Analysis of a Cutinase-Type Polyesterase from a Thermophilic Thermobifida Alba AHK119. Appl. Microbiol. Biotechnol. 2012, 95, 419–430. [Google Scholar] [CrossRef]
- Sulaiman, S.; Yamato, S.; Kanaya, E.; Kim, J.-J.; Koga, Y.; Takano, K.; Kanaya, S. Isolation of a Novel Cutinase Homolog with Polyethylene Terephthalate-Degrading Activity from Leaf-Branch Compost by Using a Metagenomic Approach. Appl. Environ. Microbiol. 2012, 78, 1556–1562. [Google Scholar] [CrossRef] [Green Version]
- Goldsmith, M.; Tawfik, D.S. Enzyme Engineering: Reaching the Maximal Catalytic Efficiency Peak. Curr. Opin. Struct. Biol. 2017, 47, 140–150. [Google Scholar] [CrossRef]
- Sharma, A.; Gupta, G.; Ahmad, T.; Mansoor, S.; Kaur, B. Enzyme Engineering: Current Trends and Future Perspectives. Food Rev. Int. 2021, 37, 121–154. [Google Scholar] [CrossRef]
- Hida, K.; Hanes, J.; Ostermeier, M. Directed Evolution for Drug and Nucleic Acid Delivery. Adv. Drug Deliv. Rev. 2007, 59, 1562–1578. [Google Scholar] [CrossRef]
- Dubey, K.K.; Pramanik, A.; Ankush; Khushboo; Yadav, J. Chapter 12—Enzyme Engineering. In Biomass, Biofuels, Biochemicals; Singh, R.S., Singhania, R.R., Pandey, A., Larroche, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 325–347. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Available online: rcsb.org (accessed on 22 September 2021).
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Planas-Iglesias, J.; Marques, S.M.; Pinto, G.P.; Musil, M.; Stourac, J.; Damborsky, J.; Bednar, D. Computational Design of Enzymes for Biotechnological Applications. Biotechnol. Adv. 2021, 47, 107696. [Google Scholar] [CrossRef]
- Labrou, N.E. Random Mutagenesis Methods for In Vitro Directed Enzyme Evolution. Curr. Protein Pept. Sci. 2010, 11, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Wolfenden, R. Benchmark Reaction Rates, the Stability of Biological Molecules in Water, and the Evolution of Catalytic Power in Enzymes. Annu. Rev. Biochem. 2011, 80, 645–667. [Google Scholar] [CrossRef]
- Wolfenden, R. Degrees of Difficulty of Water-Consuming Reactions in the Absence of Enzymes. Chem. Rev. 2006, 106, 3379–3396. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, M.; Wiuf, C. The CATH Database. Hum. Genom. 2010, 4, 207–212. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Wang, C.; Yu, K.; Kuzin, A.P.; Richter, F.; Lew, S.; Miklos, A.E.; Matthews, M.L.; Seetharaman, J.; Su, M.; et al. Design of Activated Serine-Containing Catalytic Triads with Atomic-Level Accuracy. Nat. Chem. Biol. 2014, 10, 386–391. [Google Scholar] [CrossRef]
- Ordentlich, A.; Barak, D.; Kronman, C.; Ariel, N.; Segall, Y.; Velan, B.; Shafferman, A. Functional Characteristics of the Oxyanion Hole in Human Acetylcholinesterase. J. Biol. Chem. 1998, 273, 19509–19517. [Google Scholar] [CrossRef] [Green Version]
- Daniel, R.M.; Danson, M.J. A New Understanding of How Temperature Affects the Catalytic Activity of Enzymes. Trends Biochem. Sci. 2010, 35, 584–591. [Google Scholar] [CrossRef]
- Joo, S.; Cho, I.J.; Seo, H.; Son, H.F.; Sagong, H.-Y.; Shin, T.J.; Choi, S.Y.; Lee, S.Y.; Kim, K.-J. Structural Insight into Molecular Mechanism of Poly(Ethylene Terephthalate) Degradation. Nat. Commun. 2018, 9, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, H.P.; Allen, M.D.; Donohoe, B.S.; Rorrer, N.A.; Kearns, F.L.; Silveira, R.L.; Pollard, B.C.; Dominick, G.; Duman, R.; El Omari, K.; et al. Characterization and Engineering of a Plastic-Degrading Aromatic Polyesterase. Proc. Natl. Acad. Sci. USA 2018, 115, E4350–E4357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Liu, W.; Huang, J.-W.; Ma, J.; Zheng, Y.; Ko, T.-P.; Xu, L.; Cheng, Y.-S.; Chen, C.-C.; Guo, R.-T. Structural Insight into Catalytic Mechanism of PET Hydrolase. Nat. Commun. 2017, 8, 2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiland, M.H. Enzymatic Biodegradation by Exploring the Rational Protein Engineering of the Polyethylene Terephthalate Hydrolyzing Enzyme PETase from Ideonella Sakaiensis 201-F6. In Mechanistic Enzymology: Bridging Structure and Function; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2020; Volume 1357, pp. 161–174. [Google Scholar]
- Liu, B.; He, L.; Wang, L.; Li, T.; Li, C.; Liu, H.; Luo, Y.; Bao, R. Protein Crystallography and Site-Direct Mutagenesis Analysis of the Poly(Ethylene Terephthalate) Hydrolase PETase from Ideonella Sakaiensis. ChemBioChem 2018, 19, 1471–1475. [Google Scholar] [CrossRef] [PubMed]
- Fecker, T.; Galaz-Davison, P.; Engelberger, F.; Narui, Y.; Sotomayor, M.; Parra, L.P.; Ramírez-Sarmiento, C.A. Active Site Flexibility as a Hallmark for Efficient PET Degradation by I. Sakaiensis PETase. Biophys. J. 2018, 114, 1302–1312. [Google Scholar] [CrossRef] [Green Version]
- Palm, G.J.; Reisky, L.; Böttcher, D.; Müller, H.; Michels, E.A.P.; Walczak, M.C.; Berndt, L.; Weiss, M.S.; Bornscheuer, U.T.; Weber, G. Structure of the Plastic-Degrading Ideonella Sakaiensis MHETase Bound to a Substrate. Nat. Commun. 2019, 10, 1717. [Google Scholar] [CrossRef]
- Son, H.F.; Cho, I.J.; Joo, S.; Seo, H.; Sagong, H.-Y.; Choi, S.Y.; Lee, S.Y.; Kim, K.-J. Rational Protein Engineering of Thermo-Stable PETase from Ideonella Sakaiensis for Highly Efficient PET Degradation. ACS Catal. 2019, 9, 3519–3526. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, Y.; Liu, X.; Dong, S.; Tian, Y.; Qiao, Y.; Mitra, R.; Han, J.; Li, C.; Han, X.; et al. Computational Redesign of a PETase for Plastic Biodegradation by the GRAPE Strategy. bioRxiv 2019, 787069. [Google Scholar] [CrossRef]
- Son, H.F.; Joo, S.; Seo, H.; Sagong, H.Y.; Lee, S.H.; Hong, H.; Kim, K.J. Structural Bioinformatics-Based Protein Engineering of Thermo-Stable PETase from Ideonella Sakaiensis. Enzym. Microb. Technol. 2020, 141, 109656. [Google Scholar] [CrossRef]
- Campo, E.A. Thermal Properties of Polymeric Materials. In Selection of Polymeric Materials; Campo, E.A., Ed.; Plastics Design Library; William Andrew Publishing: Norwich, NY, USA, 2008; pp. 103–140. [Google Scholar]
- Pohl, F.; Eggenhuisen, J.T.; Kane, I.A.; Clare, M.A. Transport and Burial of Microplastics in Deep-Marine Sediments by Turbidity Currents. Environ. Sci. Technol. 2020, 54, 4180–4189. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Y.; Cheng, Y.; Wang, X.; Tong, S.; Yang, H.; Wang, Z. Efficient Biodegradation of Highly Crystallized Polyethylene Terephthalate through Cell Surface Display of Bacterial PETase. Sci. Total Environ. 2020, 709, 136138. [Google Scholar] [CrossRef] [PubMed]
- Moog, D.; Schmitt, J.; Senger, J.; Zarzycki, J.; Rexer, K.-H.; Linne, U.; Erb, T.; Maier, U.G. Using a Marine Microalga as a Chassis for Polyethylene Terephthalate (PET) Degradation. Microb. Cell Fact. 2019, 18, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Park, S.-B.; Tran, Q.-G.; Cho, D.-H.; Choi, D.-Y.; Lee, Y.J.; Kim, H.-S. Functional Expression of Polyethylene Terephthalate-Degrading Enzyme (PETase) in Green Microalgae. Microb. Cell Fact. 2020, 19, 97. [Google Scholar] [CrossRef] [PubMed]
- Knott, B.C.; Erickson, E.; Allen, M.D.; Gado, J.E.; Graham, R.; Kearns, F.L.; Pardo, I.; Topuzlu, E.; Anderson, J.J.; Austin, H.P.; et al. Characterization and Engineering of a Two-Enzyme System for Plastics Depolymerization. Proc. Natl. Acad. Sci. USA 2020, 117, 25476–25485. [Google Scholar] [CrossRef]
- Papadopoulou, A.; Hecht, K.; Buller, R. Enzymatic PET Degradation. CHIMIA Int. J. Chem. 2019, 73, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Song, C.; Gräsing, D.; Schneider, T.; Bielytskyi, P.; Böttcher, D.; Matysik, J.; Bornscheuer, U.T.; Zimmermann, W. Conformational Fitting of a Flexible Oligomeric Substrate Does Not Explain the Enzymatic PET Degradation. Nat. Commun. 2019, 10, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Cho, I.J.; Joo, S.; Son, H.F.; Sagong, H.-Y.; Choi, S.Y.; Lee, S.Y.; Kim, K.-J. Reply to “Conformational Fitting of a Flexible Oligomeric Substrate Does Not Explain the Enzymatic PET Degradation”. Nat. Commun. 2019, 10, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Kawai, F.; Kawabata, T.; Oda, M. Current Knowledge on Enzymatic PET Degradation and Its Possible Application to Waste Stream Management and Other Fields. Appl. Microbiol. Biotechnol. 2019, 103, 4253–4268. [Google Scholar] [CrossRef] [Green Version]
- Cousin, X.; Hotelier, T.; Giles, K.; Lievin, P.; Toutant, J.-P.; Chatonnet, A. The α/β Fold Family of Proteins Database and the Cholinesterase Gene Server ESTHER. Nucleic Acids Res. 1997, 25, 143–146. [Google Scholar] [CrossRef] [Green Version]
- Sagong, H.-Y.; Seo, H.; Kim, T.; Son, H.F.; Joo, S.; Lee, S.H.; Kim, S.; Woo, J.-S.; Hwang, S.Y.; Kim, K.-J. Decomposition of the PET Film by MHETase Using Exo-PETase Function. ACS Catal. 2020, 10, 4805–4812. [Google Scholar] [CrossRef]
- Suzuki, K.; Hori, A.; Kawamoto, K.; Thangudu, R.R.; Ishida, T.; Igarashi, K.; Samejima, M.; Yamada, C.; Arakawa, T.; Wakagi, T.; et al. Crystal Structure of a Feruloyl Esterase Belonging to the Tannase Family: A Disulfide Bond near a Catalytic Triad. Proteins Struct. Funct. Bioinform. 2014, 82, 2857–2867. [Google Scholar] [CrossRef]
- Bollinger, A.; Thies, S.; Knieps-Grünhagen, E.; Gertzen, C.; Kobus, S.; Höppner, A.; Ferrer, M.; Gohlke, H.; Smits, S.H.J.; Jaeger, K.E. A Novel Polyester Hydrolase from the Marine Bacterium Pseudomonas Aestusnigri—Structural and Functional Insights. Front. Microbiol. 2020, 11, 114. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, D.; Mulet, M.; Rodríguez, A.C.; David, Z.; Lalucat, J.; García-Valdés, E. Pseudomonas Aestusnigri Sp. Nov. Isolated from Crude Oil-Contaminated Intertidal Sand Samples after the Prestige Oil Spill. Syst. Appl. Microbiol. 2014, 37, 89–94. [Google Scholar] [CrossRef]
- Molitor, R.; Bollinger, A.; Kubicki, S.; Loeschcke, A.; Jaeger, K.-E.; Thies, S. Agar Plate-Based Screening Methods for the Identification of Polyester Hydrolysis by Pseudomonas Species. Microb. Biotechnol. 2020, 13, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Gomila, M.; Mulet, M.; Lalucat, J.; García-Valdés, E. Draft Genome Sequence of the Marine Bacterium Pseudomonas Aestusnigri VGXO14(T). Genome Announc. 2017, 5, e00765-17. [Google Scholar] [CrossRef] [Green Version]
- Bollinger, A.; Thies, S.; Katzke, N.; Jaeger, K.-E. The Biotechnological Potential of Marine Bacteria in the Novel Lineage of Pseudomonas Pertucinogena. Microb. Biotechnol. 2020, 13, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Truhlar, D.G. QM/MM: What Have We Learned, Where Are We, and Where Do We Go from Here? Theor. Chem. Acc. 2007, 117, 185–199. [Google Scholar] [CrossRef] [Green Version]
- Magalhães, R.P.; Fernandes, H.S.; Sousa, S.F. Modelling Enzymatic Mechanisms with QM/MM Approaches: Current Status and Future Challenges. Isr. J. Chem. 2020, 60, 655–666. [Google Scholar] [CrossRef]
- Senn, H.M.; Thiel, W. QM/MM Methods for Biomolecular Systems. Angew. Chem. Int. Ed. 2009, 48, 1198–1229. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, S.; You, D.-J.; Kanaya, E.; Koga, Y.; Kanaya, S. Crystal Structure and Thermodynamic and Kinetic Stability of Metagenome-Derived LC-Cutinase. Biochemistry 2014, 53, 1858–1869. [Google Scholar] [CrossRef]
- Tournier, V.; Topham, C.M.; Gilles, A.; David, B.; Folgoas, C.; Moya-Leclair, E.; Kamionka, E.; Desrousseaux, M.-L.; Texier, H.; Gavalda, S.; et al. An Engineered PET Depolymerase to Break down and Recycle Plastic Bottles. Nature 2020, 580, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Shirke, A.N.; White, C.; Englaender, J.A.; Zwarycz, A.; Butterfoss, G.L.; Linhardt, R.J.; Gross, R.A. Stabilizing Leaf and Branch Compost Cutinase (LCC) with Glycosylation: Mechanism and Effect on PET Hydrolysis. Biochemistry 2018, 57, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Wei, R.; Cui, Q.; Bornscheuer, U.T.; Liu, Y.J. Thermophilic Whole-Cell Degradation of Polyethylene Terephthalate Using Engineered Clostridium Thermocellum. Microb. Biotechnol. 2020, 14, 374–385. [Google Scholar] [CrossRef]
- Kleeberg, I.; Hetz, C.; Kroppenstedt, R.M.; Müller, R.J.; Deckwer, W.D. Biodegradation of Aliphatic-Aromatic Copolyesters by Thermomonospora Fusca and Other Thermophilic Compost Isolates. Appl. Environ. Microbiol. 1998, 64, 1731–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Tong, X.; Woodard, R.W.; Du, G.; Wu, J.; Chen, J. Identification and Characterization of Bacterial Cutinase. J. Biol. Chem. 2008, 283, 25854–25862. [Google Scholar] [CrossRef] [Green Version]
- Lao, G.; Ghangas, G.S.; Jung, E.D.; Wilson, D.B. DNA Sequences of Three Beta-1,4-Endoglucanase Genes from Thermomonospora Fusca. J. Bacteriol. 1991, 173, 3397–3407. [Google Scholar] [CrossRef] [Green Version]
- Dresler, K.; Van Den Heuvel, J.; Müller, R.J.; Deckwer, W.D. Production of a Recombinant Polyester-Cleaving Hydrolase from Thermobifida Fusca in Escherichia Coli. Bioprocess Biosyst. Eng. 2006, 29, 169–183. [Google Scholar] [CrossRef] [Green Version]
- Kleeberg, I.; Welzel, K.; VandenHeuvel, J.; Müller, R.J.; Deckwer, W.D. Characterization of a New Extracellular Hydrolase from Thermobifida Fusca Degrading Aliphatic-Aromatic Copolyesters. Biomacromolecules 2005, 6, 262–270. [Google Scholar] [CrossRef]
- Then, J.; Wei, R.; Oeser, T.; Barth, M.; Belisário-Ferrari, M.R.; Schmidt, J.; Zimmermann, W. Ca2+ and Mg2+ Binding Site Engineering Increases the Degradation of Polyethylene Terephthalate Films by Polyester Hydrolases from Thermobifida Fusca. Biotechnol. J. 2015, 10, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Yuan, S.; Wu, L.; Su, L.; Zhao, Q.; Wu, J.; Huang, W.; Zhou, J. Structure-Guided Engineering of a Thermobifida Fusca Cutinase for Enhanced Hydrolysis on Natural Polyester Substrate. Bioresour. Bioprocess. 2020, 7, 37. [Google Scholar] [CrossRef]
- Alisch-Mark, M.; Herrmann, A.; Zimmermann, W. Increase of the Hydrophilicity of Polyethylene Terephthalate Fibres by Hydrolases from Thermomonospora Fusca and Fusarium Solani f. Sp. Pisi. Biotechnol. Lett. 2006, 28, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Woodard, R.W.; Chen, J.; Wu, J. Extracellular Location of Thermobifida Fusca Cutinase Expressed in Escherichia Coli BL21(DE3) without Mediation of a Signal Peptide. Appl. Environ. Microbiol. 2013, 79, 4192–4198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Malten, M.; Grote, A.; Jahn, D.; Deckwer, W.-D. Codon Optimized Thermobifida Fusca Hydrolase Secreted by Bacillus Megaterium. J. Anat. 2006, 189 Pt 3, 503–505. [Google Scholar]
- Sinsereekul, N.; Wangkam, T.; Thamchaipenet, A.; Srikhirin, T.; Eurwilaichitr, L.; Champreda, V. Recombinant Expression of BTA Hydrolase in Streptomyces Rimosus and Catalytic Analysis on Polyesters by Surface Plasmon Resonance. Appl. Microbiol. Biotechnol. 2010, 86, 1775–1784. [Google Scholar] [CrossRef]
- Sousa, S.F.; Ribeiro, A.J.M.; Coimbra, J.T.S.; Neves, R.P.P.; Martins, S.A.; Moorthy, N.S.H.N.; Fernandes, P.A.; Ramos, M.J. Protein-Ligand Docking in the New Millennium—A Retrospective of 10 Years in the Field. Curr. Med. Chem. 2013, 20, 2296–2314. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein–Ligand Docking: Current Status and Future Challenges. Proteins Struct. Funct. Bioinform. 2006, 65, 15–26. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Halperin, I.; Ma, B.; Wolfson, H.; Nussinov, R. Principles of Docking: An Overview of Search Algorithms and a Guide to Scoring Functions. Proteins: Struct. Funct. Bioinform. 2002, 443, 409–443. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; Teixeira, C.S.S.; Cooper, D.N.; Carneiro, J.; Lopes-Marques, M.; Stenson, P.D.; Amorim, A.; Prata, M.J.; Sousa, S.F.; Azevedo, L. Compensatory Epistasis Explored by Molecular Dynamics Simulations. Hum. Genet. 2021, 140, 1329–1342. [Google Scholar] [CrossRef] [PubMed]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular Dynamics Simulations: Advances and Applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar] [PubMed] [Green Version]
- Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int. J. Mol. Sci. 2020, 21, 6339. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; McCammon, J.A. Molecular Dynamics Simulations of Biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Klepeis, J.L.; Lindorff-Larsen, K.; Dror, R.O.; Shaw, D.E. Long-Timescale Molecular Dynamics Simulations of Protein Structure and Function. Curr. Opin. Struct. Biol. 2009, 19, 120–127. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Pati, A.; Sikorski, J.; Nolan, M.; Lapidus, A.; Copeland, A.; Glavina Del Rio, T.; Lucas, S.; Chen, F.; Tice, H.; Pitluck, S.; et al. Complete Genome Sequence of Saccharomonospora Viridis Type Strain (P101T). Stand. Genom. Sci. 2009, 1, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Miyakawa, T.; Mizushima, H.; Ohtsuka, J.; Oda, M.; Kawai, F.; Tanokura, M. Structural Basis for the Ca2+-Enhanced Thermostability and Activity of PET-Degrading Cutinase-like Enzyme from Saccharomonospora Viridis AHK190. Appl. Microbiol. Biotechnol. 2015, 99, 4297–4307. [Google Scholar] [CrossRef]
- Numoto, N.; Kamiya, N.; Bekker, G.-J.; Yamagami, Y.; Inaba, S.; Ishii, K.; Uchiyama, S.; Kawai, F.; Ito, N.; Oda, M. Structural Dynamics of the PET-Degrading Cutinase-like Enzyme from Saccharomonospora Viridis AHK190 in Substrate-Bound States Elucidates the Ca2+-Driven Catalytic Cycle. Biochemistry 2018, 57, 5289–5300. [Google Scholar] [CrossRef]
- Oda, M.; Yamagami, Y.; Inaba, S.; Oida, T.; Yamamoto, M.; Kitajima, S.; Kawai, F. Enzymatic Hydrolysis of PET: Functional Roles of Three Ca2+ Ions Bound to a Cutinase-like Enzyme, Cut190*, and Its Engineering for Improved Activity. Appl. Microbiol. Biotechnol. 2018, 102, 10067–10077. [Google Scholar] [CrossRef]
- Senga, A.; Numoto, N.; Yamashita, M.; Iida, A.; Ito, N.; Kawai, F.; Oda, M. Multiple Structural States of Ca2+-Regulated PET Hydrolase, Cut190, and Its Correlation with Activity and Stability. J. Biochem. 2021, 169, 207–213. [Google Scholar] [CrossRef]
- Emori, M.; Numoto, N.; Senga, A.; Bekker, G.-J.; Kamiya, N.; Kobayashi, Y.; Ito, N.; Kawai, F.; Oda, M. Structural Basis of Mutants of PET-Degrading Enzyme from Saccharomonospora Viridis AHK190 with High Activity and Thermal Stability. Proteins Struct. Funct. Bioinform. 2021, 89, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T.; Oda, M.; Kawai, F. Mutational Analysis of Cutinase-like Enzyme, Cut190, Based on the 3D Docking Structure with Model Compounds of Polyethylene Terephthalate. J. Biosci. Bioeng. 2017, 124, 28–35. [Google Scholar] [CrossRef]
- Hantani, Y.; Imamura, H.; Yamamoto, T.; Senga, A.; Yamagami, Y.; Kato, M.; Kawai, F.; Oda, M. Functional Characterizations of Polyethylene Terephthalate-Degrading Cutinase-like Enzyme Cut190 Mutants Using Bis(2-Hydroxyethyl) Terephthalate as the Model Substrate. AIMS Biophys. 2018, 5, 290–302. [Google Scholar] [CrossRef]
- Herrero Acero, E.; Ribitsch, D.; Steinkellner, G.; Gruber, K.; Greimel, K.; Eiteljoerg, I.; Trotscha, E.; Wei, R.; Zimmermann, W.; Zinn, M.; et al. Enzymatic Surface Hydrolysis of PET: Effect of Structural Diversity on Kinetic Properties of Cutinases from Thermobifida. Macromolecules 2011, 44, 4632–4640. [Google Scholar] [CrossRef] [Green Version]
- Ribitsch, D.; Acero, E.H.; Greimel, K.; Eiteljoerg, I.; Trotscha, E.; Freddi, G.; Schwab, H.; Guebitz, G.M. Characterization of a New Cutinase from Thermobifida Alba for PET-Surface Hydrolysis. Biocatal. Biotransformation 2012, 30, 2–9. [Google Scholar] [CrossRef]
- Roth, C.; Wei, R.; Oeser, T.; Then, J.; Föllner, C.; Zimmermann, W.; Sträter, N. Structural and Functional Studies on a Thermostable Polyethylene Terephthalate Degrading Hydrolase from Thermobifida Fusca. Appl. Microbiol. Biotechnol. 2014, 98, 7815–7823. [Google Scholar] [CrossRef] [PubMed]
- Ribitsch, D.; Hromic, A.; Zitzenbacher, S.; Zartl, B.; Gamerith, C.; Pellis, A.; Jungbauer, A.; Łyskowski, A.; Steinkellner, G.; Gruber, K.; et al. Small Cause, Large Effect: Structural Characterization of Cutinases from Thermobifida Cellulosilytica. Biotechnol. Bioeng. 2017, 114, 2481–2488. [Google Scholar] [CrossRef]
- Gamerith, C.; Zartl, B.; Pellis, A.; Guillamot, F.; Marty, A.; Acero, E.H.; Guebitz, G.M. Enzymatic Recovery of Polyester Building Blocks from Polymer Blends. Process Biochem. 2017, 59, 58–64. [Google Scholar] [CrossRef]
- Barth, M.; Oeser, T.; Wei, R.; Then, J.; Schmidt, J.; Zimmermann, W. Effect of Hydrolysis Products on the Enzymatic Degradation of Polyethylene Terephthalate Nanoparticles by a Polyester Hydrolase from Thermobifida Fusca. Biochem. Eng. J. 2015, 93, 222–228. [Google Scholar] [CrossRef]
- Wei, R.; Oeser, T.; Schmidt, J.; Meier, R.; Barth, M.; Then, J.; Zimmermann, W. Engineered Bacterial Polyester Hydrolases Efficiently Degrade Polyethylene Terephthalate Due to Relieved Product Inhibition. Biotechnol. Bioeng. 2016, 113, 1658–1665. [Google Scholar] [CrossRef]
- Barth, M.; Wei, R.; Oeser, T.; Then, J.; Schmidt, J.; Wohlgemuth, F.; Zimmermann, W. Enzymatic Hydrolysis of Polyethylene Terephthalate Films in an Ultrafiltration Membrane Reactor. J. Memb. Sci. 2015, 494, 182–187. [Google Scholar] [CrossRef]
- Then, J.; Wei, R.; Oeser, T.; Gerdts, A.; Schmidt, J.; Barth, M.; Zimmermann, W. A Disulfide Bridge in the Calcium Binding Site of a Polyester Hydrolase Increases Its Thermal Stability and Activity against Polyethylene Terephthalate. FEBS Open Bio 2016, 6, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Herrero Acero, E.; Ribitsch, D.; Dellacher, A.; Zitzenbacher, S.; Marold, A.; Steinkellner, G.; Gruber, K.; Schwab, H.; Guebitz, G.M. Surface Engineering of a Cutinase from Thermobifida Cellulosilytica for Improved Polyester Hydrolysis. Biotechnol. Bioeng. 2013, 110, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Ribitsch, D.; Yebra, A.O.; Zitzenbacher, S.; Wu, J.; Nowitsch, S.; Steinkellner, G.; Greimel, K.; Doliska, A.; Oberdorfer, G.; Gruber, C.C.; et al. Fusion of Binding Domains to Thermobifida Cellulosilytica Cutinase to Tune Sorption Characteristics and Enhancing PET Hydrolysis. Biomacromolecules 2013, 14, 1769–1776. [Google Scholar] [CrossRef]
- Ribitsch, D.; Herrero Acero, E.; Przylucka, A.; Zitzenbacher, S.; Marold, A.; Gamerith, C.; Tscheließnig, R.; Jungbauer, A.; Rennhofer, H.; Lichtenegger, H.; et al. Enhanced Cutinase-Catalyzed Hydrolysis of Polyethylene Terephthalate by Covalent Fusion to Hydrophobins. Appl. Environ. Microbiol. 2015, 81, 3586–3592. [Google Scholar] [CrossRef] [Green Version]
- Pellis, A.; Gamerith, C.; Ghazaryan, G.; Ortner, A.; Herrero Acero, E.; Guebitz, G.M. Ultrasound-Enhanced Enzymatic Hydrolysis of Poly(Ethylene Terephthalate). Bioresour. Technol. 2016, 218, 1298–1302. [Google Scholar] [CrossRef] [PubMed]
- Gott, K.P.; Burgess, L.W.; Balmas, V.; Duff, J. Mycogeography of Fusarium: Fusarium Species in Soils from Palm Valley, Central Australia. Australas. Plant Pathol. 1994, 23, 112–117. [Google Scholar] [CrossRef]
- Dimarogona, M.; Nikolaivits, E.; Kanelli, M.; Christakopoulos, P.; Sandgren, M.; Topakas, E. Structural and Functional Studies of a Fusarium Oxysporum Cutinase with Polyethylene Terephthalate Modification Potential. Biochim. Biophys. Acta 2015, 1850, 2308–2317. [Google Scholar] [CrossRef]
- Nikolaivits, E.; Kokkinou, A.; Karpusas, M.; Topakas, E. Microbial Host Selection and Periplasmic Folding in Escherichia Coli Affect the Biochemical Characteristics of a Cutinase from Fusarium Oxysporum. Protein Expr. Purif. 2016, 127, 1–7. [Google Scholar] [CrossRef]
- Kanelli, M.; Vasilakos, S.; Nikolaivits, E.; Ladas, S.; Christakopoulos, P.; Topakas, E. Surface Modification of Poly(Ethylene Terephthalate) (PET) Fibers by a Cutinase from Fusarium Oxysporum. Process Biochem. 2015, 50, 1885–1892. [Google Scholar] [CrossRef]
- Maheshwari, R.; Bharadwaj, G.; Bhat, M.K. Thermophilic Fungi: Their Physiology and Enzymes. Microbiol. Mol. Biol. Rev. 2000, 64, 461–488. [Google Scholar] [CrossRef] [Green Version]
- Ronkvist, Å.M.; Xie, W.; Lu, W.; Gross, R.A. Cutinase-Catalyzed Hydrolysis of Poly(Ethylene Terephthalate). Macromolecules 2009, 42, 5128–5138. [Google Scholar] [CrossRef]
- de Castro, A.M.; Carniel, A.; Nicomedes Junior, J.; da Conceição Gomes, A.; Valoni, É. Screening of Commercial Enzymes for Poly(Ethylene Terephthalate) (PET) Hydrolysis and Synergy Studies on Different Substrate Sources. J. Ind. Microbiol. Biotechnol. 2017, 44, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Kold, D.; Dauter, Z.; Laustsen, A.K.; Brzozowski, A.M.; Turkenburg, J.P.; Nielsen, A.D.; Koldsø, H.; Petersen, E.; Schiøtt, B.; De Maria, L.; et al. Thermodynamic and Structural Investigation of the Specific SDS Binding of Humicola Insolens Cutinase. Protein Sci. 2014, 23, 1023–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazenwadel, C.; Eiben, S.; Maurer, S.; Beuttler, H.; Wetzl, D.; Hauer, B.; Koschorreck, K. Thiol-Functionalization of Acrylic Ester Monomers Catalyzed by Immobilized Humicola Insolens Cutinase. Enzym. Microb. Technol. 2012, 51, 9–15. [Google Scholar] [CrossRef]
- UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef] [PubMed]
- Kobayashi, S.; Makino, A. Enzymatic Polymer Synthesis: An Opportunity for Green Polymer Chemistry. Chem. Rev. 2009, 109, 5288–5353. [Google Scholar] [CrossRef]
- Fabbri, F.; Bertolini, F.A.; Guebitz, G.M.; Pellis, A. Biocatalyzed Synthesis of Flavor Esters and Polyesters: A Design of Experiments (DoE) Approach. Int. J. Mol. Sci. 2021, 22, 8493. [Google Scholar] [CrossRef] [PubMed]
- Feder, D.; Gross, R.A. Exploring Chain Length Selectivity in HIC-Catalyzed Polycondensation Reactions. Biomacromolecules 2010, 11, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Carniel, A.; Valoni, É.; Nicomedes, J.; da Conceição Gomes, A.; de Castro, A.M. Lipase from Candida Antarctica (CALB) and Cutinase from Humicola Insolens Act Synergistically for PET Hydrolysis to Terephthalic Acid. Process Biochem. 2017, 59, 84–90. [Google Scholar] [CrossRef]
- Greimel, K.; Marold, A.; Sohar, C.; Feola, R.; Temel, A.; Schoenbacher, T.; Herrero Acero, E.; Guebitz, G.M. Enzymatic Hydrolysis of Polyester Based Coatings. React. Funct. Polym. 2013, 73, 1335–1339. [Google Scholar] [CrossRef]
- Quartinello, F.; Vajnhandl, S.; Volmajer Valh, J.; Farmer, T.J.; Vončina, B.; Lobnik, A.; Herrero Acero, E.; Pellis, A.; Guebitz, G.M. Synergistic Chemo-Enzymatic Hydrolysis of Poly(Ethylene Terephthalate) from Textile Waste. Microb. Biotechnol. 2017, 10, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Carniel, A.; da Conceição Gomes, A.; Coelho, M.A.Z.; de Castro, A.M. Process Strategies to Improve Biocatalytic Depolymerization of Post-Consumer PET Packages in Bioreactors, and Investigation on Consumables Cost Reduction. Bioprocess Biosyst. Eng. 2021, 44, 507–516. [Google Scholar] [CrossRef] [PubMed]
- de Queiros Eugenio, E.; Campisano, I.S.P.; de Castro, A.M.; Coelho, M.A.Z.; Langone, M.A.P. Experimental and Mathematical Modeling Approaches for Biocatalytic Post-Consumer Poly(Ethylene Terephthalate) Hydrolysis. J. Biotechnol. 2021, 341, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Quartinello, F.; Kremser, K.; Vecchiato, S.; Schoen, H.; Vielnascher, R.; Ploszczanski, L.; Pellis, A.; Guebitz, G.M. Increased Flame Retardancy of Enzymatic Functionalized PET and Nylon Fabrics via DNA Immobilization. Front. Chem. 2019, 7, 685. [Google Scholar] [CrossRef] [PubMed]
- de Castro, A.M.; Carniel, A.; Stahelin, D.; Chinelatto Junior, L.S.; de Angeli Honorato, H.; de Menezes, S.M.C. High-Fold Improvement of Assorted Post-Consumer Poly(Ethylene Terephthalate) (PET) Packages Hydrolysis Using Humicola Insolens Cutinase as a Single Biocatalyst. Process Biochem. 2019, 81, 85–91. [Google Scholar] [CrossRef]
- Su, L.; Hong, R.; Kong, D.; Wu, J. Enhanced Activity towards Polyacrylates and Poly(Vinyl Acetate) by Site-Directed Mutagenesis of Humicola Insolens Cutinase. Int. J. Biol. Macromol. 2020, 162, 1752–1759. [Google Scholar] [CrossRef] [PubMed]
- Shirke, A.N.; Butterfoss, G.L.; Saikia, R.; Basu, A.; de Maria, L.; Svendsen, A.; Gross, R.A. Engineered Humicola Insolens Cutinase for Efficient Cellulose Acetate Deacetylation. Biotechnol. J. 2017, 12, 1700188. [Google Scholar] [CrossRef]
- Martinez, C.; De Geus, P.; Lauwereys, M.; Matthyssens, G.; Cambillau, C. Fusarium Solani Cutinase Is a Lipolytic Enzyme with a Catalytic Serine Accessible to Solvent. Nature 1992, 356, 615–618. [Google Scholar] [CrossRef]
- Summerell, B.A.; Laurence, M.H.; Liew, E.C.Y.; Leslie, J.F. Biogeography and Phylogeography of Fusarium: A Review. Fungal Divers. 2010, 44, 3–13. [Google Scholar] [CrossRef]
- LIN, T.-S.; KOLATTUKUDY, P.E. Structural Studies on Cutinase, a Glycoprotein Containing Novel Amino Acids and Glucuronic Acid Amide at the N Terminus. Eur. J. Biochem. 1980, 106, 341–351. [Google Scholar] [CrossRef]
- Martinez, C.; Nicolas, A.; van Tilbeurgh, H.; Egloff, M.P.; Cambillau, C.; Cudrey, C.; Verger, R. Cutinase, a Lipolytic Enzyme with a Preformed Oxyanion Hole. Biochemistry 1994, 33, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.; Egmond, M.; Verrips, C.T.; De Vlieg, J.; Longhi, S.; Cambillau, C.; Martinez, C. Contribution of Cutinase Serine 42 Side Chain to the Stabilization of the Oxyanion Transition State. Biochemistry 1996, 35, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Longhi, S.; Nicolas, A.; Creveld, L.; Egmond, M.; Verrips, C.T.; De Vlieg, J.; Martinez, C.; Cambillau, C. Dynamics of Fusarium Solani Cutinase Investigated through Structural Comparison among Different Crystal Forms of Its Variants. Proteins Struct. Funct. Genet. 1996, 26, 442–458. [Google Scholar] [CrossRef]
- Longhi, S.; Mannesse, M.; Verheij, H.M.; De Haas, G.H.; Egmond, M.; Knoops-Mouthuy, E.; Cambillau, C. Crystal Structure of Cutinase Covalently Inhibited by a Triglyceride Analogue. Protein Sci. 1997, 6, 275–286. [Google Scholar] [CrossRef]
- Longhi, S.; Czjzek, M.; Lamzin, V.; Nicolas, A.; Cambillau, C. Atomic Resolution (1.0 Å) Crystal Structure of Fusarium Solani Cutinase: Stereochemical Analysis11Edited by R. Huber. J. Mol. Biol. 1997, 268, 779–799. [Google Scholar] [CrossRef] [PubMed]
- Rutten, L.; Wieczorek, B.; Mannie, J.-P.B.A.; Kruithof, C.A.; Dijkstra, H.P.; Egmond, M.R.; Lutz, M.; Klein Gebbink, R.J.M.; Gros, P.; van Koten, G. Solid-State Structural Characterization of Cutinase–ECE-Pincer–Metal Hybrids. Chem.-A Eur. J. 2009, 15, 4270–4280. [Google Scholar] [CrossRef] [PubMed]
- Heumann, S.; Eberl, A.; Pobeheim, H.; Liebminger, S.; Fischer-Colbrie, G.; Almansa, E.; Cavaco-Paulo, A.; Gübitz, G.M. New Model Substrates for Enzymes Hydrolysing Polyethyleneterephthalate and Polyamide Fibres. J. Biochem. Biophys. Methods 2006, 69, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araújo, R.; Silva, C.; O’Neill, A.; Micaelo, N.; Guebitz, G.; Soares, C.M.; Casal, M.; Cavaco-Paulo, A. Tailoring Cutinase Activity towards Polyethylene Terephthalate and Polyamide 6,6 Fibers. J. Biotechnol. 2007, 128, 849–857. [Google Scholar] [CrossRef] [Green Version]
- Eberl, A.; Heumann, S.; Brückner, T.; Araujo, R.; Cavaco-Paulo, A.; Kaufmann, F.; Kroutil, W.; Guebitz, G.M. Enzymatic Surface Hydrolysis of Poly(Ethylene Terephthalate) and Bis(Benzoyloxyethyl) Terephthalate by Lipase and Cutinase in the Presence of Surface Active Molecules. J. Biotechnol. 2009, 143, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Sugiyama, J.; Iizuka, H. A Taxonomic Study of Antarctic Yeasts. Mycologia 1969, 61, 748–774. [Google Scholar] [CrossRef] [PubMed]
- Heldt-Hansen, H.P.; Ishii, M.; Patkar, S.A.; Hansen, T.T.; Eigtved, P. A New Immobilized Positional Nonspecific Lipase for Fat Modification and Ester Synthesis. In Biocatalysis in Agricultural Biotechnology; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1989; Volume 389, pp. 11–158. [Google Scholar]
- Uppenberg, J.; Ohmer, N.; Norin, M.; Hult, K.; Kleywegt, G.J.; Patkar, S.; Waagen, V.; Anthonsen, T.; Jones, T.A. Crystallographic and Molecular-Modeling Studies of Lipase B from Candida Antarctica Reveal a Stereospecificity Pocket for Secondary Alcohols. Biochemistry 1995, 34, 16838–16851. [Google Scholar] [CrossRef] [PubMed]
- Strzelczyk, P.; Bujacz, G.D.; Kiełbasiński, P.; Błaszczyk, J. Crystal and Molecular Structure of Hexagonal Form of Lipase B from Candida Antarctica. Acta Biochim. Pol. 2016, 63, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.M.; Larsson, K.M.; Kirk, O. One Biocatalyst–Many Applications: The Use of Candida Antarctica B-Lipase in Organic Synthesis. Biocatal. Biotransformation 1998, 16, 181–204. [Google Scholar] [CrossRef]
- Stauch, B.; Fisher, S.J.; Cianci, M. Open and Closed States of Candida Antarctica Lipase B: Protonation and the Mechanism of Interfacial Activation. J. Lipid Res. 2015, 56, 2348–2358. [Google Scholar] [CrossRef] [Green Version]
- The Sequence, Crystal Structure Determination and Refinement of Two Crystal Forms of Lipase B from Candida Antarctica. Int. J. Biol. Macromol. 1994, 140, 761–770.
- Park, A.; Kim, S.; Park, J.; Joe, S.; Min, B.; Oh, J.; Song, J.; Park, S.Y.; Park, S.; Lee, H. Structural and Experimental Evidence for the Enantiomeric Recognition toward a Bulky Sec-Alcohol by Candida Antarctica Lipase B. ACS Catal. 2016, 6, 7458–7465. [Google Scholar] [CrossRef]
- Qian, Z.; Horton, J.R.; Cheng, X.; Lutz, S. Structural Redesign of Lipase B from Candida Antarctica by Circular Permutation and Incremental Truncation. J. Mol. Biol. 2009, 393, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; An, J.; Yang, G.; Wu, G.; Zhang, Y.; Cui, L.; Feng, Y. Enhanced Enzyme Kinetic Stability by Increasing Rigidity within the Active Site. J. Biol. Chem. 2014, 289, 7994–8006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cen, Y.; Singh, W.; Arkin, M.; Moody, T.S.; Huang, M.; Zhou, J.; Wu, Q.; Reetz, M.T. Artificial Cysteine-Lipases with High Activity and Altered Catalytic Mechanism Created by Laboratory Evolution. Nat. Commun. 2019, 10, 3198. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cen, Y.; Singh, W.; Fan, J.; Wu, L.; Lin, X.; Zhou, J.; Huang, M.; Reetz, M.T.; Wu, Q. Stereodivergent Protein Engineering of a Lipase to Access All Possible Stereoisomers of Chiral Esters with Two Stereocenters. J. Am. Chem. Soc. 2019, 141, 7934–7945. [Google Scholar] [CrossRef]
- Silvestrini, L.; Cianci, M. Principles of Lipid–Enzyme Interactions in the Limbus Region of the Catalytic Site of Candida Antarctica Lipase B. Int. J. Biol. Macromol. 2020, 158, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Korpecka, J.; Heumann, S.; Billig, S.; Zimmermann, W.; Zinn, M.; Ihssen, J.; Cavaco-Paulo, A.; Guebitz, G.M. Hydrolysis of Cutin by PET-Hydrolases. Macromol. Symp. 2010, 296, 342–346. [Google Scholar] [CrossRef]
- Vertommen, M.; Nierstrasz, V.A.; van der Veer, M.; Warmoeskerken, M.M. Enzymatic Surface Modification of Poly(Ethylene Terephthalate). J. Biotechnol. 2005, 120, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Yoo, Y.J. Activity Enhancement of Candida Antarctica Lipase B by Flexibility Modulation in Helix Region Surrounding the Active Site. Appl. Biochem. Biotechnol. 2013, 170, 925–933. [Google Scholar] [CrossRef]
- Park, H.J.; Park, K.; Kim, Y.H.; Yoo, Y.J. Computational Approach for Designing Thermostable Candida Antarctica Lipase B by Molecular Dynamics Simulation. J. Biotechnol. 2014, 192, 66–70. [Google Scholar] [CrossRef]
- Le, Q.A.T.; Joo, J.C.; Yoo, Y.J.; Kim, Y.H. Development of Thermostable Candida Antarctica Lipase B through Novel in Silico Design of Disulfide Bridge. Biotechnol. Bioeng. 2012, 109, 867–876. [Google Scholar] [CrossRef]
- Suen, W.; Zhang, N.; Xiao, L.; Madison, V.; Zaks, A. Improved Activity and Thermostability of Candida Antarctica Lipase B by DNA Family Shuffling. Protein Eng. Des. Sel. 2004, 17, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Suen, W.; Windsor, W.; Xiao, L.; Madison, V.; Zaks, A. Improving Tolerance of Candida Antarctica Lipase B towards Irreversible Thermal Inactivation through Directed Evolution. Protein Eng. Des. Sel. 2003, 16, 599–605. [Google Scholar] [CrossRef] [Green Version]
- Rotticci, D.; Rotticci-Mulder, J.C.; Denman, S.; Norin, T.; Hult, K. Improved Enantioselectivity of a Lipase by Rational Protein Engineering. ChemBioChem 2001, 2, 766–770. [Google Scholar] [CrossRef]
- Magnusson, A.O.; Rotticci-Mulder, J.C.; Santagostino, A.; Hult, K. Creating Space for Large Secondary Alcohols by Rational Redesign of Candida Antarctica Lipase B. ChemBioChem 2005, 6, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Idris, A.; Bukhari, A. Immobilized Candida Antarctica Lipase B: Hydration, Stripping off and Application in Ring Opening Polyester Synthesis. Biotechnol. Adv. 2012, 30, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Maniar, D.; Jiang, Y.; Woortman, A.J.J.; van Dijken, J.; Loos, K. Furan-Based Copolyesters from Renewable Resources: Enzymatic Synthesis and Properties. ChemSusChem 2019, 12, 990–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thumarat, U.; Kawabata, T.; Nakajima, M.; Nakajima, H.; Sugiyama, A.; Yazaki, K.; Tada, T.; Waku, T.; Tanaka, N.; Kawai, F. Comparison of Genetic Structures and Biochemical Properties of Tandem Cutinase-Type Polyesterases from Thermobifida Alba AHK119. J. Biosci. Bioeng. 2015, 120, 491–497. [Google Scholar] [CrossRef]
- Kitadokoro, K.; Matsui, S.; Osokoshi, R.; Nakata, K.; Kamitani, S. Expression, Purification and Crystallization of Thermostable Mutant of Cutinase Est1 from Thermobifida alba. Adv. Biosci. Biotechnol. 2018, 9, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Tsiklinsky, P. Sur Les Mucedinees Thermophiles. Ann. Inst. Pasteur 1899, 13, 501–505. [Google Scholar]
- Arima, K.; Liu, W.-H.; Beppu, T. Studies on the Lipase of Thermophilic Fungus Humicola Lanuginosa. Agric. Biol. Chem. 1972, 36, 893–895. [Google Scholar] [CrossRef]
- Chew, Y.H.; Chua, L.S.; Cheng, K.K.; Sarmidi, M.R.; Aziz, R.A.; Lee, C.T. Kinetic Study on the Hydrolysis of Palm Olein Using Immobilized Lipase. Biochem. Eng. J. 2008, 39, 516–520. [Google Scholar] [CrossRef] [Green Version]
- Tangkam, K.; Weber, N.; Wiege, B. Solvent-Free Lipase-Catalyzed Preparation of Diglycerides from Co-Products of Vegetable Oil Refining. Grasas Y Aceites 2008, 59, 245–253. [Google Scholar]
- Sivalingam, G.; Chattopadhyay, S.; Madras, G. Solvent Effects on the Lipase Catalyzed Biodegradation of Poly (??-Caprolactone) in Solution. Polym. Degrad. Stab. 2003, 79, 413–418. [Google Scholar] [CrossRef]
- Reddy, J.R.C.; Vijeeta, T.; Karuna, M.S.L.; Rao, B.V.S.K.; Prasad, R.B.N. Lipase-Catalyzed Preparation of Palmitic and Stearic Acid-Rich Phosphatidylcholine. J. Am. Oil Chem. Soc. 2005, 82, 727–730. [Google Scholar] [CrossRef]
- Brueckner, T.; Eberl, A.; Heumann, S.; Rabe, M.; Guebitz, G.M. Enzymatic and Chemical Hydrolysis of Poly(Ethylene Terephthalate) Fabrics. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 6435–6443. [Google Scholar] [CrossRef]
- Lawson, D.M.; Brzozowski, A.M.; Rety, S.; Verma, C.; Dodson, G.G. Probing the Nature of Substrate Binding in Humicola Lanuginosa Lipase through X-Ray Crystallography and Intuitive Modelling. Protein Eng. Des. Sel. 1994, 7, 543–550. [Google Scholar] [CrossRef]
- Derewenda, U.; Swenson, L.; Wei, Y.; Green, R.; Kobos, P.M.; Joerger, R.; Haas, M.J.; Derewenda, Z.S. Conformational Lability of Lipases Observed in the Absence of an Oil-Water Interface: Crystallographic Studies of Enzymes from the Fungi Humicola Lanuginosa and Rhizopus Delemar. J. Lipid Res. 1994, 35, 524–534. [Google Scholar] [CrossRef]
- Holmquist, M.; Clausen, I.G.; Patkar, S.; Svendsen, A.; Hult, K. Probing a Functional Role of Glu87 and Trp89 in the Lid OfHumicola Lanuginosa Lipase through Transesterification Reactions in Organic Solvent. J. Protein Chem. 1995, 14, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Martinelle, M.; Holmquist, M.; Clausen, I.G.; Patkar, S.; Svendsen, A.; Hult, K. The Role of Glu87 and Trp89 in the Lid of Humicola Lanuginosa Lipase. Protein Eng. Des. Sel. 1996, 9, 519–524. [Google Scholar] [CrossRef] [Green Version]
- Holmquist, M.; Martinelle, M.; Clausen, I.G.; Patkar, S.; Svendsen, A.; Hult, K. Trp89 in the Lid OfHumicola Lanuginosa Lipase Is Important for Efficient Hydrolysis of Tributyrin. Lipids 1994, 29, 599–603. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Savage, H.; Verma, C.S.; Turkenburg, J.P.; Lawson, D.M.; Svendsen, A.; Patkar, S. Structural Origins of the Interfacial Activation in Thermomyces (Humicola) Lanuginosa Lipase. Biochemistry 2000, 39, 15071–15082. [Google Scholar] [CrossRef] [PubMed]
- Yapoudjian, S.; Ivanova, M.G.; Brzozowski, A.M.; Patkar, S.A.; Vind, J.; Svendsen, A.; Verger, R. Binding of Thermomyces (Humicola) Lanuginosa Lipase to the Mixed Micelles of Cis-Parinaric Acid/NaTDC. Eur. J. Biochem. 2002, 269, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Mukherjee, J.; Sinha, M.; Kaur, P.; Sharma, S.; Gupta, M.N.; Singh, T.P. Enhancement of Stability of a Lipase by Subjecting to Three Phase Partitioning (TPP): Structures of Native and TPP-Treated Lipase from Thermomyces Lanuginosa. Sustain. Chem. Process. 2015, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Skjold-Jørgensen, J.; Vind, J.; Moroz, O.V.; Blagova, E.; Bhatia, V.K.; Svendsen, A.; Wilson, K.S.; Bjerrum, M.J. Controlled Lid-Opening in Thermomyces Lanuginosus Lipase- An Engineered Switch for Studying Lipase Function. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 20–27. [Google Scholar] [CrossRef]
- de Wijn, R.; Hennig, O.; Roche, J.; Engilberge, S.; Rollet, K.; Fernandez-Millan, P.; Brillet, K.; Betat, H.; Mörl, M.; Roussel, A.; et al. A Simple and Versatile Microfluidic Device for Efficient Biomacromolecule Crystallization and Structural Analysis by Serial Crystallography. IUCrJ 2019, 6, 454–464. [Google Scholar] [CrossRef]
- McPherson, A.; Larson, S.B.; Kalasky, A. The Crystal Structures of Thermomyces (Humicola) Lanuginosa Lipase in Complex with Enzymatic Reactants. Curr. Enzym. Inhib. 2020, 16, 199–213. [Google Scholar] [CrossRef]
- Omar, I.C.; Nishio, N.; Nagai, S. Production of a Thermostable Lipase by Humicola Lanuginosa Grown on Sorbitol-Corn Steep Liquor Medium. Agric. Biol. Chem. 1987, 51, 2145–2151. [Google Scholar] [CrossRef]
- Peters, G.H.; Svendsen, A.; Langberg, H.; Vind, J.; Patkar, S.A.; Toxvaerd, S.; Kinnunen, P.K.J. Active Serine Involved in the Stabilization of the Active Site Loop in the Humicola Lanuginosa Lipase. Biochemistry 1998, 37, 12375–12383. [Google Scholar] [CrossRef] [PubMed]
- Oeser, T.; Wei, R.; Baumgarten, T.; Billig, S.; Föllner, C.; Zimmermann, W. High Level Expression of a Hydrophobic Poly(Ethylene Terephthalate)-Hydrolyzing Carboxylesterase from Thermobifida Fusca KW3 in Escherichia Coli BL21(DE3). J. Biotechnol. 2010, 146, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Billig, S.; Oeser, T.; Birkemeyer, C.; Zimmermann, W. Hydrolysis of Cyclic Poly(Ethylene Terephthalate) Trimers by a Carboxylesterase from Thermobifida Fusca KW3. Appl. Microbiol. Biotechnol. 2010, 87, 1753–1764. [Google Scholar] [CrossRef]
- Barth, M.; Honak, A.; Oeser, T.; Wei, R.; Belisário-Ferrari, M.R.; Then, J.; Schmidt, J.; Zimmermann, W. A Dual Enzyme System Composed of a Polyester Hydrolase and a Carboxylesterase Enhances the Biocatalytic Degradation of Polyethylene Terephthalate Films. Biotechnol. J. 2016, 11, 1082–1087. [Google Scholar] [CrossRef]
- Belisário-Ferrari, M.R.; Wei, R.; Schneider, T.; Honak, A.; Zimmermann, W. Fast Turbidimetric Assay for Analyzing the Enzymatic Hydrolysis of Polyethylene Terephthalate Model Substrates. Biotechnol. J. 2019, 14, 1800272. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.; Yin, X.; Liu, T.; Zhang, H.; Chen, G.; Wu, S. Biodegradation of Bis(2-Hydroxyethyl) Terephthalate by a Newly Isolated Enterobacter Sp. HY1 and Characterization of Its Esterase Properties. J. Basic Microbiol. 2020, 60, 699–711. [Google Scholar] [CrossRef]
- Xi, X.; Ni, K.; Hao, H.; Shang, Y.; Zhao, B.; Qian, Z. Secretory Expression in Bacillus Subtilis and Biochemical Characterization of a Highly Thermostable Polyethylene Terephthalate Hydrolase from Bacterium HR29. Enzym. Microb. Technol. 2021, 143, 109715. [Google Scholar] [CrossRef]
- Kato, S.; Sakai, S.; Hirai, M.; Tasumi, E.; Nishizawa, M.; Suzuki, K.; Takai, K. Long-Term Cultivation and Metagenomics Reveal Ecophysiology of Previously Uncultivated Thermophiles Involved in Biogeochemical Nitrogen Cycle. Microbes Environ. 2018, 33, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Eggert, T.; Pencreac‘h, G.; Douchet, I.; Verger, R.; Jaeger, K.-E. A Novel Extracellular Esterase from Bacillus Subtilis and Its Conversion to a Monoacylglycerol Hydrolase. Eur. J. Biochem. 2000, 267, 6459–6469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabloune, R.; Khalil, M.; Ben Moussa, I.E.; Simao-Beaunoir, A.-M.; Lerat, S.; Brzezinski, R.; Beaulieu, C. Enzymatic Degradation of P-Nitrophenyl Esters, Polyethylene Terephthalate, Cutin, and Suberin by Sub1, a Suberinase Encoded by the Plant Pathogen Streptomyces Scabies. Microbes Environ. 2020, 35, ME19086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komeil, D.; Simao-Beaunoir, A.-M.; Beaulieu, C. Detection of Potential Suberinase-Encoding Genes in Streptomyces Scabiei Strains and Other Actinobacteria. Can. J. Microbiol. 2013, 59, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Lambert, C.; Léonard, N.; De Bolle, X.; Depiereux, E. ESyPred3D: Prediction of Proteins 3D Structures. Bioinformatics 2002, 18, 1250–1256. [Google Scholar] [CrossRef]
- Yoon, M.-Y.; Kellis, J.; Poulose, A.J. Enzymatic Modification of Polyester. AATCC Rev. 2002, 2, 33–36. [Google Scholar]
- Danso, D.; Schmeisser, C.; Chow, J.; Zimmermann, W.; Wei, R.; Leggewie, C.; Li, X.; Hazen, T.; Streit, W.R. New Insights into the Function and Global Distribution of Polyethylene Terephthalate (PET)-Degrading Bacteria and Enzymes in Marine and Terrestrial Metagenomes. Appl. Environ. Microbiol. 2018, 84, e02773-17. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.; Oeser, T.; Then, J.; Kühn, N.; Barth, M.; Schmidt, J.; Zimmermann, W. Functional Characterization and Structural Modeling of Synthetic Polyester-Degrading Hydrolases from Thermomonospora Curvata. AMB Express 2014, 4, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henssen, A. Beiträge Zur Morphologie Und Systematik Der Thermophilen Actinomyceten. Arch. Mikrobiol. 1957, 26, 373–414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, Y.; Ruan, J. Reclassification of Thermomonospora and Microtetraspora. Int. J. Syst. Bacteriol. 1998, 48 Pt 2, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Chertkov, O.; Sikorski, J.; Nolan, M.; Lapidus, A.; Lucas, S.; Del Rio, T.G.; Tice, H.; Cheng, J.-F.; Goodwin, L.; Pitluck, S.; et al. Complete Genome Sequence of Thermomonospora Curvata Type Strain (B9). Stand. Genom. Sci. 2011, 4, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, S.; Apitius, L.; Jakob, F.; Schwaneberg, U. Targeting Microplastic Particles in the Void of Diluted Suspensions. Environ. Int. 2019, 123, 428–435. [Google Scholar] [CrossRef]
- Schmidt, J.; Wei, R.; Oeser, T.; Dedavid e Silva, L.A.; Breite, D.; Schulze, A.; Zimmermann, W. Degradation of Polyester Polyurethane by Bacterial Polyester Hydrolases. Polymers 2017, 9, 65. [Google Scholar] [CrossRef] [Green Version]
- Liebminger, S.; Eberl, A.; Sousa, F.; Heumann, S.; Fischer-Colbrie, G.; Cavaco-Paulo, A.; Guebitz, G.M. Hydrolysis of PET and Bis-(Benzoyloxyethyl) Terephthalate with a New Polyesterase from Penicillium Citrinum. Biocatal. Biotransform. 2007, 25, 171–177. [Google Scholar] [CrossRef]
- da Costa, A.M.; de Oliveira Lopes, V.R.; Vidal, L.; Nicaud, J.M.; de Castro, A.M.; Coelho, M.A.Z. Poly(Ethylene Terephthalate) (PET) Degradation by Yarrowia Lipolytica: Investigations on Cell Growth, Enzyme Production and Monomers Consumption. Process Biochem. 2020, 95, 81–90. [Google Scholar] [CrossRef]
- Roberts, C.; Edwards, S.; Vague, M.; León-Zayas, R.; Scheffer, H.; Chan, G.; Swartz, N.A.; Mellies, J.L. Environmental Consortium Containing Pseudomonas and Bacillus Species Synergistically Degrades Polyethylene Terephthalate Plastic. MSphere 2020, 5, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Shirke, A.N.; Basore, D.; Butterfoss, G.L.; Bonneau, R.; Bystroff, C.; Gross, R.A. Toward Rational Thermostabilization of Aspergillus Oryzae Cutinase: Insights into Catalytic and Structural Stability. Proteins Struct. Funct. Bioinform. 2016, 84, 60–72. [Google Scholar] [CrossRef] [Green Version]
- Zhiqiang, L.; Gosser, Y.; Baker, P.J.; Ravee, Y.; Ziying, L.; Alemu, G.; Huiguang, L.; Butterfoss, G.L.; Kong, X.P.; Gross, R.; et al. Structural and Functional Studies of Aspergillus Oryzae Cutinase: Enhanced Thermostability and Hydrolytic Activity of Synthetic Ester and Polyester Degradation. J. Am. Chem. Soc. 2009, 131, 15711–15716. [Google Scholar]
- Almeida, E.L.; Rincón, A.F.C.; Jackson, S.A.; Dobson, A.D.W. In Silico Screening and Heterologous Expression of a Polyethylene Terephthalate Hydrolase (PETase)-Like Enzyme (SM14est) With Polycaprolactone (PCL)-Degrading Activity, From the Marine Sponge-Derived Strain Streptomyces Sp. SM14. Front. Microbiol. 2019, 10, 2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-C.; Chen, G.-H.; Chen, Y.-F.; Chen, W.-L.; Yang, C.-H. Heterologous Expression of Thermostable Acetylxylan Esterase Gene from Thermobifida Fusca and Its Synergistic Action with Xylanase for the Production of Xylooligosaccharides. Biochem. Biophys. Res. Commun. 2010, 400, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Thumarat, U.; Zhang, X.; Tang, M.; Kawai, F. Diversity of Polyester-Degrading Bacteria in Compost and Molecular Analysis of a Thermoactive Esterase from Thermobifida Alba AHK119. Appl. Microbiol. Biotechnol. 2010, 87, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Kitadokoro, K.; Thumarat, U.; Nakamura, R.; Nishimura, K.; Karatani, H.; Suzuki, H.; Kawai, F. Crystal Structure of Cutinase Est119 from Thermobifida Alba AHK119 That Can Degrade Modified Polyethylene Terephthalate at 1.76Å Resolution. Polym. Degrad. Stab. 2012, 97, 771–775. [Google Scholar] [CrossRef]
- Kitadokoro, K.; Kakara, M.; Matsui, S.; Osokoshi, R.; Thumarat, U.; Kawai, F.; Kamitani, S. Structural Insights into the Unique Polylactate-Degrading Mechanism of Thermobifida Alba Cutinase. FEBS J. 2019, 286, 2087–2098. [Google Scholar] [CrossRef]
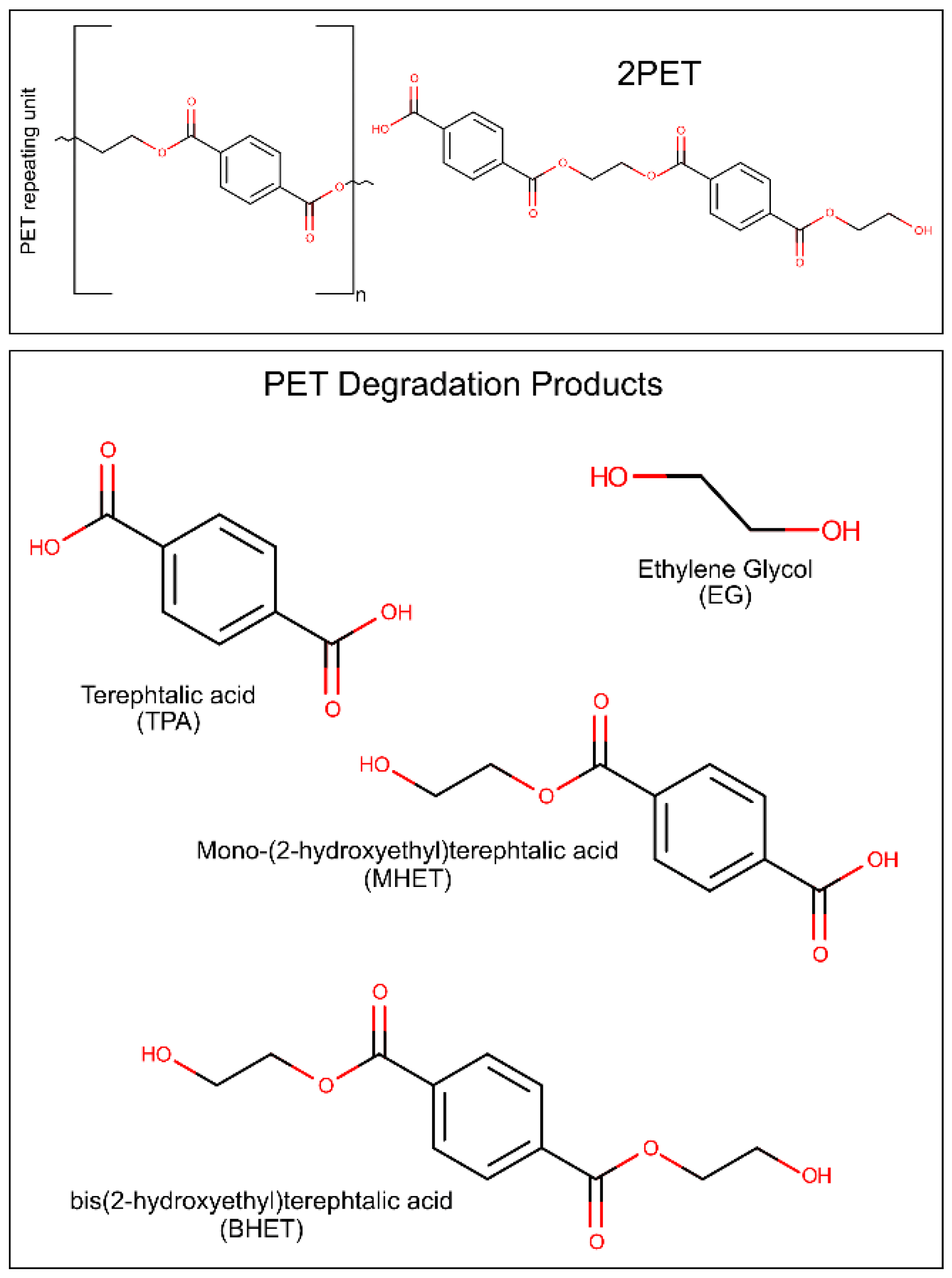













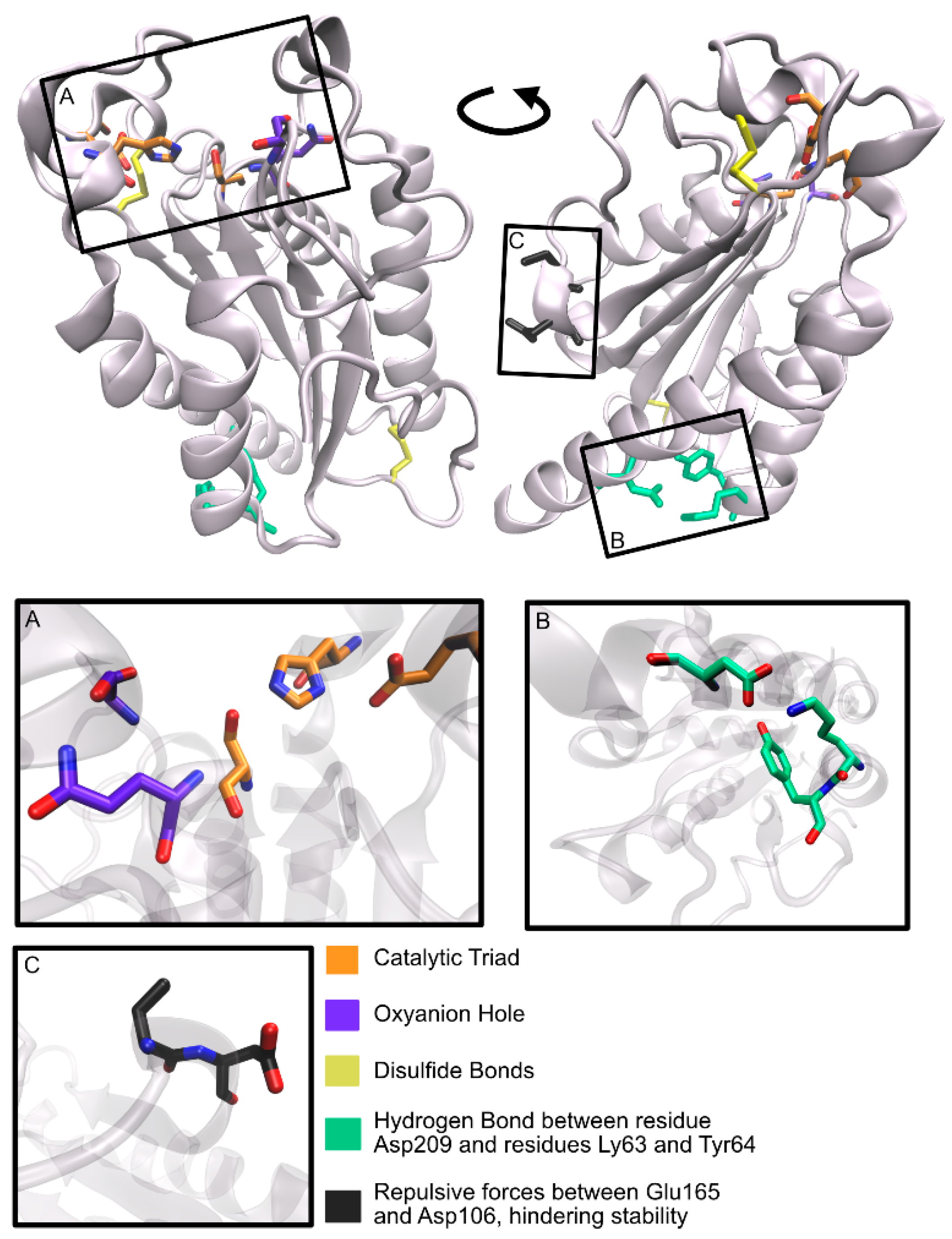
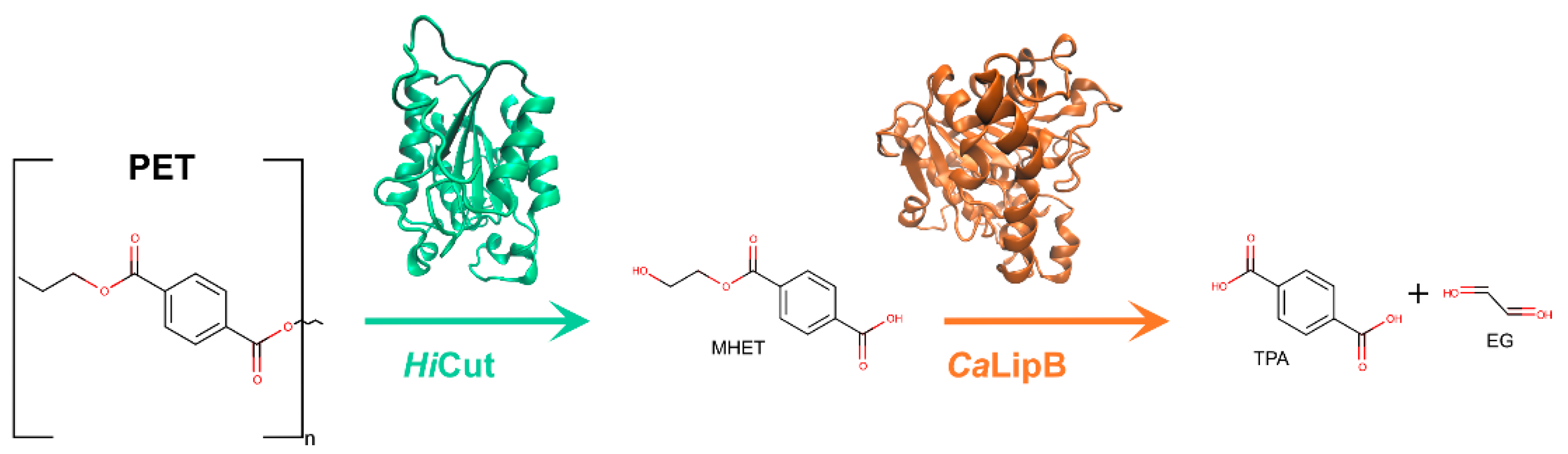

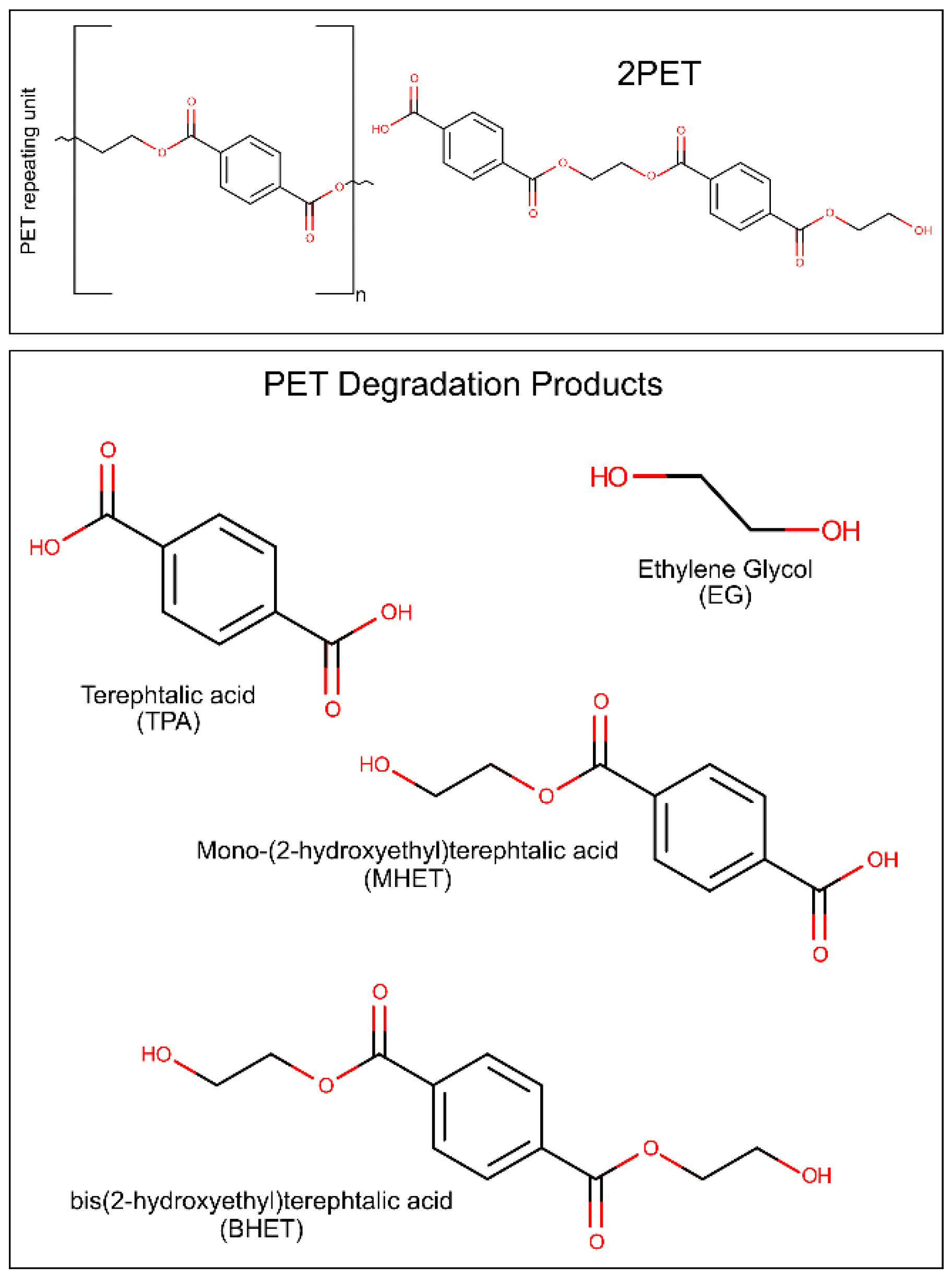{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Code | Resolution (Å) | Ligand | Mutations | Year of Deposition | Ref. |
|---|---|---|---|---|---|
| 5XG0 | 1.58 | Free | - | 2017 | [76] |
| 5XFY | 1.40 | Free | S131A | 2017 | [76] |
| 5XFZ | 1.55 | Free | R103G/S131A | 2017 | [76] |
| 5ZH3 | 1.30 | HEMT | R103G/S131A | 2017 | [76] |
| 5XH2 | 1.20 | pNP | R103G/S131A | 2017 | [76] |
| 5XJH | 1.54 | Free | - | 2017 | [74] |
| 5YNS | 1.36 | Free | R280A | 2017 | [74] |
| 6EQD | 1.70 | Free | - | 2017 | [75] |
| 6EQH | 1.58 | Free | - | 2017 | [75] |
| 6EQG | 1.799 | Free | - | 2017 | [75] |
| 6EQF | 1.70 | Free | - | 2017 | [75] |
| 6EQE | 0.92 | Free | - | 2017 | [75] |
| 6ANE | 2.02 | Free | - | 2017 | [79] |
| 5YFE | 1.39 | Free | - | 2017 | [78] |
| 6ILW | 1.575 | Free | - | 2018 | [34] |
| 6ILX | 1.45 | Free | W159F | 2018 | [34] |
| 6QGC | 2.0 | Free | - | 2019 | [80] |
| 6IJ3 | 1.40 | Free | S121D/D186H | 2019 | [81] |
| 6IJ4 | 1.86 | Free | S121D/D186H | 2019 | [81] |
| 6IJ5 | 1.72 | Free | P181A | 2019 | [81] |
| 6IJ6 | 1.95 | Free | S121E/D186H/R280A | 2019 | [81] |
| 6KY5 | 1.63 | Free | S214H/I168R/W159H/S188Q/R280A/A180I/G165A/Q119Y/L117F/T140D | 2019 | [82] |
| 6KUO | 1.90 | Free | N246D | 2019 | [83] |
| 6KUQ | 1.91 | Free | A248D/R280K | 2019 | [83] |
| 6KUS | 2.00 | Free | S121E/D186H/S242T/N246D | 2019 | [83] |
| PDB Code | Resolution (Å) | Ligand | Mutations | Year of Deposition | Ref |
|---|---|---|---|---|---|
| 6QGA | 2.1 | MHETA | - | 2019 | [80] |
| 6QGB | 2.2 | Benzoic acid | - | 2019 | [80] |
| 6QG9 | 2.05 | Free | - | 2019 | [80] |
| GJTU | 2.1 | Free | - | 2019 | [95] |
| 6JTT | 2.51 | BHET | - | 2019 | [95] |
| 6QZ1 | 1.7 | Benzoic acid | - | 2019 | [89] |
| 6QZ2 | 1.9 | Benzoic acid | - | 2019 | [89] |
| 6QZ3 | 1.6 | Free | - | 2019 | [89] |
| 6QZ4 | 1.8 | Free | - | 2019 | [89] |
| PDB Code | Resolution (Å) | Ligand | Mutation | Year of Deposition | Ref. |
|---|---|---|---|---|---|
| 6SBN | 1.09 | - | - | 2019 | [97] |
| 6SCD | 1.35 | - | Y250S | 2019 | [97] |
| PDB Code | Resolution (Å) | Ligand | Mutations | Year of Deposition | Ref. |
|---|---|---|---|---|---|
| 4EB0 | 1.5 | Free | - | 2012 | [105] |
| 6THS | 1.10 | Free | S165A | 2019 | [106] |
| 6THT | 1.14 | Free | Y127G/S165A/D238C/F243I/S283C | 2019 | [106] |
| PDB Code | Resolution (Å) | Ligand | Mutation | Year of Deposition | Ref. |
|---|---|---|---|---|---|
| 4WFK | 2.35 | Free | S226P Ca2+ bound | 2014 | [132] |
| 4WFI | 1.45 | Free | S226P Ca2+ free | 2014 | [132] |
| 4WFJ | 1.75 | Free | S226P Ca2+ bound | 2014 | [132] |
| 5ZNO | 1.6 | Free | S176A/S226P/R228S | 2018 | [133] |
| 5ZRQ | 1.12 | Free | S176A/S226P/R228S | 2018 | [133] |
| 5ZRR | 1.34 | Monoethyl succinate | S176A/S226P/R228S | 2018 | [133] |
| 5ZRS | 1.4 | Monoethyl adipate | S176A/S226P/R228S | 2018 | [133] |
| 7CEH | 1.09 | Free | S176A/S226P/R228S with C-terminal three residues deletion | 2020 | [135] |
| 7CEF | 1.6 | Free | S226P/R228S with C-terminal three residues deletion | 2020 | [135] |
| 7CTR | 1.20 | Free | S226P/R228S/Q138A/D250C–E296C/Q123H/N202H | 2020 | [136] |
| 7CTS | 1.10 | Free | S176A/S226P/R228S/Q138A/D250C–E296C/Q123H/N202H | 2020 | [136] |
| PDB Code | Enzyme | Resolution (Å) | Ligand | Mutation | Year of Deposition | Ref. |
|---|---|---|---|---|---|---|
| 4CG1 | TfCut2 | 1.4 | Free | - | 2013 | [141] |
| 4CG2 | TfCut2 | 1.44 | PMSF | - | 2013 | [141] |
| 4CG3 | TfCut2 | 4.55 | Free | - | 2013 | [141] |
| 5LUK | TcCut2 | 1.45 | Free | R28N/A30V | 2016 | [142] |
| 5LUJ | TcCut2 | 2.2 | Free | - | 2016 | [142] |
| 5LUL | TcCut2 | 1.9 | Free | R19S/R29N/A30V | 2016 | [142] |
| 5LUI | TcCut1 | 1.5 | Free | - | 2016 | [142] |
| PDB Code | Resolution (Å) | Ligand | Mutation | Year of Deposition | Ref. |
|---|---|---|---|---|---|
| 4OYY | 3 | - | - | 2014 | [159] |
| 4OYL | 2.05 | Mono-ethyl phosphate | - | 2014 | [159] |
| PDB Code | Resolution (Å) | Ligand | Mutations | Year of Deposition | Ref. |
|---|---|---|---|---|---|
| 1CUS | 1.25 | - | - | 1994 | [174] |
| 2CUT | 1.90 | Diethyl Phosphonate | - | 1994 | [177] |
| 1FFA | 1.69 | - | N84A | 1995 | [178] |
| 1FFB | 1.75 | - | N84D | 1995 | [178] |
| 1FFC | 1.75 | - | N84L | 1995 | [178] |
| 1FFD | 1.69 | - | N84W | 1995 | [178] |
| 1FFE | 1.69 | - | S42A | 1995 | [178] |
| 1XZA | 1.80 | - | S129C | 1995 | [179] |
| 1XZB | 1.75 | Mercury Acetate | S129C | 1995 | [179] |
| 1XZC | 1.75 | Para-Sulfurous phenyl mercury | S129C | 1995 | [179] |
| 1XZD | 2.70 | - | S213C | 1995 | [179] |
| 1XZE | 1.75 | - | S92C | 1995 | [179] |
| 1XZF | 1.69 | - | T144C | 1995 | [179] |
| 1XZG | 1.69 | - | T45A | 1995 | [179] |
| 1XZH | 1.69 | - | T80P | 1995 | [179] |
| 1XZI | 1.69 | - | Y119H | 1995 | [179] |
| 1XZJ | 1.69 | - | T38F | 1995 | [179] |
| 1CUA | 1.80 | - | N172K | 1995 | [179] |
| 1CUB | 1.75 | - | N172K/R196D | 1995 | [179] |
| 1CUC | 1.75 | - | N172K/R196D | 1995 | [179] |
| 1CUD | 2.70 | - | N172K/R196D | 1995 | [179] |
| 1CUE | 2.10 | - | Q121L | 1995 | [179] |
| 1CUF | 1.75 | - | R156L | 1995 | [179] |
| 1CUG | 1.75 | - | R17E/N172K | 1995 | [179] |
| 1CUH | 1.75 | - | R196E | 1995 | [179] |
| 1CUI | 2.50 | - | S120A | 1995 | [179] |
| 1CUJ | 1.60 | - | S120C | 1995 | [179] |
| 1CUU | 1.69 | - | A199C | 1995 | [179] |
| 1CUV | 2.01 | - | A85F | 1995 | [179] |
| 1CUW | 2.70 | - | G82A/A85F/V184I/L189F | 1995 | [179] |
| 1CUX | 1.75 | - | L114Y | 1995 | [179] |
| 1CUY | 1.69 | - | L189F | 1995 | [179] |
| 1CUZ | 2.10 | - | L81G/L182G | 1995 | [179] |
| 1XZK | 2.01 | - | 1995 | [179] | |
| 1XZL | 1.69 | N-hexylphosphonate ethyl ester | - | 1995 | [179] |
| 1XZM | 1.75 | N-undecyl o-methyl chloro Phosphonate ester | - | 1995 | [179] |
| 1OXM | 2.30 | triglyceride analogue ([(2R)-2-(butylcarbamoyloxy)-3-butylphosphonoyloxypropyl] N-butylcarbamate) | - | 1996 | [180] |
| 1AGY | 1.15 | - | - | 1997 | [181] |
| 1CEX | 1.00 | - | - | 1997 | [181] |
| 3EF3 | 1.50 | - | N172K | 2008 | [182] |
| 3ESA | 2.00 | - | N172K | 2008 | [182] |
| 3ESB | 2.30 | - | N172K | 2008 | [182] |
| 3ESC | 1.20 | triglyceride analogue (ethyl 4-nitrophenyl P-[3-(4-(bromopallado)-1,3-bis[(methylthio)methyl]-phenyl)propyl]phosphonate) | N172K | 2008 | [182] |
| 3ESD | 1.22 | triglyceride analogue (ethyl 4-nitrophenyl P-[3-(4-(bromopallado)-1,3-bis[(methylthio)methyl]-phenyl)propyl]phosphonate) | N172K | 2008 | [182] |
| 3QPA | 0.85 | - | - | 2011 | N/A |
| 3QPC | 0.98 | - | - | 2011 | N/A |
| PDB Code | Resolution (Å) | Ligand | Mutations | Year of Deposition | Ref. |
|---|---|---|---|---|---|
| 1TCA | 1.55 | - | - | 1994 | [192] |
| 1TCB | 2.10 | - | - | 1994 | [192] |
| 1TCC | 2.5 | - | - | 1994 | [192] |
| 1LBS | 2.60 | Phosphonate inhibitor | - | 1995 | [188] |
| 1LBT | 2.50 | Tween 80 | - | 1995 | [188] |
| 3ICV | 1.49 | - | - | 2009 | [194] |
| 3ICW | 1.69 | Inhibitor (methyl hydrogen (R)-hexylphosphonate) | - | 2009 | [194] |
| 3W9B | 2.90 | - | - | 2013 | N/A |
| 4K5Q | 1.49 | - | D223G/L278M | 2013 | [195] |
| 4K6K | 1.60 | - | D223G | 2013 | [195] |
| 4K6H | 1.60 | - | L278M | 2013 | [195] |
| 4K6G | 1.50 | - | - | 2013 | [195] |
| 4ZV7 | 2.00 | - | - | 2015 | [195] |
| 5A6V | 2.28 | - | - | 2015 | [191] |
| 5A71 | 0.91 | - | - | 2015 | [191] |
| 5GV5 | 2.89 | - | - | 2016 | [193] |
| 6ISP | 1.88 | - | W104V/A281Y/A282Y/V149G | 2018 | [196] |
| 6ISQ | 1.86 | - | W104V/S105C/A281Y/A282Y/V149G | 2018 | [196] |
| 6ISR | 2.6 | - | W104V/S105C/A281Y/A282Y/V149G | 2018 | [196] |
| 6J1P | 1.76 | - | A281G/A282V/V190C | 2018 | [197] |
| 6J1R | 1.6 | - | Q157L/I189A | 2018 | [197] |
| 6J1Q | 1.6 | - | W104A/I189V | 2018 | [197] |
| 6J1T | 1.78 | Synthesized product 3a’ ((2S)-2-phenyl-N-[(1R)-1-phenylethyl]propanamide) | A281G/A282V/V190C | 2018 | [197] |
| 6J1S | 1.83 | - | W104A/I189M/V190C/D134L | 2018 | [197] |
| 6TP8 | 1.55 | - | - | 2019 | [198] |
| PDB Code | Resolution (Å) | Ligand | Mutation | Year of Deposition | Ref. |
|---|---|---|---|---|---|
| 1TIB | 1.84 | Free | - | 1993 | [220] |
| 1DT3 | 2.6 | Free | - | 2000 | [224] |
| 1DT5 | 2.4 | Free | - | 2000 | [224] |
| 1DTE | 2.35 | Free | - | 2000 | [224] |
| 1DU4 | 2.5 | Free | - | 2000 | [224] |
| 1EIN | 3 | didodecyl phosphatidylcholine | - | 2000 | [224] |
| 1GT6 | 2.2 | Oleic acid | S146A | 2002 | [225] |
| 4GYH | 2 | Free | - | 2012 | N/A |
| 4EA6 | 2.3 | Free | - | 2012 | N/A |
| 4FLF | 2.15 | TPP | - | 2012 | [226] |
| 4GBG | 2.9 | Free | - | 2012 | N/A |
| 4GHW | 2.6 | decanoic acid | - | 2012 | N/A |
| 4GI1 | 2.43 | 16-hydroxypalmitic acid | - | 2012 | N/A |
| 4GLB | 2.69 | p-nitrobenzaldehyde | - | 2012 | N/A |
| 4GWL | 2.55 | Free | - | 2012 | N/A |
| 4KJX | 2.1 | p-nitrobenzaldehyde and lauric acid | - | 2013 | N/A |
| 4N8S | 2.3 | p-nitrobenzaldehyde and ethyl acetoacetate | - | 2013 | N/A |
| 4S0X | 2.1 | Lauric acid | - | 2015 | N/A |
| 4ZGB | 2.3 | Free | - | 2015 | [226] |
| 5AP9 | 1.8 | Free | I186C/I255C | 2015 | [227] |
| 6HW1 | 2.5 | Free | - | 2018 | [228] |
| 6OR3 | 1.45 | Palmitic acid | - | 2020 | [229] |
| 6XOK | 1.3 | 2-hydroxy-3-(octadecanoyloxy)propyl pentacosanoate | - | 2020 | [229] |
| 6XRV | 1.43 | 2-hydroxy-3-(octadecanoyloxy)propyl pentacosanoate and caprylic acid | - | 2020 | [229] |
| 6XS3 | 2.48 | 2-hydroxy-3-(octadecanoyloxy)propyl pentacosanoate and caprylic acid | - | 2020 | [229] |
| Pet No. | UniProt Code | Sequence Length | Organism |
|---|---|---|---|
| 1 | E8U721 | 315 | Deinococcus maricopensis |
| 2 | C3RYL0 | 308 | uncultured bacterium |
| 3 | A0A0F9X315 | 300 | marine sediment metagenome |
| 4 | N6VY44 | 295 | Marinobacter nanhaiticus |
| 5 | R4YKL9 | 310 | Oleispira antarctica |
| 6 | A0A1Z2SIQ1 | 298 | Vibrio gazogenes |
| 7 | Q8RR62 | 304 | Acidovorax delafieldii |
| 8 | P19833 | 319 | Moraxella sp. |
| 9 | A0A0D4L7E6 | 313 | Psychrobacter sp. |
| 10 | UPI00064655D2 | 292 | Methylibium sp. |
| 11 | UPI0003660256 | 292 | Caldimonas manganoxidans |
| 12 | A0A0G3BI90 | 298 | [Polyangium] brachysporum |
| 13 | A0A1F4G492 | 283 | Burkholderiales bacterium |
| PDB Code | Resolution (Å) | Ligand | Mutation | Year of Deposition | Ref. |
|---|---|---|---|---|---|
| 3VIS | 1.76 | - | - | 2011 | [259] |
| 3WYN | 1.68 | - | - | 2014 | [45] |
| 6AID | 1.3 | Ethyl lactate | - | 2018 | [260] |
| Enzyme | Best Performing Mutation |
|---|---|
| IsPETase | S214H/I168R/W159H/S188Q/R280A/A180I/G165A/Q119Y/L117F/T140D |
| IsMHETase | W397A |
| PaPETase | Y250S |
| LCC | F243I/D238C/S283D/Y127G |
| F243I/D238C/S283C/T96M | |
| F243W/D238C/S283C/Y127G | |
| F243W/D238C/S283C/T96M | |
| TfHCut | Q132A/T101A |
| SvCut190 | S226P/R228S/Q138A/D250C–E296C/Q123H/N202H |
| TfCut2 | D204C/E253C/D174R |
| TcCut2 | R29N/A30V |
| FsCut | L182A |
| L81A | |
| V184A | |
| TaEst1 | A68V/T253P |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magalhães, R.P.; Cunha, J.M.; Sousa, S.F. Perspectives on the Role of Enzymatic Biocatalysis for the Degradation of Plastic PET. Int. J. Mol. Sci. 2021, 22, 11257. https://doi.org/10.3390/ijms222011257
Magalhães RP, Cunha JM, Sousa SF. Perspectives on the Role of Enzymatic Biocatalysis for the Degradation of Plastic PET. International Journal of Molecular Sciences. 2021; 22(20):11257. https://doi.org/10.3390/ijms222011257
Chicago/Turabian StyleMagalhães, Rita P., Jorge M. Cunha, and Sérgio F. Sousa. 2021. "Perspectives on the Role of Enzymatic Biocatalysis for the Degradation of Plastic PET" International Journal of Molecular Sciences 22, no. 20: 11257. https://doi.org/10.3390/ijms222011257