
Functional Depletion of HSP72 by siRNA and Quercetin Enhances Vorinostat-Induced Apoptosis in an HSP72-Overexpressing Cutaneous T-Cell Lymphoma Cell Line, Hut78


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
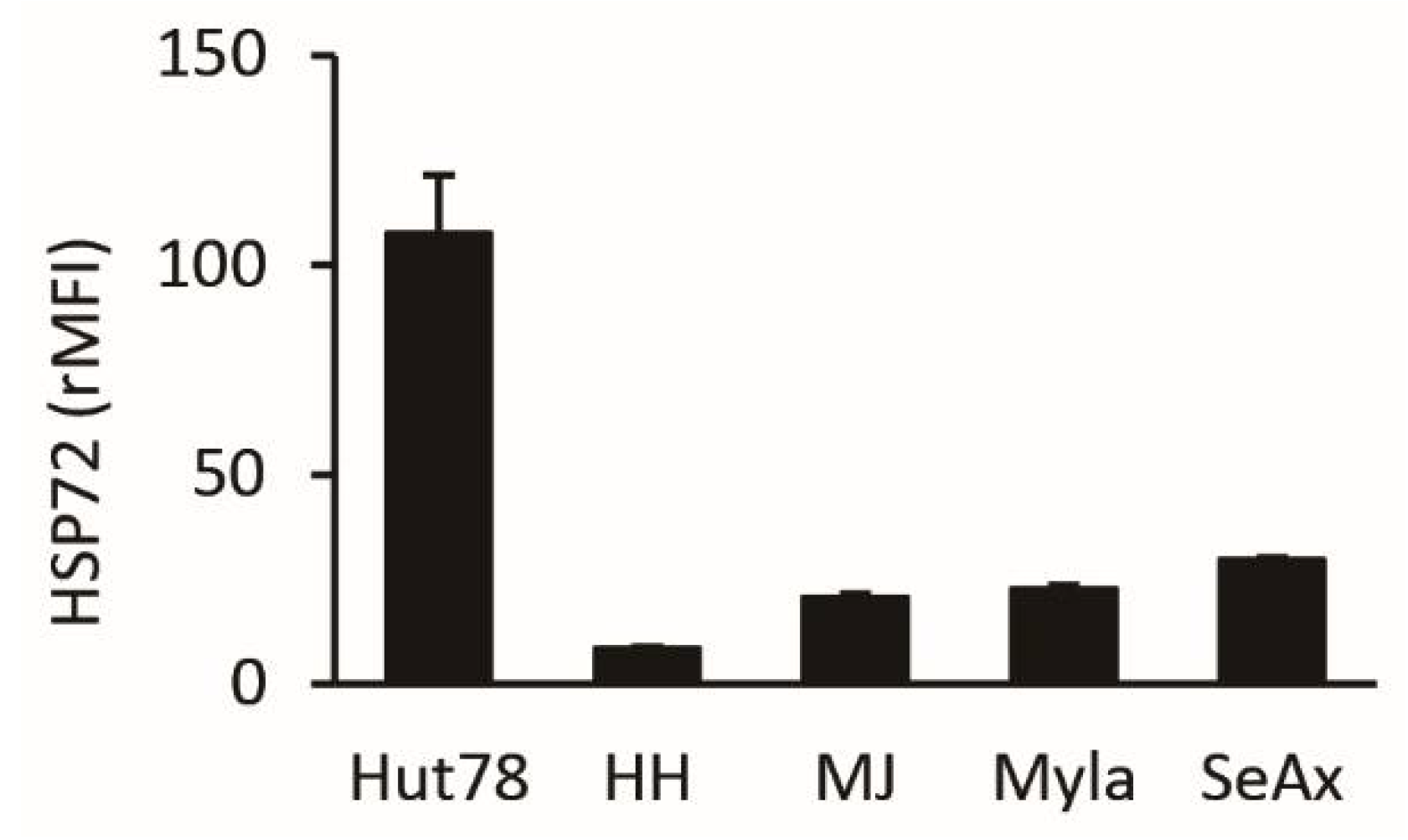
2.1. HSP72 Expression in CTCL Cell Lines
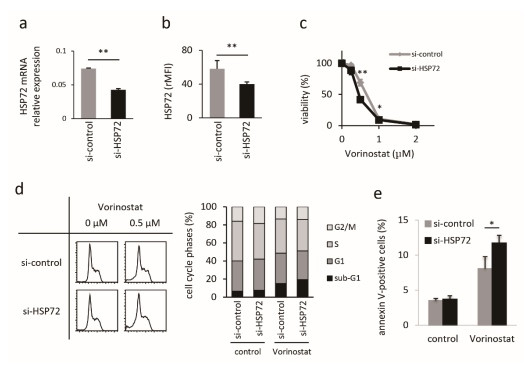
2.2. Establishment and Characterization of HSP72-Knockdown Hut78 Cells
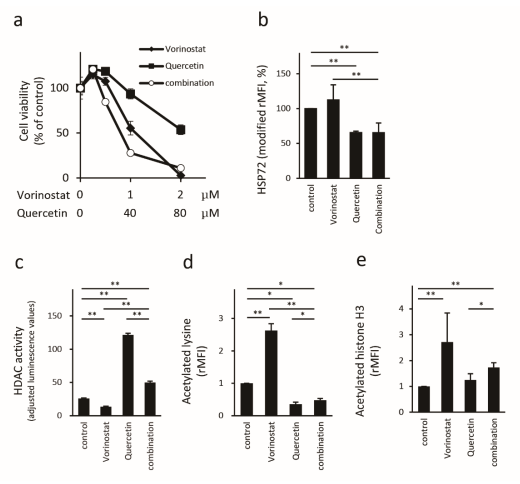
2.3. Quercetin Reduces HSP72 Expression and Enhances HDAC Activity
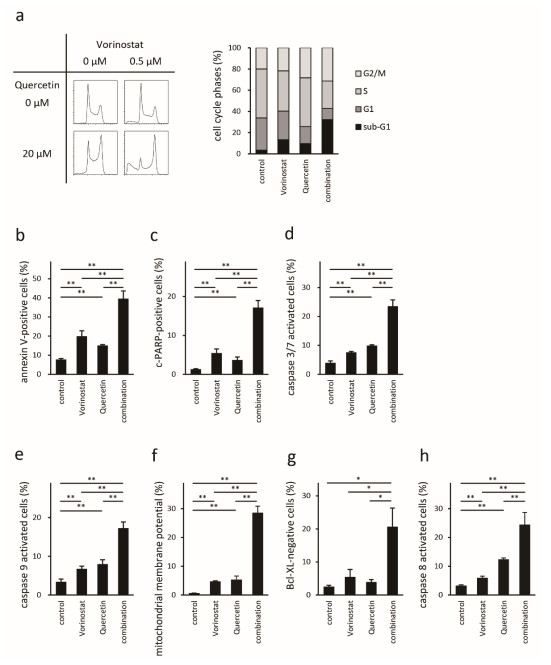
2.4. Quercetin Enhances Vorinostat-Induced Apoptosis
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Agent
4.2. Assessment of HSP72 Expression
4.3. Proliferation Assays
4.4. Assessment of HDAC Activity and Acetylation Status
4.5. Analysis of Cell Cycle and Apoptosis
4.6. Caspase Activity Assays
4.7. Assessment of the Mitochondrial Membrane Potential (MMP)
4.8. Flow Cytometric Analysis
4.9. Statistics and Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Scarisbrick, J.J.; Kim, Y.H.; Whittaker, S.J.; Wood, G.S.; Vermeer, M.H.; Prince, H.M.; Quaglino, P. Prognostic factors, prognostic indices and staging in mycosis fungoides and Sezary syndrome: Where are we now? Br. J. Dermatol. 2014, 170, 1226–1236. [Google Scholar] [CrossRef]
- Hughes, C.F.; Khot, A.; McCormack, C.; Lade, S.; Westerman, D.A.; Twigger, R.; Buelens, O.; Newland, K.; Tam, C.; Dickinson, M.; et al. Lack of durable disease control with chemotherapy for mycosis fungoides and Sezary syndrome: A comparative study of systemic therapy. Blood 2015, 125, 71–81. [Google Scholar] [CrossRef]
- Akilov, O.E.; Geskin, L. Therapeutic advances in cutaneous T-cell lymphoma. Skin Ther. Lett. 2011, 16, 1–5. [Google Scholar] [PubMed]
- Blaizot, R.; Ouattara, E.; Fauconneau, A.; Beylot-Barry, M.; Pham-Ledard, A. Infectious events and associated risk factors in mycosis fungoides/Sezary syndrome: A retrospective cohort study. Br. J. Dermatol. 2018, 179, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K. New Therapies and Immunological Findings in Cutaneous T-Cell Lymphoma. Front. Oncol. 2018, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef]
- Piekarz, R.L.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.L.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J.; et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 2009, 27, 5410–5417. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.J.; Demierre, M.F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef]
- Richardson, P.G.; Schlossman, R.L.; Alsina, M.; Weber, D.M.; Coutre, S.E.; Gasparetto, C.; Mukhopadhyay, S.; Ondovik, M.S.; Khan, M.; Paley, C.S.; et al. PANORAMA 2: Panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 2013, 122, 2331–2337. [Google Scholar] [CrossRef]
- Wozniak, M.B.; Villuendas, R.; Bischoff, J.R.; Aparicio, C.B.; Martinez Leal, J.F.; de La Cueva, P.; Rodriguez, M.E.; Herreros, B.; Martin-Perez, D.; Longo, M.I.; et al. Vorinostat interferes with the signaling transduction pathway of T-cell receptor and synergizes with phosphoinositide-3 kinase inhibitors in cutaneous T-cell lymphoma. Haematologica 2010, 95, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Sanli, H.; Akay, B.N.; Anadolu, R.; Ozcan, M.; Saral, S.; Akyol, A. The efficacy of vorinostat in combination with interferon alpha and extracorporeal photopheresis in late stage mycosis fungoides and Sezary syndrome. J. Drugs Dermatol. 2011, 10, 403–408. [Google Scholar] [PubMed]
- Dummer, R.; Beyer, M.; Hymes, K.; Epping, M.T.; Bernards, R.; Steinhoff, M.; Sterry, W.; Kerl, H.; Heath, K.; Ahern, J.D.; et al. Vorinostat combined with bexarotene for treatment of cutaneous T-cell lymphoma: In vitro and phase I clinical evidence supporting augmentation of retinoic acid receptor/retinoid X receptor activation by histone deacetylase inhibition. Leuk. Lymphoma 2012, 53, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Rozati, S.; Cheng, P.F.; Widmer, D.S.; Fujii, K.; Levesque, M.P.; Dummer, R. Romidepsin and Azacitidine Synergize in their Epigenetic Modulatory Effects to Induce Apoptosis in CTCL. Clin. Cancer Res. 2016, 22, 2020–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosenza, M.; Civallero, M.; Fiorcari, S.; Pozzi, S.; Marcheselli, L.; Bari, A.; Ferri, P.; Sacchi, S. The histone deacetylase inhibitor romidepsin synergizes with lenalidomide and enhances tumor cell death in T-cell lymphoma cell lines. Cancer Biol. Ther. 2016, 17, 1094–1106. [Google Scholar] [CrossRef] [Green Version]
- Uehara, J.; Honma, M.; Ohishi, Y.; Ishida-Yamamoto, A. Successful combination therapy of low-dose vorinostat, etretinate and narrowband ultraviolet B irradiation for Sezary syndrome. J. Dermatol. 2017, 44, e30–e31. [Google Scholar] [CrossRef] [Green Version]
- Jimura, N.; Fujii, K.; Qiao, Z.; Tsuchiya, R.; Yoshimatsu, Y.; Kondo, T.; Kanekura, T. Kinome profiling analysis identified Src pathway as a novel therapeutic target in combination with histone deacetylase inhibitors for cutaneous T-cell lymphoma. J. Dermatol. Sci. 2021, 101, 194–201. [Google Scholar] [CrossRef]
- Khan, O.; Fotheringham, S.; Wood, V.; Stimson, L.; Zhang, C.; Pezzella, F.; Duvic, M.; Kerr, D.J.; La Thangue, N.B. HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 6532–6537. [Google Scholar] [CrossRef] [Green Version]
- Qu, K.; Zaba, L.C.; Satpathy, A.T.; Giresi, P.G.; Li, R.; Jin, Y.; Armstrong, R.; Jin, C.; Schmitt, N.; Rahbar, Z.; et al. Chromatin Accessibility Landscape of Cutaneous T Cell Lymphoma and Dynamic Response to HDAC Inhibitors. Cancer Cell 2017, 32, 27–41.e4. [Google Scholar] [CrossRef] [Green Version]
- Andrews, J.M.; Schmidt, J.A.; Carson, K.R.; Musiek, A.C.; Mehta-Shah, N.; Payton, J.E. Novel cell adhesion/migration pathways are predictive markers of HDAC inhibitor resistance in cutaneous T cell lymphoma. EBioMedicine 2019, 46, 170–183. [Google Scholar] [CrossRef] [Green Version]
- Angelika Ihle, M.; Merkelbach-Bruse, S.; Hartmann, W.; Bauer, S.; Ratner, N.; Sonobe, H.; Nishio, J.; Larsson, O.; Aman, P.; Pedeutour, F.; et al. HR23b expression is a potential predictive biomarker for HDAC inhibitor treatment in mesenchymal tumours and is associated with response to vorinostat. J. Pathol. Clin. Res. 2016, 2, 59–71. [Google Scholar] [CrossRef]
- Fujii, K.; Suzuki, N.; Ikeda, K.; Hamada, T.; Yamamoto, T.; Kondo, T.; Iwatsuki, K. Proteomic study identified HSP 70 kDa protein 1A as a possible therapeutic target, in combination with histone deacetylase inhibitors, for lymphoid neoplasms. J. Proteomics 2012, 75, 1401–1410. [Google Scholar] [CrossRef]
- Fujii, K.; Suzuki, N.; Jimura, N.; Idogawa, M.; Kondo, T.; Iwatsuki, K.; Kanekura, T. HSP72 functionally inhibits the anti-neoplastic effects of HDAC inhibitors. J. Dermatol. Sci. 2018, 90, 82–89. [Google Scholar] [CrossRef] [Green Version]
- De Bono, J.S.; Kristeleit, R.; Tolcher, A.; Fong, P.; Pacey, S.; Karavasilis, V.; Mita, M.; Shaw, H.; Workman, P.; Kaye, S.; et al. Phase I pharmacokinetic and pharmacodynamic study of LAQ824, a hydroxamate histone deacetylase inhibitor with a heat shock protein-90 inhibitory profile, in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 6663–6673. [Google Scholar] [CrossRef] [Green Version]
- Stuhmer, T.; Arts, J.; Chatterjee, M.; Borawski, J.; Wolff, A.; King, P.; Einsele, H.; Leo, E.; Bargou, R.C. Preclinical anti-myeloma activity of the novel HDAC-inhibitor JNJ-26481585. Br. J. Haematol. 2010, 149, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Gabai, V.L.; Budagova, K.R.; Sherman, M.Y. Increased expression of the major heat shock protein Hsp72 in human prostate carcinoma cells is dispensable for their viability but confers resistance to a variety of anticancer agents. Oncogene 2005, 24, 3328–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef]
- Multhoff, G.; Botzler, C.; Wiesnet, M.; Muller, E.; Meier, T.; Wilmanns, W.; Issels, R.D. A stress-inducible 72-kDa heat-shock protein (HSP72) is expressed on the surface of human tumor cells, but not on normal cells. Int. J. Cancer 1995, 61, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Remesy, C.; Jimenez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [Green Version]
- Baron, B.W.; Thirman, M.J.; Giurcanu, M.C.; Baron, J.M. Quercetin Therapy for Selected Patients with PIM1 Kinase-Positive Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma: A Pilot Study. Acta Haematol. 2018, 139, 132–139. [Google Scholar] [CrossRef]
- Larocca, L.M.; Teofili, L.; Leone, G.; Sica, S.; Pierelli, L.; Menichella, G.; Scambia, G.; Benedetti Panici, P.; Ricci, R.; Piantelli, M.; et al. Antiproliferative activity of quercetin on normal bone marrow and leukaemic progenitors. Br. J. Haematol. 1991, 79, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Imran, M.; Khan, I.A.; Ur-Rehman, M.; Gilani, S.A.; Mehmood, Z.; Mubarak, M.S. Anticancer potential of quercetin: A comprehensive review. Phytother. Res. 2018, 32, 2109–2130. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Cui, M.; Lee, J.; Gong, W.; Wang, S.; Fu, J.; Wu, G.; Yan, K. Heat shock protein inhibitor, quercetin, as a novel adjuvant agent to improve radiofrequency ablation-induced tumor destruction and its molecular mechanism. Chin. J. Cancer Res. 2016, 28, 19–28. [Google Scholar] [PubMed]
- Brito, A.F.; Ribeiro, M.; Abrantes, A.M.; Pires, A.S.; Teixo, R.J.; Tralhao, J.G.; Botelho, M.F. Quercetin in Cancer Treatment, Alone or in Combination with Conventional Therapeutics? Curr. Med. Chem. 2015, 22, 3025–3039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliutz, G.; Karlseder, J.; Tempfer, C.; Orel, L.; Holzer, G.; Simon, M.M. Drug resistance against gemcitabine and topotecan mediated by constitutive hsp70 overexpression in vitro: Implication of quercetin as sensitiser in chemotherapy. Br. J. Cancer 1996, 74, 172–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobori, M.; Masumoto, S.; Akimoto, Y.; Takahashi, Y. Dietary quercetin alleviates diabetic symptoms and reduces streptozotocin-induced disturbance of hepatic gene expression in mice. Mol. Nutr. Food Res. 2009, 53, 859–868. [Google Scholar] [CrossRef]
- Yoshida, M.; Yamamoto, M.; Nikaido, T. Quercetin arrests human leukemic T-cells in late G1 phase of the cell cycle. Cancer Res. 1992, 52, 6676–6681. [Google Scholar]
- Lee, T.J.; Kim, O.H.; Kim, Y.H.; Lim, J.H.; Kim, S.; Park, J.W.; Kwon, T.K. Quercetin arrests G2/M phase and induces caspase-dependent cell death in U937 cells. Cancer Lett. 2006, 240, 234–242. [Google Scholar] [CrossRef]
- Jia, J.; Chen, J. Histone hyperacetylation is involved in the quercetin-induced human leukemia cell death. Pharmazie 2008, 63, 379–383. [Google Scholar]
- Lee, W.J.; Chen, Y.R.; Tseng, T.H. Quercetin induces FasL-related apoptosis, in part, through promotion of histone H3 acetylation in human leukemia HL-60 cells. Oncol. Rep. 2011, 25, 583–591. [Google Scholar]
- Chan, S.T.; Yang, N.C.; Huang, C.S.; Liao, J.W.; Yeh, S.L. Quercetin enhances the antitumor activity of trichostatin A through upregulation of p53 protein expression in vitro and in vivo. PLoS ONE 2013, 8, e54255. [Google Scholar] [CrossRef] [Green Version]
- Bishayee, K.; Khuda-Bukhsh, A.R.; Huh, S.O. PLGA-Loaded Gold-Nanoparticles Precipitated with Quercetin Downregulate HDAC-Akt Activities Controlling Proliferation and Activate p53-ROS Crosstalk to Induce Apoptosis in Hepatocarcinoma Cells. Mol. Cells 2015, 38, 518–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godoy, L.D.; Lucas, J.E.; Bender, A.J.; Romanick, S.S.; Ferguson, B.S. Targeting the epigenome: Screening bioactive compounds that regulate histone deacetylase activity. Mol. Nutr. Food Res. 2017, 61, 1600744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, M.C.; Maso, V.; Torello, C.O.; Ferro, K.P.; Saad, S.T.O. The polyphenol quercetin induces cell death in leukemia by targeting epigenetic regulators of pro-apoptotic genes. Clin. Epigenetics 2018, 10, 139. [Google Scholar] [CrossRef]
- Kim, Y.H.; Lee, D.H.; Jeong, J.H.; Guo, Z.S.; Lee, Y.J. Quercetin augments TRAIL-induced apoptotic death: Involvement of the ERK signal transduction pathway. Biochem. Pharmacol. 2008, 75, 1946–1958. [Google Scholar] [CrossRef] [Green Version]
- Doulias, P.T.; Kotoglou, P.; Tenopoulou, M.; Keramisanou, D.; Tzavaras, T.; Brunk, U.; Galaris, D.; Angelidis, C. Involvement of heat shock protein-70 in the mechanism of hydrogen peroxide-induced DNA damage: The role of lysosomes and iron. Free Radic Biol. Med. 2007, 42, 567–577. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujii, K.; Idogawa, M.; Suzuki, N.; Iwatsuki, K.; Kanekura, T. Functional Depletion of HSP72 by siRNA and Quercetin Enhances Vorinostat-Induced Apoptosis in an HSP72-Overexpressing Cutaneous T-Cell Lymphoma Cell Line, Hut78. Int. J. Mol. Sci. 2021, 22, 11258. https://doi.org/10.3390/ijms222011258
Fujii K, Idogawa M, Suzuki N, Iwatsuki K, Kanekura T. Functional Depletion of HSP72 by siRNA and Quercetin Enhances Vorinostat-Induced Apoptosis in an HSP72-Overexpressing Cutaneous T-Cell Lymphoma Cell Line, Hut78. International Journal of Molecular Sciences. 2021; 22(20):11258. https://doi.org/10.3390/ijms222011258
Chicago/Turabian StyleFujii, Kazuyasu, Masashi Idogawa, Norihiro Suzuki, Keiji Iwatsuki, and Takuro Kanekura. 2021. "Functional Depletion of HSP72 by siRNA and Quercetin Enhances Vorinostat-Induced Apoptosis in an HSP72-Overexpressing Cutaneous T-Cell Lymphoma Cell Line, Hut78" International Journal of Molecular Sciences 22, no. 20: 11258. https://doi.org/10.3390/ijms222011258
APA StyleFujii, K., Idogawa, M., Suzuki, N., Iwatsuki, K., & Kanekura, T. (2021). Functional Depletion of HSP72 by siRNA and Quercetin Enhances Vorinostat-Induced Apoptosis in an HSP72-Overexpressing Cutaneous T-Cell Lymphoma Cell Line, Hut78. International Journal of Molecular Sciences, 22(20), 11258. https://doi.org/10.3390/ijms222011258