
Smart Vitamin Micelles as Cancer Nanomedicines for Enhanced Intracellular Delivery of Doxorubicin

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results and Discussion
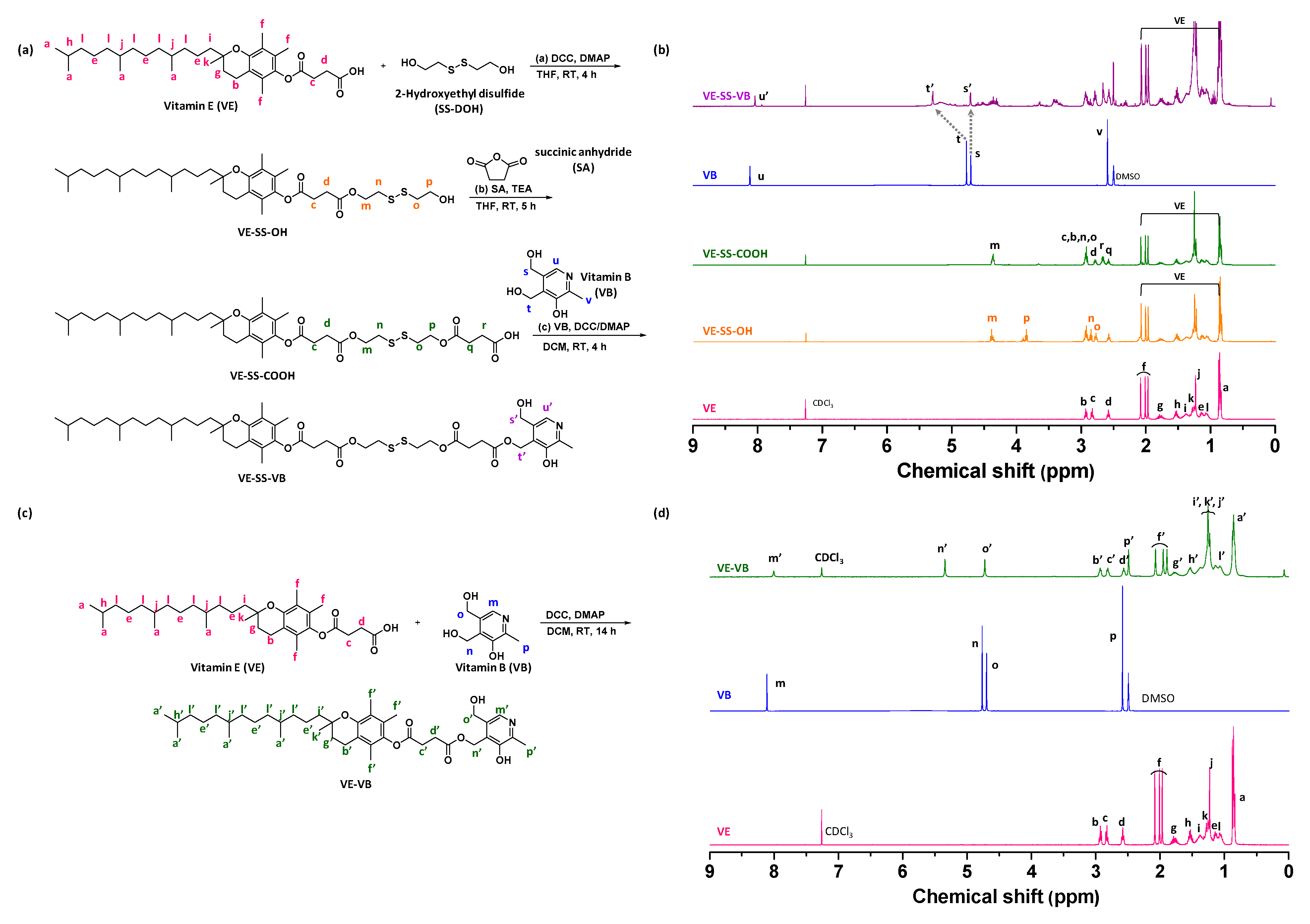
2.1. Synthesis and Characterization of Stimuli-Responsive Degradable Vitamin Conjugates
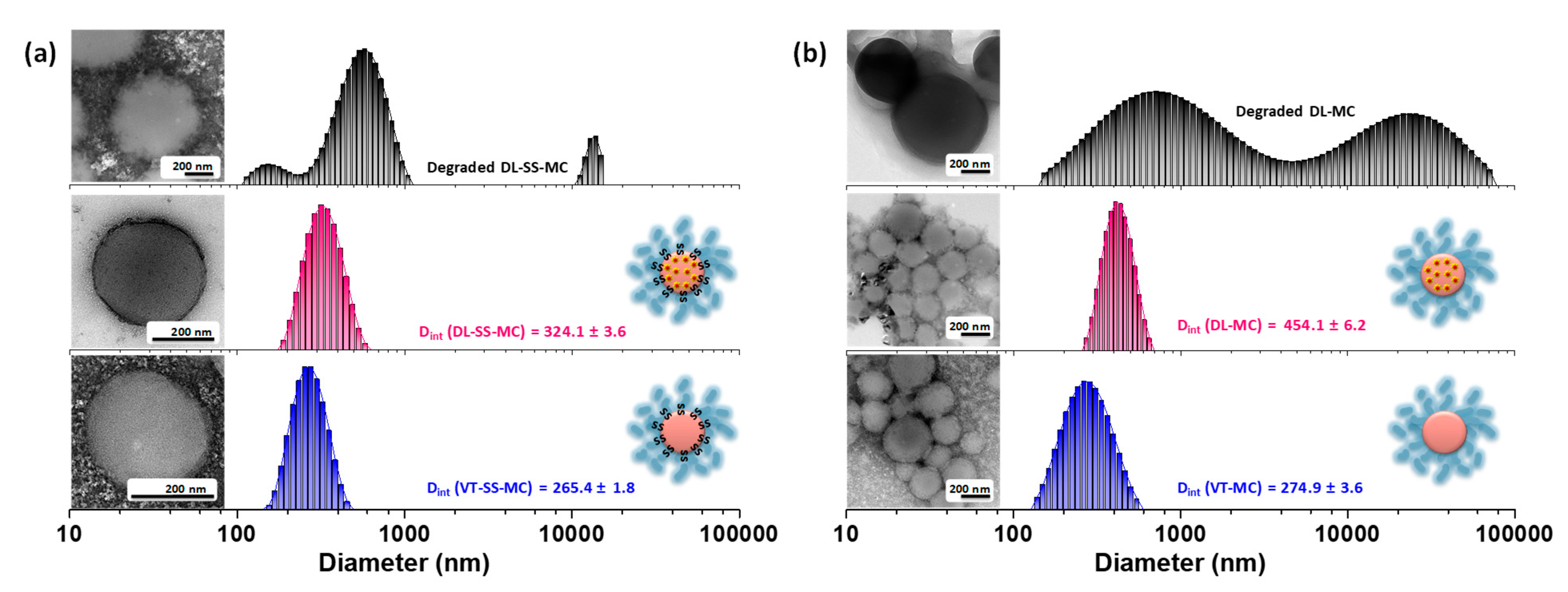
2.2. Self-Assembly and Micellar Formulation of Vitamicelles
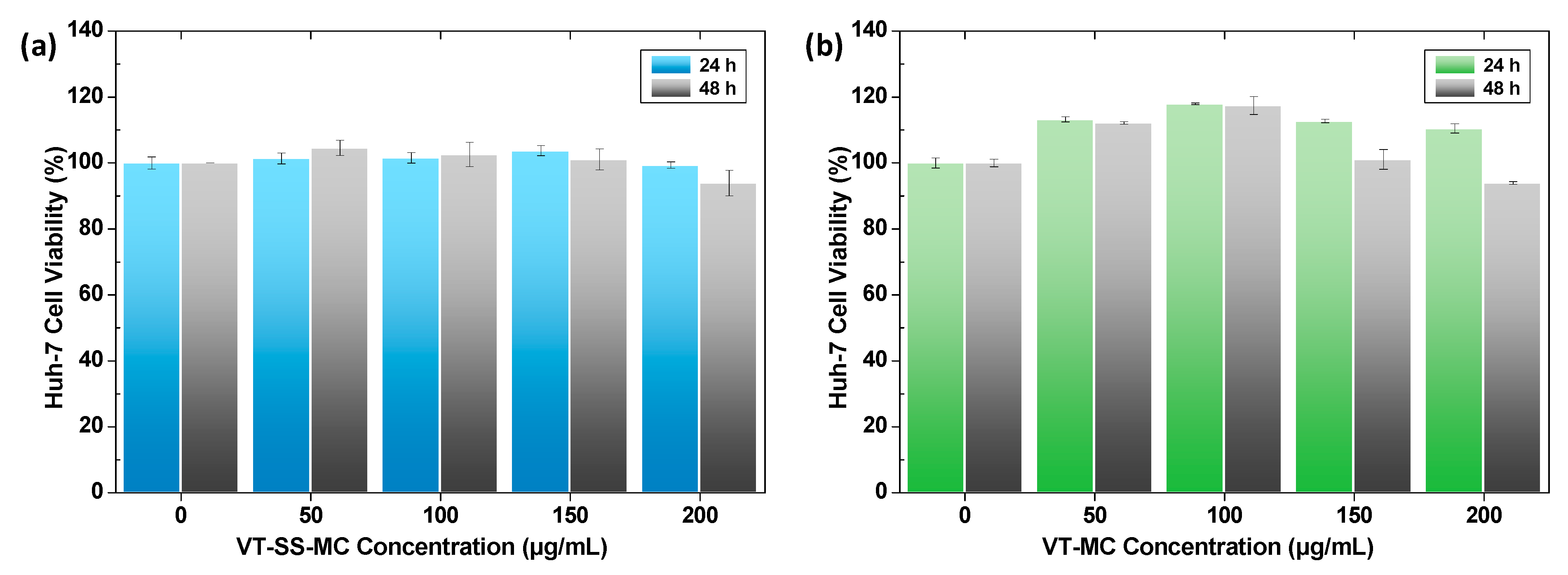
2.3. Biocompatibility of Empty Vitamicelles
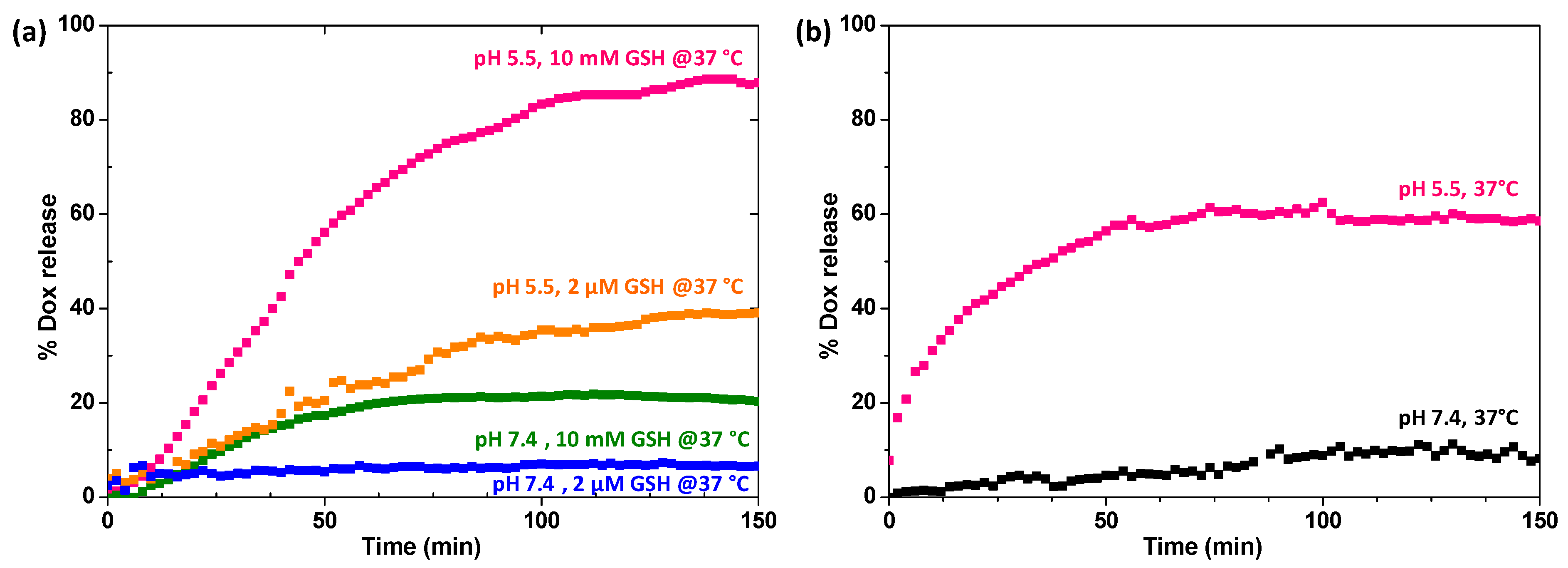
2.4. Loading and Stimuli-Responsive Release of DOX
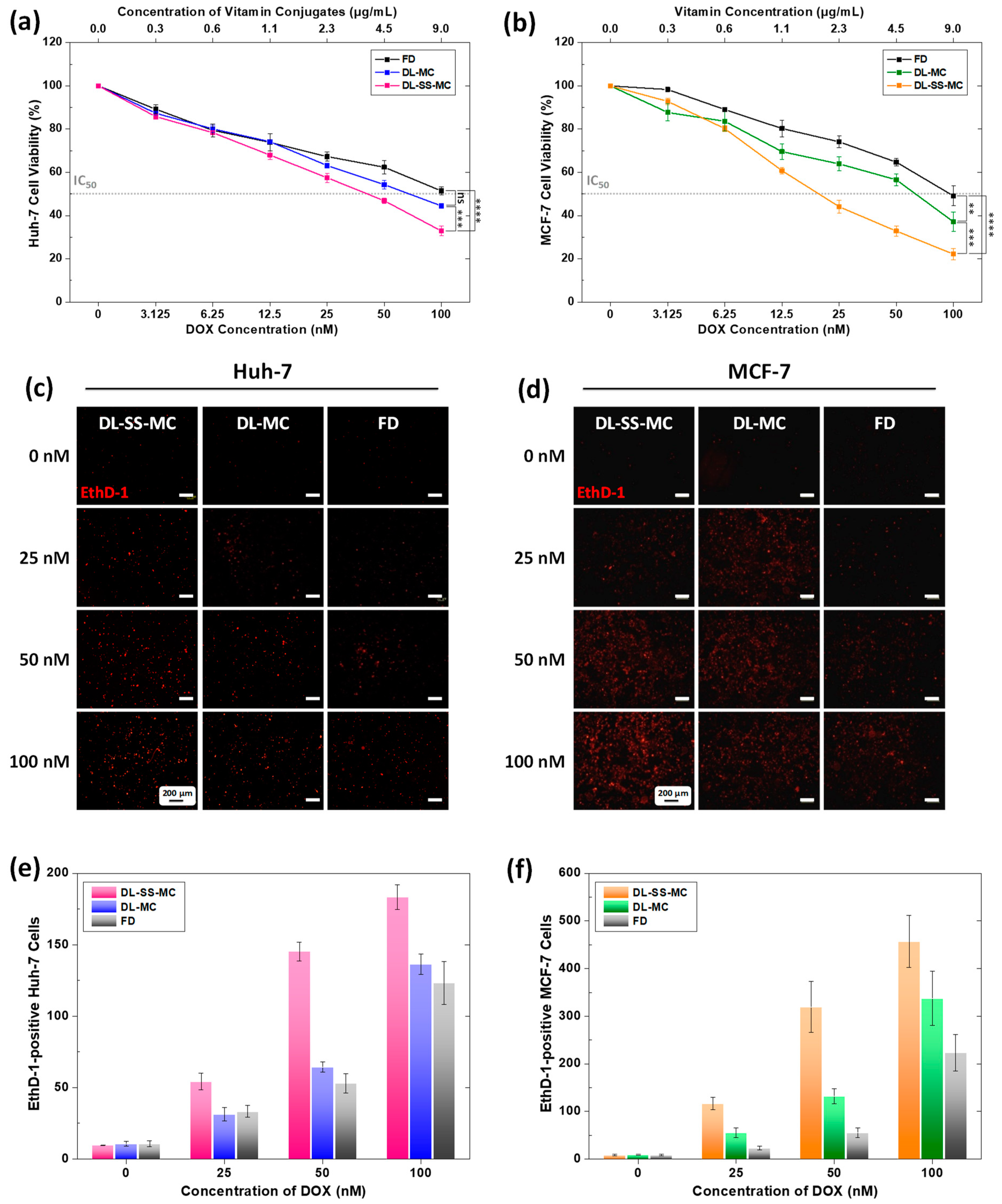
2.5. Effective Anticancer Activity of DL-SS-MC
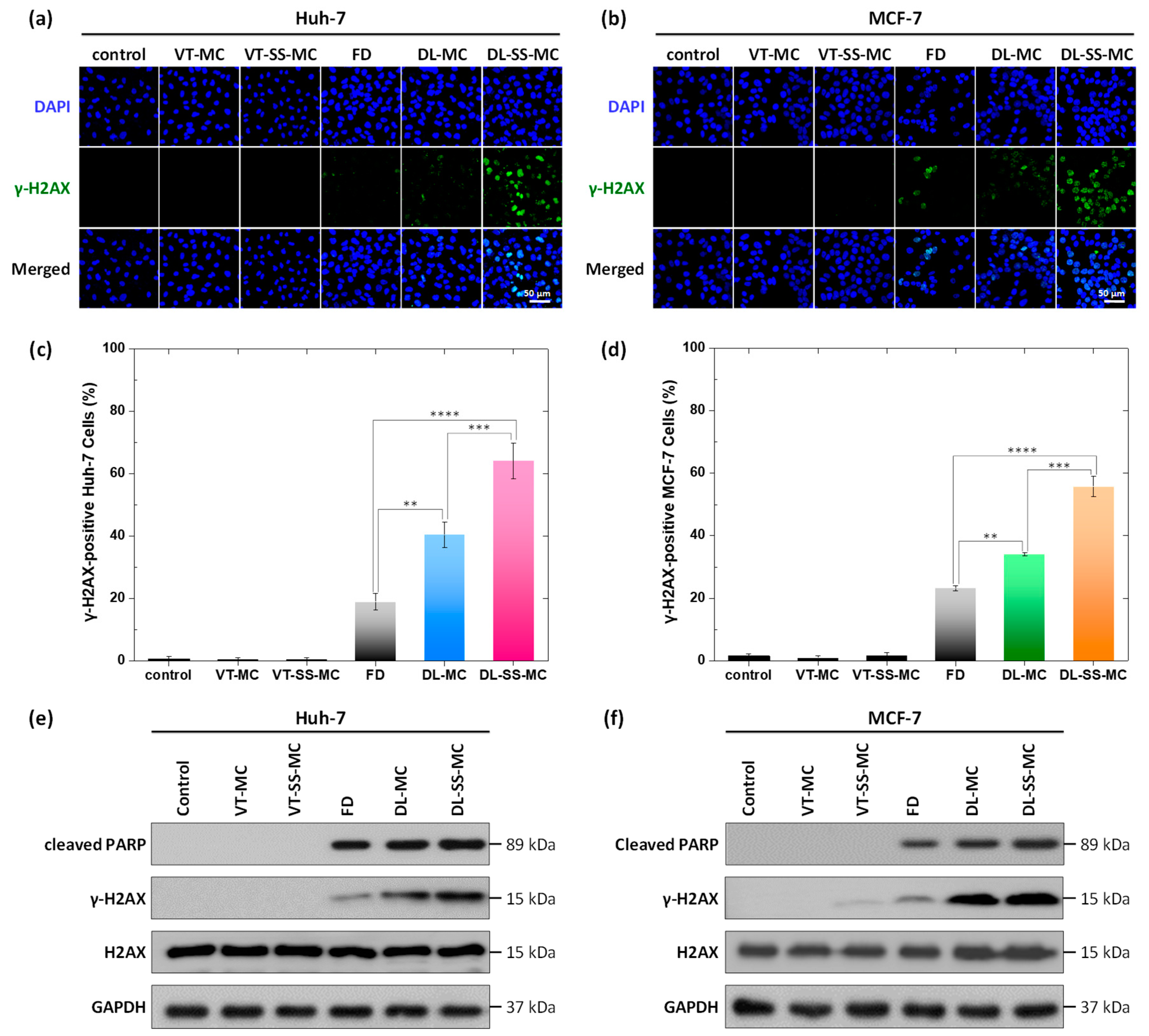
2.6. Increased DOX-Induced DNA Damage and Apoptosis
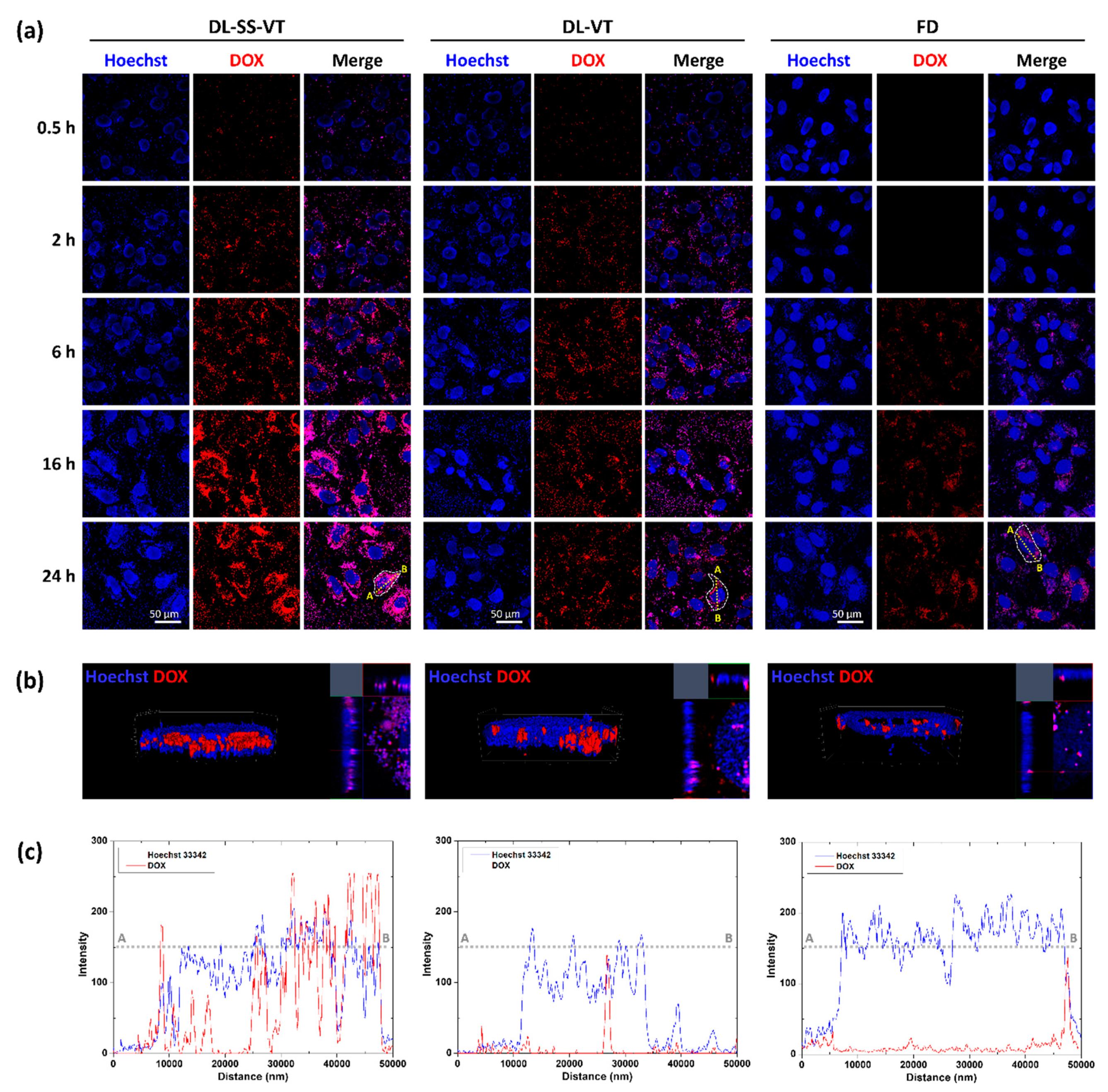
2.7. Enhanced Intracellular Trafficking and Colocalization of DOX
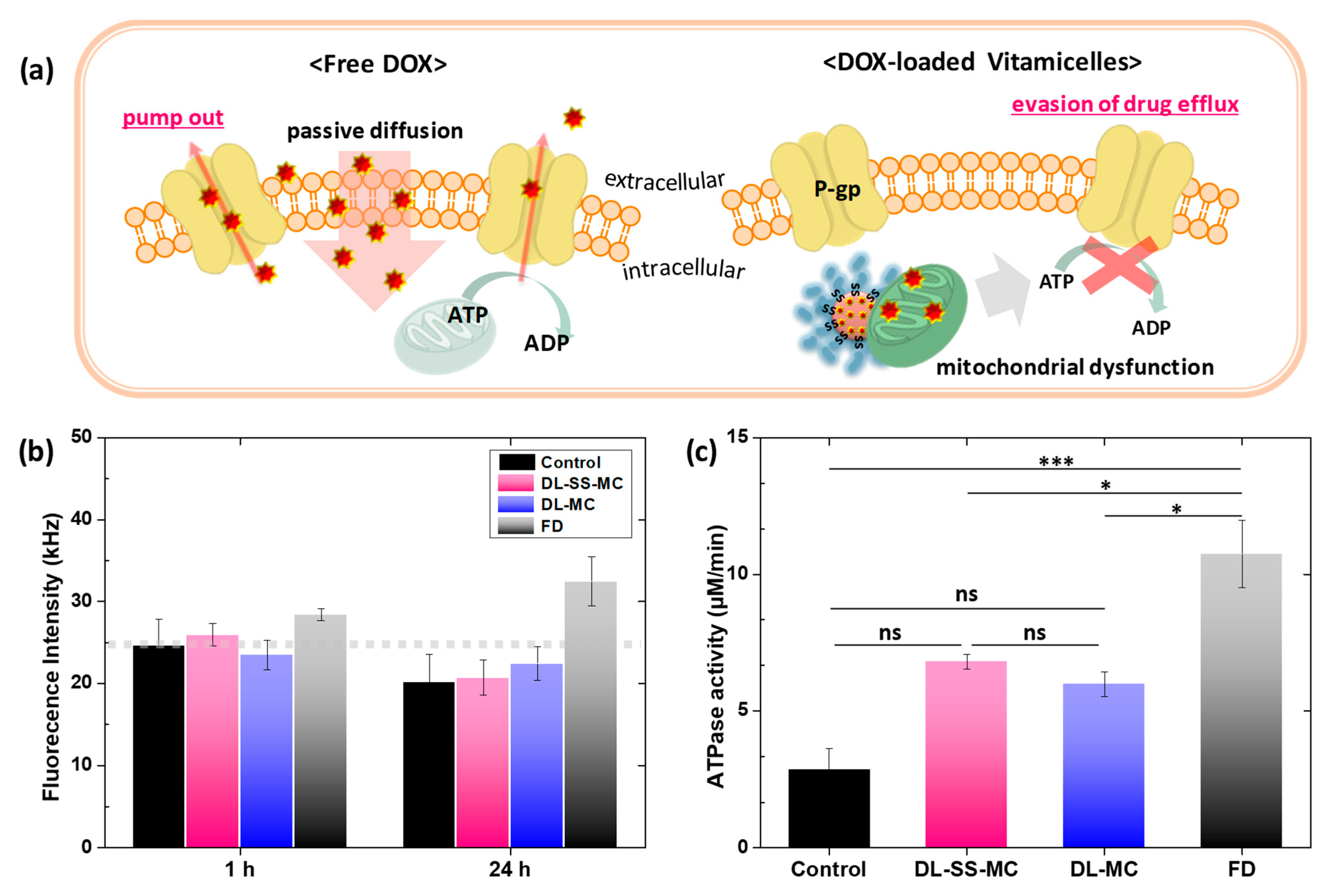
2.8. Inhibition of Drug Efflux by Decreasing ATPase Activity
3. Experimental Section
3.1. Materials
3.2. Instrumentation
3.3. Synthesis of VE-SS-OH
3.4. Synthesis of VE-SS-COOH
3.5. Synthesis of VE-SS-VB Conjugates
3.6. Synthesis of VE-VB Conjugates
3.7. Preparation of Empty Vitamicelles Using the Solvent Evaporation Method
3.8. Preparation of DOX-Loaded Vitamicelles Using the Dialysis Method
3.9. DOX Release from DOX-Loaded Vitamicelles
3.10. Cell Culture
3.11. Cell Viability
3.12. Dead Cell Staining Using Ethidium Homodimer-1
3.13. γ-H2AX Foci Formation Assay
3.14. Western Blot Analysis
3.15. Confocal Laser Scanning Microscopy (CLSM)
3.16. Fluorescence Correlation Spectroscopy (FCS)
3.17. ATPase Activity Assay
3.18. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Buechler, S.A.; Gökmen–Polar, Y.; Badve, S.S. EarlyR signature predicts response to neoadjuvant chemotherapy in breast cancer. Breast 2019, 43, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Bogani, G.; Vinti, D.; Murgia, F.; Chiappa, V.; Maggiore, U.L.R.; Martinelli, F.; Ditto, A.; Raspagliesi, F. Burden of lymphatic disease predicts efficacy of adjuvant radiation and chemotherapy in FIGO 2018 stage IIICp cervical cancer. Int. J. Gynecol. Cancer 2019, 29, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Larkin, J.; Tabernero, J.; Bonini, C. Enhancing anti-tumour efficacy with immunotherapy combinations. Lancet 2020, 397, 1010–1022. [Google Scholar] [CrossRef]
- Frank, M.M.; Fries, L.F. The role of complement in inflammation and phagocytosis. Immunol. Today 1991, 12, 322–326. [Google Scholar] [CrossRef]
- Sugano, K.; Kansy, M.; Artursson, P.; Avdeef, A.; Bendels, S.; Di, L.; Ecker, G.F.; Faller, B.; Fischer, H.; Gerebtzoff, G. Coexistence of passive and carrier-mediated processes in drug transport. Nat. Rev. Drug Discov. 2010, 9, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, H.M.; Al-Abd, A.M.; El-Dine, R.S.; El-Halawany, A.M. P-glycoprotein inhibitors of natural origin as potential tumor chemo-sensitizers: A review. J. Adv. Res. 2015, 6, 45–62. [Google Scholar] [CrossRef]
- Crommelin, D.J.A.; van Hoogevest, P.; Storm, G. The role of liposomes in clinical nanomedicine development. What now? Now what? J. Control. Release 2020, 318, 256–263. [Google Scholar] [CrossRef]
- Hong, S.H.; Larocque, K.; Jaunky, D.B.; Piekny, A.; Oh, J.K. Dual disassembly and biological evaluation of enzyme/oxidation-responsive polyester-based nanoparticulates for tumor-targeting delivery. Colloids Surf. B Biointerfaces 2018, 172, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Panagi, M.; Voutouri, C.; Mpekris, F.; Papageorgis, P.; Martin, M.R.; Martin, J.D.; Demetriou, P.; Pierides, C.; Polydorou, C.; Stylianou, A. TGF-β inhibition combined with cytotoxic nanomedicine normalizes triple negative breast cancer microenvironment towards anti-tumor immunity. Theranostics 2020, 10, 1910. [Google Scholar] [CrossRef]
- Ko, N.R.; Hong, S.H.; Nafiujjaman, M.; An, S.Y.; Revuri, V.; Lee, S.J.; Kwon, I.K.; Lee, Y.-k.; Oh, S.J. Glutathione-responsive PEGylated GQD-based nanomaterials for diagnosis and treatment of breast cancer. J. Ind. Eng. Chem. 2019, 71, 301–307. [Google Scholar] [CrossRef]
- Alemayehu, Y.A.; Ilhami, F.B.; Manayia, A.H.; Cheng, C.-C. Mercury-Containing Supramolecular Micelles with Highly Sensitive pH-Responsiveness for Selective Cancer Therapy. Acta Biomater. 2021, 129, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Pillai, G. Nanomedicines for cancer therapy: An update of fda approved and those under various stages of development. SOJ Pharm. Pharm. Sci. 2014, 1, 13. [Google Scholar]
- Aghebati-Maleki, A.; Dolati, S.; Ahmadi, M.; Baghbanzhadeh, A.; Asadi, M.; Fotouhi, A.; Yousefi, M.; Aghebati-Maleki, L. Nanoparticles and cancer therapy: Perspectives for application of nanoparticles in the treatment of cancers. J. Cell. Physiol. 2020, 235, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Ray, L.; Pal, M.K.; Ray, R.S. Synergism of co-delivered nanosized antioxidants displayed enhanced anticancer efficacy in human colon cancer cell lines. Bioact. Mater. 2017, 2, 82–95. [Google Scholar] [CrossRef]
- Ernest, U.; Chen, H.-Y.; Xu, M.-J.; Taghipour, Y.D.; Asad, M.H.H.B.; Rahimi, R.; Murtaza, G. Anti-Cancerous Potential of Polyphenol-Loaded Polymeric Nanotherapeutics. Molecules 2018, 23, 2787. [Google Scholar] [CrossRef]
- Zafar, M.S.; Quarta, A.; Marradi, M.; Ragusa, A. Recent Developments in the Reduction of Oxidative Stress through Antioxidant Polymeric Formulations. Pharmaceutics 2019, 11, 505. [Google Scholar] [CrossRef]
- de Arriba, G.; de Hornedo, J.P.; Rubio, S.R.; Fernández, M.C.; Martínez, S.B.; Camarero, M.M.; Cid, T.P. Vitamin E protects against the mitochondrial damage caused by cyclosporin A in LLC-PK1 cells. Toxicol. Appl. Pharm. 2009, 239, 241–250. [Google Scholar] [CrossRef]
- Mehata, A.K.; Bharti, S.; Singh, P.; Viswanadh, M.K.; Kumari, L.; Agrawal, P.; Singh, S.; Koch, B.; Muthu, M.S. Trastuzumab decorated TPGS-g-chitosan nanoparticles for targeted breast cancer therapy. Colloids Surf. B Biointerfaces 2019, 173, 366–377. [Google Scholar] [CrossRef]
- Xiong, S.; Wang, Z.; Liu, J.; Deng, X.; Xiong, R.; Cao, X.; Xie, Z.; Lei, X.; Chen, Y.; Tang, G. A pH-sensitive prodrug strategy to co-deliver DOX and TOS in TPGS nanomicelles for tumor therapy. Colloids Surf. B Biointerfaces 2019, 173, 346–355. [Google Scholar] [CrossRef]
- Cheng, F.; Peng, X.; Meng, G.; Pu, Y.; Luo, K.; He, B. Poly(ester-thioether) microspheres co-loaded with erlotinib and α-tocopheryl succinate for combinational therapy of non-small cell lung cancer. J. Mater. Chem. B 2020, 8, 1728–1738. [Google Scholar] [CrossRef]
- Jacinto, T.A.; Rodrigues, C.F.; Moreira, A.F.; Miguel, S.P.; Costa, E.C.; Ferreira, P.; Correia, I.J. Hyaluronic acid and vitamin E polyethylene glycol succinate functionalized gold-core silica shell nanorods for cancer targeted photothermal therapy. Colloids Surf. B Biointerfaces 2020, 188, 110778. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; McAllister, D.M.; Mackinnon, A.C.; Joseph, J.; Dwinell, M.B.; Kalyanaraman, B. Mitochondria-targeted vitamin E analogs inhibit breast cancer cell energy metabolism and promote cell death. BMC Cancer 2013, 13, 285. [Google Scholar] [CrossRef]
- Ehrenshaft, M.; Bilski, P.; Li, M.Y.; Chignell, C.F.; Daub, M.E. A highly conserved sequence is a novel gene involved in de novo vitamin B6 biosynthesis. Proc. Natl. Acad. Sci. USA 1999, 96, 9374–9378. [Google Scholar] [CrossRef]
- Gamov, G.A.; Zavalishin, M.N.; Khokhlova, A.Y.; Sharnin, V.A. Influence of aqueous dimethyl sulfoxide on pyridoxine protonation and tautomerization. J. Mol. Liq. 2016, 221, 457–462. [Google Scholar] [CrossRef]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Khan, J.M.; Haque, S. Strategies in the design of endosomolytic agents for facilitating endosomal escape in nanoparticles. Biochimie 2019, 160, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Said, Z.M.; Subramanian, V.S.; Vaziri, N.D.; Said, H.M. Pyridoxine uptake by colonocytes: A specific and regulated carrier-mediated process. Am. J. Physiol.-Cell Physiol. 2008, 294, C1192–C1197. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Stein, S. Preparation of vitamin B6-conjugated peptides at the amino terminus and of vitamin B6-peptide-oligonucleotide conjugates. Bioconjug. Chem. 1994, 5, 312–315. [Google Scholar] [CrossRef]
- Pandey, S.; Garg, P.; Lim, K.T.; Kim, J.; Choung, Y.-H.; Choi, Y.-J.; Choung, P.-H.; Cho, C.-S.; Chung, J.H. The efficiency of membrane transport of vitamin B6 coupled to poly(ester amine) gene transporter and transfection in cancer cells. Biomaterials 2013, 34, 3716–3728. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Verma, A.; Singh, J.; Teja, B.V.; Mittapelly, N.; Pandey, G.; Urandur, S.; Shukla, R.P.; Konwar, R.; Mishra, P.R. Vitamin B6 Tethered Endosomal pH Responsive Lipid Nanoparticles for Triggered Intracellular Release of Doxorubicin. ACS Appl. Mater. Interfaces 2016, 8, 30407–30421. [Google Scholar] [CrossRef]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar]
- Manickam, D.S.; Li, J.; Putt, D.A.; Zhou, Q.-H.; Wu, C.; Lash, L.H.; Oupický, D. Effect of innate glutathione levels on activity of redox-responsive gene delivery vectors. J. Control. Release 2010, 141, 77–84. [Google Scholar] [CrossRef][Green Version]
- Lu, Y.; Zhang, E.; Yang, J.; Cao, Z. Strategies to improve micelle stability for drug delivery. Nano Res. 2018, 11, 4985–4998. [Google Scholar] [CrossRef]
- Luo, Y.; Yin, X.; Yin, X.; Chen, A.; Zhao, L.; Zhang, G.; Liao, W.; Huang, X.; Li, J.; Zhang, C.Y. Dual pH/redox-responsive mixed polymeric micelles for anticancer drug delivery and controlled release. Pharmaceutics 2019, 11, 176. [Google Scholar] [CrossRef]
- Huynh, V.T.; de Souza, P.; Stenzel, M.H. Polymeric Micelles with Pendant Dicarboxylato Chelating Ligands Prepared via a Michael Addition for cis-Platinum Drug Delivery. Macromolecules 2011, 44, 7888–7900. [Google Scholar] [CrossRef]
- Cheng, F.R.; Su, T.; Cao, J.; Luo, X.L.; Li, L.; Pu, Y.; He, B. Environment-stimulated nanocarriers enabling multi-active sites for high drug encapsulation as an “on demand” drug release system. J. Mater. Chem. B 2018, 6, 2258–2273. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Nie, L.; Zou, P.; Suo, J. Effects of drug and polymer molecular weight on drug release from PLGA-mPEG microspheres. J. Appl. Polym. Sci. 2015, 132, 41431. [Google Scholar] [CrossRef]
- Brauner, B.; Schuster, C.; Wirth, M.; Gabor, F. Trimethoprim-Loaded Microspheres Prepared from Low-Molecular-Weight PLGA as a Potential Drug Delivery System for the Treatment of Urinary Tract Infections. ACS Omega 2020, 5, 9013–9022. [Google Scholar] [CrossRef]
- Abelha, T.F.; Neumann, P.R.; Holthof, J.; Dreiss, C.A.; Alexander, C.; Green, M.; Dailey, L.A. Low molecular weight PEG–PLGA polymers provide a superior matrix for conjugated polymer nanoparticles in terms of physicochemical properties, biocompatibility and optical/photoacoustic performance. J. Mater. Chem. B 2019, 7, 5115–5124. [Google Scholar] [CrossRef]
- Gewirtz, D.A. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharm. 1999, 57, 727–741. [Google Scholar] [CrossRef]
- Yang, F.; Teves, S.S.; Kemp, C.J.; Henikoff, S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2014, 1845, 84–89. [Google Scholar] [CrossRef]
- Varmark, H.; Sparks, C.A.; Nordberg, J.J.; Koppetsch, B.S.; Theurkauf, W.E. DNA damage-induced cell death is enhanced by progression through mitosis. Cell Cycle 2009, 8, 2952–2964. [Google Scholar] [CrossRef][Green Version]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef]
- Kim, T.-K.; Lee, B.-W.; Fujii, F.; Lee, K.-H.; Lee, S.; Park, Y.; Kim, J.K.; Lee, S.-W.; Pack, C.-G. Mitotic chromosomes in live cells characterized using high-speed and label-free optical diffraction tomography. Cells 2019, 8, 1368. [Google Scholar] [CrossRef]
- Kim, J.K.; Kim, J.K.; Pack, C.-G. (Eds.) Advances in Experimental Medicine and Biology; Springer: Singapore, 2021; Volume 1310, pp. 1–30. ISBN 978-981-336-063-1. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VE-SS-VB:VE-VB Ratio (w/w) | CMC (μg/mL) | Stability | Vitamicelle |
|---|---|---|---|
| 0:1 (VE-VB only) | 43.2 | Stable | VT-MC |
| 1:1 | 138.9 | Stable | VT-SS-MC |
| 1:2 | 170.3 | Unstable | N/A |
| 1:0 (VE-SS-VB only) | 201.1 | Unstable | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, N.R.; Lee, S.J.; Chandrasekaran, A.P.; Tyagi, A.; Ramakrishna, S.; Kim, S.-Y.; Kim, D.W.; Pack, C.-G.; Oh, S.J. Smart Vitamin Micelles as Cancer Nanomedicines for Enhanced Intracellular Delivery of Doxorubicin. Int. J. Mol. Sci. 2021, 22, 11298. https://doi.org/10.3390/ijms222011298
Ko NR, Lee SJ, Chandrasekaran AP, Tyagi A, Ramakrishna S, Kim S-Y, Kim DW, Pack C-G, Oh SJ. Smart Vitamin Micelles as Cancer Nanomedicines for Enhanced Intracellular Delivery of Doxorubicin. International Journal of Molecular Sciences. 2021; 22(20):11298. https://doi.org/10.3390/ijms222011298
Chicago/Turabian StyleKo, Na Re, Sang Ju Lee, Arun Pandian Chandrasekaran, Apoorvi Tyagi, Suresh Ramakrishna, Seog-Young Kim, Do Won Kim, Chan-Gi Pack, and Seung Jun Oh. 2021. "Smart Vitamin Micelles as Cancer Nanomedicines for Enhanced Intracellular Delivery of Doxorubicin" International Journal of Molecular Sciences 22, no. 20: 11298. https://doi.org/10.3390/ijms222011298
APA StyleKo, N. R., Lee, S. J., Chandrasekaran, A. P., Tyagi, A., Ramakrishna, S., Kim, S.-Y., Kim, D. W., Pack, C.-G., & Oh, S. J. (2021). Smart Vitamin Micelles as Cancer Nanomedicines for Enhanced Intracellular Delivery of Doxorubicin. International Journal of Molecular Sciences, 22(20), 11298. https://doi.org/10.3390/ijms222011298