
Common Pathogenic Mechanisms in Centronuclear and Myotubular Myopathies and Latest Treatment Advances


Abstract
1. Introduction
2. MTM1 and Myotubular Myopathy
2.1. Disease Presentation and Genetics
2.2. Myotubularin Function and Animal Models for XLMTM
2.2.1. Myotubularin
2.2.2. Mouse Models for XLMTM
2.2.3. Other Animal Models for XLMTM
3. DNM2 and Autosomal Dominant Centronuclear Myopathy
3.1. Disease Presentation and Genetics
3.2. Dynamin 2 Function and Animal Models for ADCNM
3.2.1. Dynamin 2
3.2.2. Mouse Models for DNM2-Related ADCNM
3.2.3. Other Animal Models for DNM2-Related ADCNM
4. BIN1 and Autosomal CNM Forms
4.1. Disease Presentation and Genetics
4.2. Amphiphysin 2 Function and Animal Models for BIN1-Related ARCNM
4.2.1. Amphiphysin 2
4.2.2. Mouse Models for BIN1-Related ARCNM
4.2.3. Other Animal Models for BIN1-Related ARCNM
5. RYR1 and Autosomal Recessive Centronuclear Myopathy
5.1. Disease Presentation and Genetics
5.2. RyR1 Function and Animal Models for RYR1-Related ARCNM
5.2.1. Ryanodine Receptor 1
5.2.2. Mouse Models for RYR1-Related ARCNM
5.2.3. Other Models for RYR1-Related ARCNM
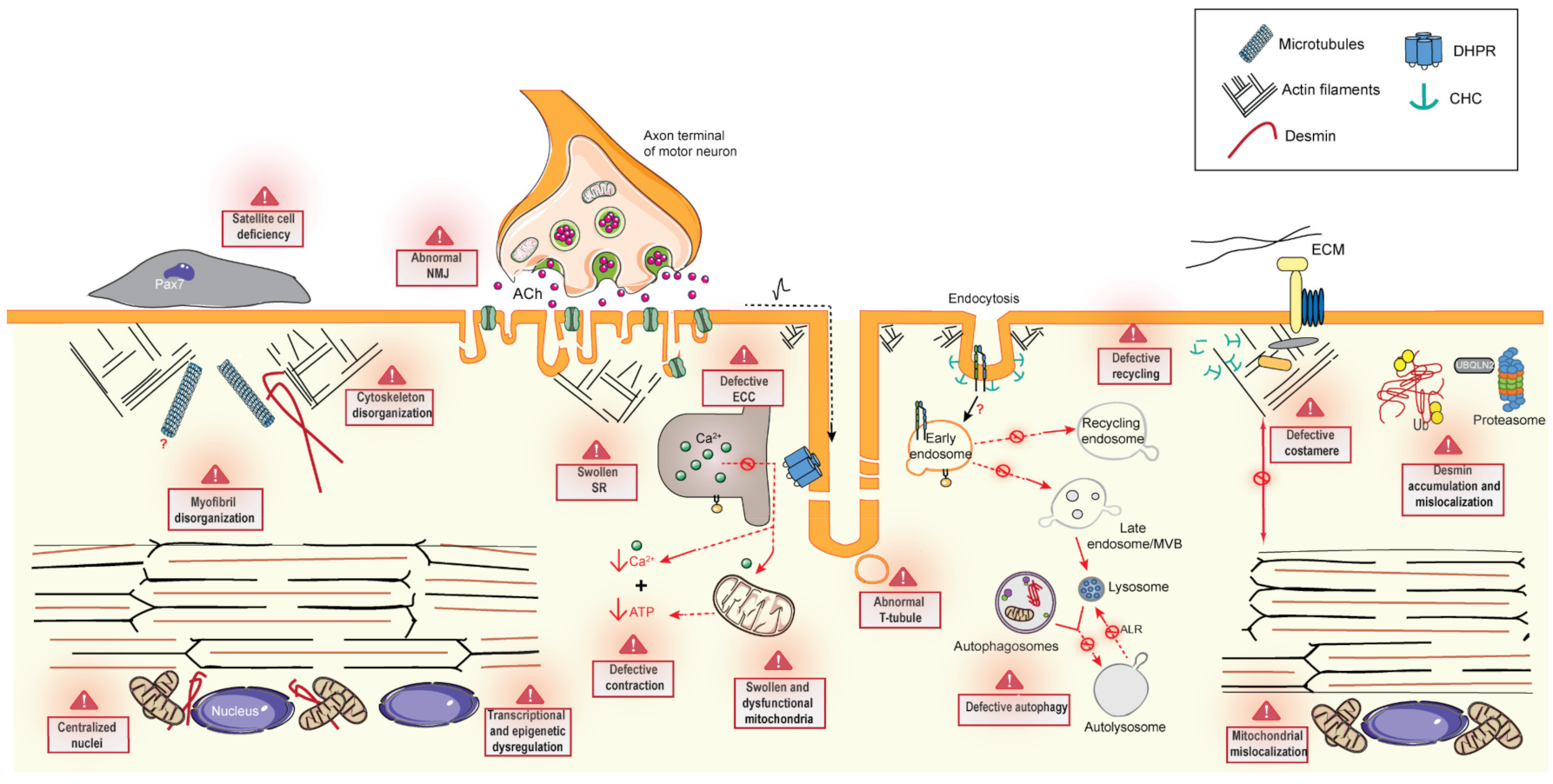
6. Common Pathomechanisms of CNMs
6.1. Excitation–Contraction Coupling and the Triad
6.2. Cytoskeleton Regulation and Organelle Positioning
6.2.1. Cytoskeleton Regulation
6.2.2. Organelle Positioning
6.3. Membrane Trafficking
6.3.1. Endocytosis
6.3.2. Endosome Recycling
6.4. Protein Homeostasis
6.4.1. Autophagy
6.4.2. Ubiquitin-Proteasome System
6.5. The Neuromuscular Junction
6.6. Muscle Regeneration
7. Therapeutic Targets in CNM
7.1. Common Therapeutic Strategies
- Gene silencing: DNM2 reduction or normalization
- Acetylcholinesterase inhibition
7.2. Specific Therapeutic Strategies
- Gene replacement: MTM1 re-expression in XLMTM
- Enzyme replacement: MTM1 delivery in XLMTM
- Gene transfer: MTMR2 expression in XLMTM
- Gene transfer delivery: BIN1 expression in XLMTM
- Allele-specific targeting of DNM2 mutations in DNM2-related ADCNM
- Exon skipping for RYR1-related myopathy
- Cell transplantation or cell therapy in XLMTM
- Myostatin inhibition in XLMTM
- Pharmacologic inhibition of mTORC1 in XLMTM
- Pharmacological inhibition of PI3K in XLMTM
- Drug repurposing: Pharmacologic inhibition of p38 MAPK in RYR1-related AR myopathy
- Drug repurposing: antioxidant therapy in RYR1-related AR myopathy
- Drug repurposing: tamoxifen treatment in XLMTM
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Romero, N.B. Centronuclear myopathies: A widening concept. Neuromuscul. Disord. 2010, 20, 223–228. [Google Scholar] [CrossRef]
- Spiro, A.J.; Shy, G.M.; Gonatas, N.K. Myotubular myopathy. Persistence of fetal muscle in an adolescent boy. Arch. Neurol. 1966, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Laporte, J.; Hu, L.J.; Kretz, C.; Mandel, J.L.; Kioschis, P.; Coy, J.F.; Klauck, S.M.; Poustka, A.; Dahl, N. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat. Genet. 1996, 13, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Bitoun, M.; Maugenre, S.; Jeannet, P.Y.; Lacene, E.; Ferrer, X.; Laforet, P.; Martin, J.J.; Laporte, J.; Lochmuller, H.; Beggs, A.H.; et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat. Genet. 2005, 37, 1207–1209. [Google Scholar] [CrossRef] [PubMed]
- Bohm, J.; Biancalana, V.; Malfatti, E.; Dondaine, N.; Koch, C.; Vasli, N.; Kress, W.; Strittmatter, M.; Taratuto, A.L.; Gonorazky, H.; et al. Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain 2014, 137, 3160–3170. [Google Scholar] [CrossRef]
- Nicot, A.S.; Toussaint, A.; Tosch, V.; Kretz, C.; Wallgren-Pettersson, C.; Iwarsson, E.; Kingston, H.; Garnier, J.M.; Biancalana, V.; Oldfors, A.; et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat. Genet. 2007, 39, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, J.A.; Monnier, N.; Bitoun, M.; Eymard, B.; Ferreiro, A.; Monges, S.; Lubieniecki, F.; Taratuto, A.L.; Laquerriere, A.; Claeys, K.G.; et al. Recessive RYR1 mutations cause unusual congenital myopathy with prominent nuclear internalization and large areas of myofibrillar disorganization. Neuropathol. Appl. Neurobiol. 2011, 37, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Wilmshurst, J.M.; Lillis, S.; Zhou, H.; Pillay, K.; Henderson, H.; Kress, W.; Muller, C.R.; Ndondo, A.; Cloke, V.; Cullup, T.; et al. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann. Neurol. 2010, 68, 717–726. [Google Scholar] [CrossRef]
- Vandersmissen, I.; Biancalana, V.; Servais, L.; Dowling, J.J.; Vander Stichele, G.; Van Rooijen, S.; Thielemans, L. An integrated modelling methodology for estimating the prevalence of centronuclear myopathy. Neuromuscul. Disord. 2018, 28, 766–777. [Google Scholar] [CrossRef]
- Ceyhan-Birsoy, O.; Agrawal, P.B.; Hidalgo, C.; Schmitz-Abe, K.; DeChene, E.T.; Swanson, L.C.; Soemedi, R.; Vasli, N.; Iannaccone, S.T.; Shieh, P.B.; et al. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology 2013, 81, 1205–1214. [Google Scholar] [CrossRef]
- Agrawal, P.B.; Pierson, C.R.; Joshi, M.; Liu, X.; Ravenscroft, G.; Moghadaszadeh, B.; Talabere, T.; Viola, M.; Swanson, L.C.; Haliloglu, G.; et al. SPEG Interacts with Myotubularin, and Its Deficiency Causes Centronuclear Myopathy with Dilated Cardiomyopathy. Am. J. Hum. Genet. 2014, 95, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Schartner, V.; Romero, N.B.; Donkervoort, S.; Treves, S.; Munot, P.; Pierson, T.M.; Dabaj, I.; Malfatti, E.; Zaharieva, I.T.; Zorzato, F.; et al. Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol. 2017, 133, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Vasli, N.; Harris, E.; Karamchandani, J.; Bareke, E.; Majewski, J.; Romero, N.B.; Stojkovic, T.; Barresi, R.; Tasfaout, H.; Charlton, R.; et al. Recessive mutations in the kinase ZAK cause a congenital myopathy with fibre type disproportion. Brain 2017, 140, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Biancalana, V.; Caron, O.; Gallati, S.; Baas, F.; Kress, W.; Novelli, G.; D’Apice, M.R.; Lagier-Tourenne, C.; Buj-Bello, A.; Romero, N.B.; et al. Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Hum. Genet. 2003, 112, 135–142. [Google Scholar] [CrossRef]
- Buj-Bello, A.; Biancalana, V.; Moutou, C.; Laporte, J.; Mandel, J.L. Identification of novel mutations in the MTM1 gene causing severe and mild forms of X-linked myotubular myopathy. Hum. Mutat. 1999, 14, 320–325. [Google Scholar] [CrossRef]
- Herman, G.E.; Kopacz, K.; Zhao, W.; Mills, P.L.; Metzenberg, A.; Das, S. Characterization of mutations in fifty North American patients with X- linked myotubular myopathy. Hum. Mutat. 2002, 19, 114–121. [Google Scholar] [CrossRef]
- Laporte, J.; Biancalana, V.; Tanner, S.M.; Kress, W.; Schneider, V.; Wallgren-Pettersson, C.; Herger, F.; Buj-Bello, A.; Blondeau, F.; Liechti-Gallati, S.; et al. MTM1 mutations in X-linked myotubular myopathy. Hum. Mutat. 2000, 15, 393–409. [Google Scholar] [CrossRef]
- Tsai, T.C.; Horinouchi, H.; Noguchi, S.; Minami, N.; Murayama, K.; Hayashi, Y.K.; Nonaka, I.; Nishino, I. Characterization of MTM1 mutations in 31 Japanese families with myotubular myopathy, including a patient carrying 240 kb deletion in Xq28 without male hypogenitalism. Neuromuscul. Disord. 2005, 15, 245–252. [Google Scholar] [CrossRef]
- Laporte, J.; Kress, W.; Mandel, J.L. Diagnosis of X-linked myotubular myopathy by detection of myotubularin. Ann. Neurol. 2001, 50, 42–46. [Google Scholar] [CrossRef]
- Tosch, V.; Vasli, N.; Kretz, C.; Nicot, A.S.; Gasnier, C.; Dondaine, N.; Oriot, D.; Barth, M.; Puissant, H.; Romero, N.B.; et al. Novel molecular diagnostic approaches for X-linked centronuclear (myotubular) myopathy reveal intronic mutations. Neuromuscul. Disord. 2010, 20, 375–381. [Google Scholar] [CrossRef]
- Jungbluth, H.; Wallgren-Pettersson, C.; Laporte, J. Centronuclear (myotubular) myopathy. Orphanet J. Rare Dis. 2008, 3, 26. [Google Scholar] [CrossRef]
- Romero, N.B.; Bitoun, M. Centronuclear myopathies. Semin. Pediatr. Neurol. 2011, 18, 250–256. [Google Scholar] [CrossRef]
- Herman, G.E.; Finegold, M.; Zhao, W.; de Gouyon, B.; Metzenberg, A. Medical complications in long-term survivors with X-linked myotubular myopathy. J. Pediatr. 1999, 134, 206–214. [Google Scholar] [CrossRef]
- D’Amico, A.; Longo, A.; Fattori, F.; Tosi, M.; Bosco, L.; Testa, M.B.C.; Paglietti, G.; Cherchi, C.; Carlesi, A.; Mizzoni, I.; et al. Hepatobiliary disease in XLMTM. A common comorbidity with potential impact on treatment strategies. Orphanet J. Rare Dis. 2021, 16, 425–431. [Google Scholar] [CrossRef]
- Molera, C.; Sarishvili, T.; Nascimento, A.; Rtskhiladze, I.; Munoz Bartolo, G.; Fernandez Cebrian, S.; Valverde Fernandez, J.; Munoz Cabello, B.; Graham, R.J.; Miller, W.; et al. Intrahepatic Cholestasis Is a Clinically Significant Feature Associated with Natural History of X-Linked Myotubular Myopathy (XLMTM): A Case Series and Biopsy Report. J. Neuromuscul. Dis. 2021. [Google Scholar] [CrossRef]
- Biancalana, V.; Scheidecker, S.; Miguet, M.; Laquerriere, A.; Romero, N.B.; Stojkovic, T.; Abath Neto, O.; Mercier, S.; Voermans, N.; Tanner, L.; et al. Affected female carriers of MTM1 mutations display a wide spectrum of clinical and pathological involvement: Delineating diagnostic clues. Acta Neuropathol. 2017, 134, 889–904. [Google Scholar] [CrossRef]
- Cocanougher, B.T.; Flynn, L.; Yun, P.; Jain, M.; Waite, M.; Vasavada, R.; Wittenbach, J.D.; de Chastonay, S.; Chhibber, S.; Innes, A.M.; et al. Adult MTM1-related myopathy carriers: Classification based on deep phenotyping. Neurology 2019, 93, e1535–e1542. [Google Scholar] [CrossRef]
- Reumers, S.F.I.; Braun, F.; Spillane, J.E.; Bohm, J.; Pennings, M.; Schouten, M.; van der Kooi, A.J.; Foley, A.R.; Bonnemann, C.G.; Kamsteeg, E.J.; et al. Spectrum of Clinical Features in X-Linked Myotubular Myopathy Carriers: An International Questionnaire Study. Neurology 2021, 97, e501–e512. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, C.; Jungbluth, H.; Muntoni, F.; Manzur, A.Y.; Zorzato, F.; Treves, S. Cellular, biochemical and molecular changes in muscles from patients with X-linked myotubular myopathy due to MTM1 mutations. Hum. Mol. Genet. 2017, 26, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, J.A.; Bitoun, M.; Biancalana, V.; Oldfors, A.; Stoltenburg, G.; Claeys, K.G.; Lacene, E.; Brochier, G.; Manere, L.; Laforet, P.; et al. “Necklace” fibers, a new histological marker of late-onset MTM1-related centronuclear myopathy. Acta Neuropathol. 2009, 117, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Hnia, K.; Tronchere, H.; Tomczak, K.K.; Amoasii, L.; Schultz, P.; Beggs, A.H.; Payrastre, B.; Mandel, J.L.; Laporte, J. Myotubularin controls desmin intermediate filament architecture and mitochondrial dynamics in human and mouse skeletal muscle. J. Clin. Investig. 2011, 121, 70–85. [Google Scholar] [CrossRef]
- Shichiji, M.; Biancalana, V.; Fardeau, M.; Hogrel, J.Y.; Osawa, M.; Laporte, J.; Romero, N.B. Extensive morphological and immunohistochemical characterization in myotubular myopathy. Brain Behav. 2013, 3, 476–486. [Google Scholar] [CrossRef]
- Toussaint, A.; Cowling, B.S.; Hnia, K.; Mohr, M.; Oldfors, A.; Schwab, Y.; Yis, U.; Maisonobe, T.; Stojkovic, T.; Wallgren-Pettersson, C.; et al. Defects in amphiphysin 2 (BIN1) and triads in several forms of centronuclear myopathies. Acta Neuropathol. 2011, 121, 253–266. [Google Scholar] [CrossRef]
- Dowling, J.J.; Vreede, A.P.; Low, S.E.; Gibbs, E.M.; Kuwada, J.Y.; Bonnemann, C.G.; Feldman, E.L. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. 2009, 5, e1000372. [Google Scholar] [CrossRef] [PubMed]
- Ketel, K.; Krauss, M.; Nicot, A.S.; Puchkov, D.; Wieffer, M.; Muller, R.; Subramanian, D.; Schultz, C.; Laporte, J.; Haucke, V. A phosphoinositide conversion mechanism for exit from endosomes. Nature 2016, 529, 408–412. [Google Scholar] [CrossRef]
- Ribeiro, I.; Yuan, L.; Tanentzapf, G.; Dowling, J.J.; Kiger, A. Phosphoinositide regulation of integrin trafficking required for muscle attachment and maintenance. PLoS Genet. 2011, 7, e1001295. [Google Scholar] [CrossRef] [PubMed]
- Bitoun, M.; Bevilacqua, J.A.; Eymard, B.; Prudhon, B.; Fardeau, M.; Guicheney, P.; Romero, N.B. A new centronuclear myopathy phenotype due to a novel dynamin 2 mutation. Neurology 2009, 72, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Bitoun, M.; Bevilacqua, J.A.; Prudhon, B.; Maugenre, S.; Taratuto, A.L.; Monges, S.; Lubieniecki, F.; Cances, C.; Uro-Coste, E.; Mayer, M.; et al. Dynamin 2 mutations cause sporadic centronuclear myopathy with neonatal onset. Ann. Neurol. 2007, 62, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, E.M.; Clarke, N.F.; Rose, K.; Oates, E.C.; Webster, R.; Feldman, E.L.; Dowling, J.J. Neuromuscular junction abnormalities in DNM2-related centronuclear myopathy. J. Mol. Med. 2013, 91, 727–737. [Google Scholar] [CrossRef]
- Bohm, J.; Vasli, N.; Maurer, M.; Cowling, B.; Shelton, G.D.; Kress, W.; Toussaint, A.; Prokic, I.; Schara, U.; Anderson, T.J.; et al. Altered Splicing of the BIN1 Muscle-Specific Exon in Humans and Dogs with Highly Progressive Centronuclear Myopathy. PLoS Genet. 2013, 9, e1003430. [Google Scholar] [CrossRef]
- D’Alessandro, M.; Hnia, K.; Gache, V.; Koch, C.; Gavriilidis, C.; Rodriguez, D.; Nicot, A.S.; Romero, N.B.; Schwab, Y.; Gomes, E.; et al. Amphiphysin 2 Orchestrates Nucleus Positioning and Shape by Linking the Nuclear Envelope to the Actin and Microtubule Cytoskeleton. Dev. Cell 2015, 35, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Abath Neto, O.; Moreno, C.A.M.; Malfatti, E.; Donkervoort, S.; Bohm, J.; Guimaraes, J.B.; Foley, A.R.; Mohassel, P.; Dastgir, J.; Bharucha-Goebel, D.X.; et al. Common and variable clinical, histological, and imaging findings of recessive RYR1-related centronuclear myopathy patients. Neuromuscul. Disord. 2017, 27, 975–985. [Google Scholar] [CrossRef]
- Pelletier, L.; Petiot, A.; Brocard, J.; Giannesini, B.; Giovannini, D.; Sanchez, C.; Travard, L.; Chivet, M.; Beaufils, M.; Kutchukian, C.; et al. In vivo RyR1 reduction in muscle triggers a core-like myopathy. Acta Neuropathol. Commun. 2020, 8, 192. [Google Scholar] [CrossRef]
- Rokach, O.; Sekulic-Jablanovic, M.; Voermans, N.; Wilmshurst, J.; Pillay, K.; Heytens, L.; Zhou, H.; Muntoni, F.; Gautel, M.; Nevo, Y.; et al. Epigenetic changes as a common trigger of muscle weakness in congenital myopathies. Hum. Mol. Genet. 2015, 24, 4636–4647. [Google Scholar] [CrossRef]
- De Craene, J.O.; Bertazzi, D.L.; Bar, S.; Friant, S. Phosphoinositides, Major Actors in Membrane Trafficking and Lipid Signaling Pathways. Int. J. Mol. Sci. 2017, 18, 634. [Google Scholar] [CrossRef]
- Vicinanza, M.; D’Angelo, G.; Di Campli, A.; De Matteis, M.A. Function and dysfunction of the PI system in membrane trafficking. EMBO J. 2008, 27, 2457–2470. [Google Scholar] [CrossRef]
- Blondeau, F.; Laporte, J.; Bodin, S.; Superti-Furga, G.; Payrastre, B.; Mandel, J.L. Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum. Mol. Genet. 2000, 9, 2223–2229. [Google Scholar] [CrossRef]
- Taylor, G.S.; Maehama, T.; Dixon, J.E. Inaugural article: Myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc. Natl. Acad. Sci. USA 2000, 97, 8910–8915. [Google Scholar] [CrossRef]
- Tronchere, H.; Laporte, J.; Pendaries, C.; Chaussade, C.; Liaubet, L.; Pirola, L.; Mandel, J.L.; Payrastre, B. Production of phosphatidylinositol 5-phosphate by the phosphoinositide 3-phosphatase myotubularin in mammalian cells. J. Biol. Chem. 2004, 279, 7304–7312. [Google Scholar] [CrossRef] [PubMed]
- Laporte, J.; Blondeau, F.; Buj-Bello, A.; Tentler, D.; Kretz, C.; Dahl, N.; Mandel, J.L. Characterization of the myotubularin dual specificity phosphatase gene family from yeast to human. Hum. Mol. Genet. 1998, 7, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Raess, M.A.; Friant, S.; Cowling, B.S.; Laporte, J. WANTED—Dead or alive: Myotubularins, a large disease-associated protein family. Adv. Biol. Regul. 2017, 63, 49–58. [Google Scholar] [CrossRef]
- Tsujita, K.; Itoh, T.; Ijuin, T.; Yamamoto, A.; Shisheva, A.; Laporte, J.; Takenawa, T. Myotubularin regulates the function of the late endosome through the gram domain-phosphatidylinositol 3,5-bisphosphate interaction. J. Biol. Chem. 2004, 279, 13817–13824. [Google Scholar] [CrossRef]
- Laporte, J.; Blondeau, F.; Gansmuller, A.; Lutz, Y.; Vonesch, J.L.; Mandel, J.L. The PtdIns3P phosphatase myotubularin is a cytoplasmic protein that also localizes to Rac1-inducible plasma membrane ruffles. J. Cell Sci. 2002, 115, 3105–3117. [Google Scholar] [CrossRef]
- Cui, X.; De Vivo, I.; Slany, R.; Miyamoto, A.; Firestein, R.; Cleary, M.L. Association of SET domain and myotubularin-related proteins modulates growth control. Nat. Genet. 1998, 18, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Amburgey, K.; Tsuchiya, E.; de Chastonay, S.; Glueck, M.; Alverez, R.; Nguyen, C.T.; Rutkowski, A.; Hornyak, J.; Beggs, A.H.; Dowling, J.J. A natural history study of X-linked myotubular myopathy. Neurology 2017, 89, 1355–1364. [Google Scholar] [CrossRef]
- Fattori, F.; Maggi, L.; Bruno, C.; Cassandrini, D.; Codemo, V.; Catteruccia, M.; Tasca, G.; Berardinelli, A.; Magri, F.; Pane, M.; et al. Centronuclear myopathies: Genotype-phenotype correlation and frequency of defined genetic forms in an Italian cohort. J. Neurol. 2015, 262, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- McEntagart, M.; Parsons, G.; Buj-Bello, A.; Biancalana, V.; Fenton, I.; Little, M.; Krawczak, M.; Thomas, N.; Herman, G.; Clarke, A.; et al. Genotype-phenotype correlations in X-linked myotubular myopathy. Neuromuscul. Disord. 2002, 12, 939–946. [Google Scholar] [CrossRef]
- Al-Qusairi, L.; Laporte, J. T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skelet. Muscle 2011, 1, 26. [Google Scholar] [CrossRef] [PubMed]
- Buj-Bello, A.; Laugel, V.; Messaddeq, N.; Zahreddine, H.; Laporte, J.; Pellissier, J.F.; Mandel, J.L. The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 15060–15065. [Google Scholar] [CrossRef] [PubMed]
- Pierson, C.R.; Dulin-Smith, A.N.; Durban, A.N.; Marshall, M.L.; Marshall, J.T.; Snyder, A.D.; Naiyer, N.; Gladman, J.T.; Chandler, D.S.; Lawlor, M.W.; et al. Modeling the human MTM1 p.R69C mutation in murine Mtm1 results in exon 4 skipping and a less severe myotubular myopathy phenotype. Hum. Mol. Genet. 2012, 21, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Fetalvero, K.M.; Yu, Y.; Goetschkes, M.; Liang, G.; Valdez, R.A.; Gould, T.; Triantafellow, E.; Bergling, S.; Loureiro, J.; Eash, J.; et al. Defective autophagy and mTORC1 signaling in myotubularin null mice. Mol. Cell Biol. 2013, 33, 98–110. [Google Scholar] [CrossRef]
- Chen, X.; Gao, Y.Q.; Zheng, Y.Y.; Wang, W.; Wang, P.; Liang, J.; Zhao, W.; Tao, T.; Sun, J.; Wei, L.; et al. The intragenic microRNA miR199A1 in the dynamin 2 gene contributes to the pathology of X-linked centronuclear myopathy. J. Biol. Chem. 2020, 295, 8656–8667. [Google Scholar] [CrossRef]
- Sabha, N.; Volpatti, J.R.; Gonorazky, H.; Reifler, A.; Davidson, A.E.; Li, X.; Eltayeb, N.M.; Dall’Armi, C.; Di Paolo, G.; Brooks, S.V.; et al. PIK3C2B inhibition improves function and prolongs survival in myotubular myopathy animal models. J. Clin. Investig. 2016, 126, 3613–3625. [Google Scholar] [CrossRef]
- Beggs, A.H.; Bohm, J.; Snead, E.; Kozlowski, M.; Maurer, M.; Minor, K.; Childers, M.K.; Taylor, S.M.; Hitte, C.; Mickelson, J.R.; et al. MTM1 mutation associated with X-linked myotubular myopathy in Labrador Retrievers. Proc. Natl. Acad. Sci. USA 2010, 107, 14697–14702. [Google Scholar] [CrossRef]
- Shelton, G.D.; Rider, B.E.; Child, G.; Tzannes, S.; Guo, L.T.; Moghadaszadeh, B.; Troiano, E.C.; Haase, B.; Wade, C.M.; Beggs, A.H. X-linked myotubular myopathy in Rottweiler dogs is caused by a missense mutation in Exon 11 of the MTM1 gene. Skelet. Muscle 2015, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Al-Qusairi, L.; Prokic, I.; Amoasii, L.; Kretz, C.; Messaddeq, N.; Mandel, J.L.; Laporte, J. Lack of myotubularin (MTM1) leads to muscle hypotrophy through unbalanced regulation of the autophagy and ubiquitin-proteasome pathways. FASEB J. 2013, 27, 3384–3394. [Google Scholar] [CrossRef] [PubMed]
- Al-Qusairi, L.; Weiss, N.; Toussaint, A.; Berbey, C.; Messaddeq, N.; Kretz, C.; Sanoudou, D.; Beggs, A.H.; Allard, B.; Mandel, J.L.; et al. T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc. Natl. Acad. Sci. USA 2009, 106, 18763–18768. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.J.; Joubert, R.; Low, S.E.; Durban, A.N.; Messaddeq, N.; Li, X.; Dulin-Smith, A.N.; Snyder, A.D.; Marshall, M.L.; Marshall, J.T.; et al. Myotubular myopathy and the neuromuscular junction: A novel therapeutic approach from mouse models. Dis. Model. Mech. 2012, 5, 852–859. [Google Scholar] [CrossRef]
- Gavriilidis, C.; Laredj, L.; Solinhac, R.; Messaddeq, N.; Viaud, J.; Laporte, J.; Sumara, I.; Hnia, K. The MTM1-UBQLN2-HSP complex mediates degradation of misfolded intermediate filaments in skeletal muscle. Nat. Cell Biol. 2018, 20, 198–210. [Google Scholar] [CrossRef]
- Kutchukian, C.; Lo Scrudato, M.; Tourneur, Y.; Poulard, K.; Vignaud, A.; Berthier, C.; Allard, B.; Lawlor, M.W.; Buj-Bello, A.; Jacquemond, V. Phosphatidylinositol 3-kinase inhibition restores Ca2+ release defects and prolongs survival in myotubularin-deficient mice. Proc. Natl. Acad. Sci. USA 2016, 113, 14432–14437. [Google Scholar] [CrossRef]
- Kutchukian, C.; Szentesi, P.; Allard, B.; Buj-Bello, A.; Csernoch, L.; Jacquemond, V. Ca(2+)-induced sarcoplasmic reticulum Ca(2+) release in myotubularin-deficient muscle fibers. Cell Calcium 2019, 80, 91–100. [Google Scholar] [CrossRef]
- Lawlor, M.W.; Alexander, M.S.; Viola, M.G.; Meng, H.; Joubert, R.; Gupta, V.; Motohashi, N.; Manfready, R.A.; Hsu, C.P.; Huang, P.; et al. Myotubularin-deficient myoblasts display increased apoptosis, delayed proliferation, and poor cell engraftment. Am. J. Pathol. 2012, 181, 961–968. [Google Scholar] [CrossRef]
- Lionello, V.M.; Nicot, A.S.; Sartori, M.; Kretz, C.; Kessler, P.; Buono, S.; Djerroud, S.; Messaddeq, N.; Koebel, P.; Prokic, I.; et al. Amphiphysin 2 modulation rescues myotubular myopathy and prevents focal adhesion defects in mice. Sci. Transl. Med. 2019, 11, 1–13. [Google Scholar] [CrossRef]
- Lawlor, M.W.; Viola, M.G.; Meng, H.; Edelstein, R.V.; Liu, F.; Yan, K.; Luna, E.J.; Lerch-Gaggl, A.; Hoffmann, R.G.; Pierson, C.R.; et al. Differential muscle hypertrophy is associated with satellite cell numbers and Akt pathway activation following activin type IIB receptor inhibition in Mtm1 p.R69C mice. Am. J. Pathol. 2014, 184, 1831–1842. [Google Scholar] [CrossRef]
- Ross, J.A.; Tasfaout, H.; Levy, Y.; Morgan, J.; Cowling, B.S.; Laporte, J.; Zanoteli, E.; Romero, N.B.; Lowe, D.A.; Jungbluth, H.; et al. rAAV-related therapy fully rescues myonuclear and myofilament function in X-linked myotubular myopathy. Acta Neuropathol. Commun. 2020, 8, 167. [Google Scholar] [CrossRef]
- Bohm, J.; Biancalana, V.; Dechene, E.T.; Bitoun, M.; Pierson, C.R.; Schaefer, E.; Karasoy, H.; Dempsey, M.A.; Klein, F.; Dondaine, N.; et al. Mutation spectrum in the large GTPase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum. Mutat. 2012, 33, 949–959. [Google Scholar] [CrossRef]
- Chin, Y.H.; Lee, A.; Kan, H.W.; Laiman, J.; Chuang, M.C.; Hsieh, S.T.; Liu, Y.W. Dynamin-2 mutations associated with centronuclear myopathy are hypermorphic and lead to T-tubule fragmentation. Hum. Mol. Genet. 2015, 24, 5542–5554. [Google Scholar] [CrossRef]
- Kenniston, J.A.; Lemmon, M.A. Dynamin GTPase regulation is altered by PH domain mutations found in centronuclear myopathy patients. EMBO J. 2010, 29, 3054–3067. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Barylko, B.; Byers, C.; Ross, J.A.; Jameson, D.M.; Albanesi, J.P. Dynamin 2 mutants linked to centronuclear myopathies form abnormally stable polymers. J. Biol. Chem. 2010, 285, 22753–22757. [Google Scholar] [CrossRef] [PubMed]
- Susman, R.D.; Quijano-Roy, S.; Yang, N.; Webster, R.; Clarke, N.F.; Dowling, J.; Kennerson, M.; Nicholson, G.; Biancalana, V.; Ilkovski, B.; et al. Expanding the clinical, pathological and MRI phenotype of DNM2-related centronuclear myopathy. Neuromuscul. Disord. 2010, 20, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Nicot, A.S.; Carre, S.; Franques, J.; Tranchant, C.; Dondaine, N.; Biancalana, V.; Mandel, J.L.; Laporte, J. Subtle central and peripheral nervous system abnormalities in a family with centronuclear myopathy and a novel dynamin 2 gene mutation. Neuromuscul. Disord. 2007, 17, 955–959. [Google Scholar] [CrossRef]
- Fischer, D.; Herasse, M.; Bitoun, M.; Barragan-Campos, H.M.; Chiras, J.; Laforet, P.; Fardeau, M.; Eymard, B.; Guicheney, P.; Romero, N.B. Characterization of the muscle involvement in dynamin 2-related centronuclear myopathy. Brain 2006, 129, 1463–1469. [Google Scholar] [CrossRef]
- Ferguson, S.M.; De Camilli, P. Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 2012, 13, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Cowling, B.S.; Prokic, I.; Tasfaout, H.; Rabai, A.; Humbert, F.; Rinaldi, B.; Nicot, A.S.; Kretz, C.; Friant, S.; Roux, A.; et al. Amphiphysin (BIN1) negatively regulates dynamin 2 for normal muscle maturation. J. Clin. Investig. 2017, 127, 4477–4487. [Google Scholar] [CrossRef] [PubMed]
- McNiven, M.A.; Cao, H.; Pitts, K.R.; Yoon, Y. The dynamin family of mechanoenzymes: Pinching in new places. Trends Biochem. Sci. 2000, 25, 115–120. [Google Scholar] [CrossRef]
- Warnock, D.E.; Baba, T.; Schmid, S.L. Ubiquitously expressed dynamin-II has a higher intrinsic GTPase activity and a greater propensity for self-assembly than neuronal dynamin-I. Mol. Biol. Cell 1997, 8, 2553–2562. [Google Scholar] [CrossRef]
- Gu, C.; Yaddanapudi, S.; Weins, A.; Osborn, T.; Reiser, J.; Pollak, M.; Hartwig, J.; Sever, S. Direct dynamin-actin interactions regulate the actin cytoskeleton. EMBO J. 2010, 29, 3593–3606. [Google Scholar] [CrossRef]
- Klein, D.E.; Lee, A.; Frank, D.W.; Marks, M.S.; Lemmon, M.A. The pleckstrin homology domains of dynamin isoforms require oligomerization for high affinity phosphoinositide binding. J. Biol. Chem. 1998, 273, 27725–27733. [Google Scholar] [CrossRef]
- Reubold, T.F.; Faelber, K.; Plattner, N.; Posor, Y.; Ketel, K.; Curth, U.; Schlegel, J.; Anand, R.; Manstein, D.J.; Noé, F.; et al. Crystal structure of the dynamin tetramer. Nature 2015, 525, 404. [Google Scholar] [CrossRef]
- Antonny, B.; Burd, C.; De Camilli, P.; Chen, E.; Daumke, O.; Faelber, K.; Ford, M.; Frolov, V.A.; Frost, A.; Hinshaw, J.E.; et al. Membrane fission by dynamin: What we know and what we need to know. EMBO J. 2016, 35, 2270–2284. [Google Scholar] [CrossRef] [PubMed]
- James, N.G.; Digman, M.A.; Ross, J.A.; Barylko, B.; Wang, L.; Li, J.; Chen, Y.; Mueller, J.D.; Gratton, E.; Albanesi, J.P.; et al. A mutation associated with centronuclear myopathy enhances the size and stability of dynamin 2 complexes in cells. Biochim. Biophys. Acta 2014, 1840, 315–321. [Google Scholar] [CrossRef][Green Version]
- Srinivasan, S.; Dharmarajan, V.; Reed, D.K.; Griffin, P.R.; Schmid, S.L. Identification and function of conformational dynamics in the multidomain GTPase dynamin. EMBO J. 2016, 35, 443–457. [Google Scholar] [CrossRef]
- Durieux, A.C.; Vignaud, A.; Prudhon, B.; Viou, M.T.; Beuvin, M.; Vassilopoulos, S.; Fraysse, B.; Ferry, A.; Laine, J.; Romero, N.B.; et al. A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum. Mol. Genet. 2010, 19, 4820–4836. [Google Scholar] [CrossRef]
- Massana Munoz, X.; Kretz, C.; Silva-Rojas, R.; Ochala, J.; Menuet, A.; Romero, N.B.; Cowling, B.S.; Laporte, J. Physiological impact and disease reversion for the severe form of centronuclear myopathy linked to dynamin. JCI Insight 2020, 5, e137899. [Google Scholar] [CrossRef]
- Tinelli, E.; Pereira, J.A.; Suter, U. Muscle-specific function of the centronuclear myopathy and Charcot-Marie-Tooth neuropathy-associated dynamin 2 is required for proper lipid metabolism, mitochondria, muscle fibers, neuromuscular junctions and peripheral nerves. Hum. Mol. Genet. 2013, 22, 4417–4429. [Google Scholar] [CrossRef]
- Cowling, B.S.; Chevremont, T.; Prokic, I.; Kretz, C.; Ferry, A.; Coirault, C.; Koutsopoulos, O.; Laugel, V.; Romero, N.B.; Laporte, J. Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J. Clin. Investig. 2014, 124, 1350–1363. [Google Scholar] [CrossRef]
- Cowling, B.S.; Toussaint, A.; Amoasii, L.; Koebel, P.; Ferry, A.; Davignon, L.; Nishino, I.; Mandel, J.L.; Laporte, J. Increased expression of wild-type or a centronuclear myopathy mutant of dynamin 2 in skeletal muscle of adult mice leads to structural defects and muscle weakness. Am. J. Pathol. 2011, 178, 2224–2235. [Google Scholar] [CrossRef] [PubMed]
- Massana Munoz, X.; Buono, S.; Koebel, P.; Laporte, J.; Cowling, B.S. Different in vivo impacts of dynamin 2 mutations implicated in Charcot-Marie-Tooth neuropathy or centronuclear myopathy. Hum. Mol. Genet. 2019, 28, 4067–4077. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Bezprozvannaya, S.; Shelton, J.M.; Frisard, M.I.; Hulver, M.W.; McMillan, R.P.; Wu, Y.; Voelker, K.A.; Grange, R.W.; Richardson, J.A.; et al. Mice lacking microRNA 133a develop dynamin 2-dependent centronuclear myopathy. J. Clin. Investig. 2011, 121, 3258–3268. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, D.T.; Grigliatti, T.; Williamson, R. Temperature-sensitive mutations in Drosophila melanogaster. VII. A mutation (para-ts) causing reversible adult paralysis. Proc. Natl. Acad. Sci. USA 1971, 68, 890–893. [Google Scholar] [CrossRef] [PubMed]
- van der Bliek, A.M.; Meyerowitz, E.M. Dynamin-like protein encoded by the Drosophila shibire gene associated with vesicular traffic. Nature 1991, 351, 411–414. [Google Scholar] [CrossRef]
- Gibbs, E.M.; Davidson, A.E.; Telfer, W.R.; Feldman, E.L.; Dowling, J.J. The myopathy-causing mutation DNM2-S619L leads to defective tubulation in vitro and in developing zebrafish. Dis. Model. Mech. 2014, 7, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Smith, L.; Volpatti, J.; Fabian, L.; Dowling, J.J. Insights into wild type dynamin 2 and the consequences of DNM2 mutations from transgenic zebrafish. Hum. Mol. Genet. 2019, 28, 4186–4196. [Google Scholar] [CrossRef] [PubMed]
- Bragato, C.; Gaudenzi, G.; Blasevich, F.; Pavesi, G.; Maggi, L.; Giunta, M.; Cotelli, F.; Mora, M. Zebrafish as a Model to Investigate Dynamin 2-Related Diseases. Sci. Rep. 2016, 6, 20466. [Google Scholar] [CrossRef]
- Böhm, J.; Barthélémy, I.; Blot, S.; Tiret, L.; Laporte, J. A dog model for centronuclear myopathy carrying the most common DNM2 mutation. Neuromuscul. Disord. 2020, 30, S75. [Google Scholar] [CrossRef]
- Almeida, C.F.; Bitoun, M.; Vainzof, M. Satellite cells deficiency and defective regeneration in dynamin 2-related centronuclear myopathy. FASEB J. 2021, 35, e21346. [Google Scholar] [CrossRef] [PubMed]
- Durieux, A.C.; Vassilopoulos, S.; Laine, J.; Fraysse, B.; Brinas, L.; Prudhon, B.; Castells, J.; Freyssenet, D.; Bonne, G.; Guicheney, P.; et al. A centronuclear myopathy--dynamin 2 mutation impairs autophagy in mice. Traffic 2012, 13, 869–879. [Google Scholar] [CrossRef]
- Fongy, A.; Falcone, S.; Laine, J.; Prudhon, B.; Martins-Bach, A.; Bitoun, M. Nuclear defects in skeletal muscle from a Dynamin 2-linked centronuclear myopathy mouse model. Sci. Rep. 2019, 9, 1580. [Google Scholar] [CrossRef] [PubMed]
- Franck, A.; Laine, J.; Moulay, G.; Lemerle, E.; Trichet, M.; Gentil, C.; Benkhelifa-Ziyyat, S.; Lacene, E.; Bui, M.T.; Brochier, G.; et al. Clathrin plaques and associated actin anchor intermediate filaments in skeletal muscle. Mol. Biol. Cell 2019, 30, 579–590. [Google Scholar] [CrossRef]
- Fraysse, B.; Guicheney, P.; Bitoun, M. Calcium homeostasis alterations in a mouse model of the Dynamin 2-related centronuclear myopathy. Biol. Open 2016, 5, 1691–1696. [Google Scholar] [CrossRef]
- Gonzalez-Jamett, A.M.; Baez-Matus, X.; Olivares, M.J.; Hinostroza, F.; Guerra-Fernandez, M.J.; Vasquez-Navarrete, J.; Bui, M.T.; Guicheney, P.; Romero, N.B.; Bevilacqua, J.A.; et al. Dynamin-2 mutations linked to Centronuclear Myopathy impair actin-dependent trafficking in muscle cells. Sci. Rep. 2017, 7, 4580. [Google Scholar] [CrossRef] [PubMed]
- Kutchukian, C.; Szentesi, P.; Allard, B.; Trochet, D.; Beuvin, M.; Berthier, C.; Tourneur, Y.; Guicheney, P.; Csernoch, L.; Bitoun, M.; et al. Impaired excitation-contraction coupling in muscle fibres from the dynamin2(R465W) mouse model of centronuclear myopathy. J. Physiol. 2017, 595, 7369–7382. [Google Scholar] [CrossRef] [PubMed]
- Puri, C.; Manni, M.M.; Vicinanza, M.; Hilcenko, C.; Zhu, Y.; Runwal, G.; Stamatakou, E.; Menzies, F.M.; Mamchaoui, K.; Bitoun, M.; et al. A DNM2 Centronuclear Myopathy Mutation Reveals a Link between Recycling Endosome Scission and Autophagy. Dev. Cell 2020, 53, 154–168.e156. [Google Scholar] [CrossRef] [PubMed]
- Rabai, A.; Reisser, L.; Reina-San-Martin, B.; Mamchaoui, K.; Cowling, B.S.; Nicot, A.S.; Laporte, J. Allele-Specific CRISPR/Cas9 Correction of a Heterozygous DNM2 Mutation Rescues Centronuclear Myopathy Cell Phenotypes. Mol. Ther. Nucleic Acids 2019, 16, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Bohm, J.; Yis, U.; Ortac, R.; Cakmakci, H.; Kurul, S.H.; Dirik, E.; Laporte, J. Case report of intrafamilial variability in autosomal recessive centronuclear myopathy associated to a novel BIN1 stop mutation. Orphanet J. Rare Dis. 2010, 5, 35. [Google Scholar] [CrossRef]
- Cabrera-Serrano, M.; Mavillard, F.; Biancalana, V.; Rivas, E.; Morar, B.; Hernandez-Lain, A.; Olive, M.; Muelas, N.; Khan, E.; Carvajal, A.; et al. A Roma founder BIN1 mutation causes a novel phenotype of centronuclear myopathy with rigid spine. Neurology 2018, 91, e339–e348. [Google Scholar] [CrossRef]
- Claeys, K.G.; Maisonobe, T.; Bohm, J.; Laporte, J.; Hezode, M.; Romero, N.B.; Brochier, G.; Bitoun, M.; Carlier, R.Y.; Stojkovic, T. Phenotype of a patient with recessive centronuclear myopathy and a novel BIN1 mutation. Neurology 2010, 74, 519–521. [Google Scholar] [CrossRef]
- Prokic, I.; Cowling, B.S.; Laporte, J. Amphiphysin 2 (BIN1) in physiology and diseases. J. Mol. Med. 2014, 92, 453–463. [Google Scholar] [CrossRef]
- Frost, A.; Unger, V.M.; De Camilli, P. The BAR domain superfamily: Membrane-molding macromolecules. Cell 2009, 137, 191–196. [Google Scholar] [CrossRef]
- Peter, B.J.; Kent, H.M.; Mills, I.G.; Vallis, Y.; Butler, P.J.; Evans, P.R.; McMahon, H.T. BAR domains as sensors of membrane curvature: The amphiphysin BAR structure. Science 2004, 303, 495–499. [Google Scholar] [CrossRef]
- Wechsler-Reya, R.; Sakamuro, D.; Zhang, J.; Duhadaway, J.; Prendergast, G.C. Structural analysis of the human BIN1 gene. Evidence for tissue-specific transcriptional regulation and alternate RNA splicing. J. Biol. Chem. 1997, 272, 31453–31458. [Google Scholar] [CrossRef] [PubMed]
- Fugier, C.; Klein, A.F.; Hammer, C.; Vassilopoulos, S.; Ivarsson, Y.; Toussaint, A.; Tosch, V.; Vignaud, A.; Ferry, A.; Messaddeq, N.; et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med. 2011, 17, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Marcucci, M.; Daniell, L.; Pypaert, M.; Weisz, O.A.; Ochoa, G.C.; Farsad, K.; Wenk, M.R.; De Camilli, P. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science 2002, 297, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Kojima, C.; Hashimoto, A.; Yabuta, I.; Hirose, M.; Hashimoto, S.; Kanaho, Y.; Sumimoto, H.; Ikegami, T.; Sabe, H. Regulation of Bin1 SH3 domain binding by phosphoinositides. Embo J. 2004, 23, 4413–4422. [Google Scholar] [CrossRef]
- Butler, M.H.; David, C.; Ochoa, G.C.; Freyberg, Z.; Daniell, L.; Grabs, D.; Cremona, O.; De Camilli, P. Amphiphysin II (SH3P9; BIN1), a member of the amphiphysin/Rvs family, is concentrated in the cortical cytomatrix of axon initial segments and nodes of Ranvier in brain and around T tubules in skeletal muscle. J. Cell Biol. 1997, 137, 1355–1367. [Google Scholar] [CrossRef]
- Ramjaun, A.R.; Micheva, K.D.; Bouchelet, I.; McPherson, P.S. Identification and characterization of a nerve terminal-enriched amphiphysin isoform. J. Biol. Chem. 1997, 272, 16700–16706. [Google Scholar] [CrossRef]
- Sakamuro, D.; Elliott, K.J.; Wechsler-Reya, R.; Prendergast, G.C. BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nat. Genet. 1996, 14, 69–77. [Google Scholar] [CrossRef]
- Yu, H.; Chen, J.K.; Feng, S.; Dalgarno, D.C.; Brauer, A.W.; Schreiber, S.L. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell 1994, 76, 933–945. [Google Scholar] [CrossRef]
- Falcone, S.; Roman, W.; Hnia, K.; Gache, V.; Didier, N.; Laine, J.; Aurade, F.; Marty, I.; Nishino, I.; Charlet-Berguerand, N.; et al. N-WASP is required for Amphiphysin-2/BIN1-dependent nuclear positioning and triad organization in skeletal muscle and is involved in the pathophysiology of centronuclear myopathy. EMBO Mol. Med. 2014, 6, 1455–1475. [Google Scholar] [CrossRef] [PubMed]
- Royer, B.; Hnia, K.; Gavriilidis, C.; Tronchere, H.; Tosch, V.; Laporte, J. The myotubularin-amphiphysin 2 complex in membrane tubulation and centronuclear myopathies. EMBO Rep. 2013, 14, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Prokic, I.; Cowling, B.S.; Kutchukian, C.; Kretz, C.; Tasfaout, H.; Gache, V.; Hergueux, J.; Wendling, O.; Ferry, A.; Toussaint, A.; et al. Differential physiological role of BIN1 isoforms in skeletal muscle development, function and regeneration. Dis. Model. Mech. 2020, 13, 1–15. [Google Scholar] [CrossRef]
- Muller, A.J.; Baker, J.F.; DuHadaway, J.B.; Ge, K.; Farmer, G.; Donover, P.S.; Meade, R.; Reid, C.; Grzanna, R.; Roach, A.H.; et al. Targeted disruption of the murine Bin1/Amphiphysin II gene does not disable endocytosis but results in embryonic cardiomyopathy with aberrant myofibril formation. Mol. Cell Biol. 2003, 23, 4295–4306. [Google Scholar] [CrossRef]
- Laury-Kleintop, L.D.; Mulgrew, J.R.; Heletz, I.; Nedelcoviciu, R.A.; Chang, M.Y.; Harris, D.M.; Koch, W.J.; Schneider, M.D.; Muller, A.J.; Prendergast, G.C. Cardiac-specific disruption of Bin1 in mice enables a model of stress- and age-associated dilated cardiomyopathy. J. Cell Biochem. 2015, 116, 2541–2551. [Google Scholar] [CrossRef]
- Tjondrokoesoemo, A.; Park, K.H.; Ferrante, C.; Komazaki, S.; Lesniak, S.; Brotto, M.; Ko, J.K.; Zhou, J.; Weisleder, N.; Ma, J. Disrupted membrane structure and intracellular Ca(2)(+) signaling in adult skeletal muscle with acute knockdown of Bin1. PLoS ONE 2011, 6, e25740. [Google Scholar] [CrossRef]
- Silva-Rojas, R.; Nattarayan, V.; Jaque-Fernandez, F.; Gomez-Oca, R.; Menuet, A.; Reiss, D.; Goret, M.; Messaddeq, N.; Lionello, V.M.; Kretz, C.; et al. Mice with muscle-specific deletion of Bin1 recapitulate centronuclear myopathy and acute downregulation of dynamin 2 improves their phenotypes. Mol. Ther. 2021, 30, 1–13. [Google Scholar] [CrossRef]
- Smith, L.L.; Gupta, V.A.; Beggs, A.H. Bridging integrator 1 (Bin1) deficiency in zebrafish results in centronuclear myopathy. Hum. Mol. Genet. 2014, 23, 3566–3578. [Google Scholar] [CrossRef]
- Davies, S.E.; Davies, D.R.; Richards, R.B.; Bruce, W.J. Inherited myopathy in a Great Dane. Aust. Vet. J. 2008, 86, 43–45. [Google Scholar] [CrossRef]
- Lujan Feliu-Pascual, A.; Shelton, G.D.; Targett, M.P.; Long, S.N.; Comerford, E.J.; McMillan, C.; Davies, D.; Rusbridge, C.; Mellor, D.; Chang, K.C.; et al. Inherited myopathy of great Danes. J. Small Anim. Pract. 2006, 47, 249–254. [Google Scholar] [CrossRef] [PubMed]
- McMillan, C.J.; Taylor, S.M.; Shelton, G.D. Inherited myopathy in a young Great Dane. Can. Vet. J. 2006, 47, 891–893. [Google Scholar] [PubMed]
- Kushnir, A.; Wajsberg, B.; Marks, A.R. Ryanodine receptor dysfunction in human disorders. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- des Georges, A.; Clarke, O.B.; Zalk, R.; Yuan, Q.; Condon, K.J.; Grassucci, R.A.; Hendrickson, W.A.; Marks, A.R.; Frank, J. Structural Basis for Gating and Activation of RyR1. Cell 2016, 167, 145–157.e117. [Google Scholar] [CrossRef] [PubMed]
- Van Petegem, F. Ryanodine receptors: Structure and function. J. Biol. Chem. 2012, 287, 31624–31632. [Google Scholar] [CrossRef] [PubMed]
- Lawal, T.A.; Wires, E.S.; Terry, N.L.; Dowling, J.J.; Todd, J.J. Preclinical model systems of ryanodine receptor 1-related myopathies and malignant hyperthermia: A comprehensive scoping review of works published 1990-2019. Orphanet J. Rare Dis. 2020, 15, 113. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.; Garcia-Castaneda, M.; Michelucci, A.; Sabha, N.; Malik, S.; Groom, L.; Wei LaPierre, L.; Dowling, J.J.; Dirksen, R.T. Mouse model of severe recessive RYR1-related myopathy. Hum. Mol. Genet. 2019, 28, 3024–3036. [Google Scholar] [CrossRef]
- Elbaz, M.; Ruiz, A.; Bachmann, C.; Eckhardt, J.; Pelczar, P.; Venturi, E.; Lindsay, C.; Wilson, A.D.; Alhussni, A.; Humberstone, T.; et al. Quantitative RyR1 reduction and loss of calcium sensitivity of RyR1Q1970fsX16+A4329D cause cores and loss of muscle strength. Hum. Mol. Genet. 2019, 28, 2987–2999. [Google Scholar] [CrossRef]
- Takeshima, H.; Iino, M.; Takekura, H.; Nishi, M.; Kuno, J.; Minowa, O.; Takano, H.; Noda, T. Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature 1994, 369, 556–559. [Google Scholar] [CrossRef]
- Takekura, H.; Nishi, M.; Noda, T.; Takeshima, H.; Franzini-Armstrong, C. Abnormal junctions between surface membrane and sarcoplasmic reticulum in skeletal muscle with a mutation targeted to the ryanodine receptor. Proc. Natl. Acad. Sci. USA 1995, 92, 3381–3385. [Google Scholar] [CrossRef]
- Garibaldi, M.; Rendu, J.; Brocard, J.; Lacene, E.; Faure, J.; Brochier, G.; Beuvin, M.; Labasse, C.; Madelaine, A.; Malfatti, E.; et al. ‘Dusty core disease’ (DuCD): Expanding morphological spectrum of RYR1 recessive myopathies. Acta Neuropathol. Commun. 2019, 7, 3. [Google Scholar] [CrossRef]
- Cacheux, M.; Blum, A.; Sebastien, M.; Wozny, A.S.; Brocard, J.; Mamchaoui, K.; Mouly, V.; Roux-Buisson, N.; Rendu, J.; Monnier, N.; et al. Functional Characterization of a Central Core Disease RyR1 Mutation (p.Y4864H) Associated with Quantitative Defect in RyR1 Protein. J. Neuromuscul. Dis. 2015, 2, 421–432. [Google Scholar] [CrossRef]
- Hirata, H.; Watanabe, T.; Hatakeyama, J.; Sprague, S.M.; Saint-Amant, L.; Nagashima, A.; Cui, W.W.; Zhou, W.; Kuwada, J.Y. Zebrafish relatively relaxed mutants have a ryanodine receptor defect, show slow swimming and provide a model of multi-minicore disease. Development 2007, 134, 2771–2781. [Google Scholar] [CrossRef]
- Dowling, J.J.; Arbogast, S.; Hur, J.; Nelson, D.D.; McEvoy, A.; Waugh, T.; Marty, I.; Lunardi, J.; Brooks, S.V.; Kuwada, J.Y.; et al. Oxidative stress and successful antioxidant treatment in models of RYR1-related myopathy. Brain 2012, 135, 1115–1127. [Google Scholar] [CrossRef]
- Chagovetz, A.A.; Klatt Shaw, D.; Ritchie, E.; Hoshijima, K.; Grunwald, D.J. Interactions among ryanodine receptor isotypes contribute to muscle fiber type development and function. Dis. Model. Mech. 2019, 13, dmm038844. [Google Scholar] [CrossRef]
- Djeddi, S.; Reiss, D.; Menuet, A.; Freismuth, S.; de Carvalho Neves, J.; Djerroud, S.; Massana-Munoz, X.; Sosson, A.S.; Kretz, C.; Raffelsberger, W.; et al. Multi-omics comparisons of different forms of centronuclear myopathies and the effects of several therapeutic strategies. Mol. Ther. 2021, 29, 2514–2534. [Google Scholar] [CrossRef] [PubMed]
- Dupont, J.B.; Guo, J.; Renaud-Gabardos, E.; Poulard, K.; Latournerie, V.; Lawlor, M.W.; Grange, R.W.; Gray, J.T.; Buj-Bello, A.; Childers, M.K.; et al. AAV-Mediated Gene Transfer Restores a Normal Muscle Transcriptome in a Canine Model of X-Linked Myotubular Myopathy. Mol. Ther. 2020, 28, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Razzaq, A.; Robinson, I.M.; McMahon, H.T.; Skepper, J.N.; Su, Y.; Zelhof, A.C.; Jackson, A.P.; Gay, N.J.; O’Kane, C.J. Amphiphysin is necessary for organization of the excitation-contraction coupling machinery of muscles, but not for synaptic vesicle endocytosis in Drosophila. Genes Dev. 2001, 15, 2967–2979. [Google Scholar] [CrossRef] [PubMed]
- Takekura, H.; Flucher, B.E.; Franzini-Armstrong, C. Sequential docking, molecular differentiation, and positioning of T-Tubule/SR junctions in developing mouse skeletal muscle. Dev. Biol. 2001, 239, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Sarnat, H.B. Myotubular myopathy: Arrest of morphogenesis of myofibres associated with persistence of fetal vimentin and desmin. Four cases compared with fetal and neonatal muscle. Can. J. Neurol. Sci. 1990, 17, 109–123. [Google Scholar] [CrossRef]
- Amoasii, L.; Hnia, K.; Chicanne, G.; Brech, A.; Cowling, B.S.; Muller, M.M.; Schwab, Y.; Koebel, P.; Ferry, A.; Payrastre, B.; et al. Myotubularin and PtdIns3P remodel the sarcoplasmic reticulum in muscle in vivo. J. Cell Sci. 2013, 126, 1806–1819. [Google Scholar] [CrossRef]
- Dirksen, R.T.; Avila, G. Altered ryanodine receptor function in central core disease: Leaky or uncoupled Ca(2+) release channels? Trends Cardiovasc. Med. 2002, 12, 189–197. [Google Scholar] [CrossRef]
- Klatt Shaw, D.; Gunther, D.; Jurynec, M.J.; Chagovetz, A.A.; Ritchie, E.; Grunwald, D.J. Intracellular Calcium Mobilization Is Required for Sonic Hedgehog Signaling. Dev. Cell 2018, 45, 512–525.e5. [Google Scholar] [CrossRef]
- Fujise, K.; Okubo, M.; Abe, T.; Yamada, H.; Nishino, I.; Noguchi, S.; Takei, K.; Takeda, T. Mutant BIN1-Dynamin 2 complexes dysregulate membrane remodeling in the pathogenesis of centronuclear myopathy. J. Biol. Chem. 2021, 296, 100077. [Google Scholar] [CrossRef]
- Di Biase, V.; Tuluc, P.; Campiglio, M.; Obermair, G.J.; Heine, M.; Flucher, B.E. Surface traffic of dendritic CaV1.2 calcium channels in hippocampal neurons. J. Neurosci. 2011, 31, 13682–13694. [Google Scholar] [CrossRef]
- Yang, T.; Xu, X.; Kernan, T.; Wu, V.; Colecraft, H.M. Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J. Physiol. 2010, 588, 1665–1681. [Google Scholar] [CrossRef]
- Shen, J.; Yu, W.M.; Brotto, M.; Scherman, J.A.; Guo, C.; Stoddard, C.; Nosek, T.M.; Valdivia, H.H.; Qu, C.K. Deficiency of MIP/MTMR14 phosphatase induces a muscle disorder by disrupting Ca(2+) homeostasis. Nat. Cell Biol. 2009, 11, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Tomasevic, N.; Jia, Z.; Russell, A.; Fujii, T.; Hartman, J.J.; Clancy, S.; Wang, M.; Beraud, C.; Wood, K.W.; Sakowicz, R. Differential regulation of WASP and N-WASP by Cdc42, Rac1, Nck, and PI(4,5)P2. Biochemistry 2007, 46, 3494–3502. [Google Scholar] [CrossRef]
- Neukomm, L.J.; Nicot, A.S.; Kinchen, J.M.; Almendinger, J.; Pinto, S.M.; Zeng, S.; Doukoumetzidis, K.; Tronchere, H.; Payrastre, B.; Laporte, J.F.; et al. The phosphoinositide phosphatase MTM-1 regulates apoptotic cell corpse clearance through CED-5-CED-12 in C. elegans. Development 2011, 138, 2003–2014. [Google Scholar] [CrossRef]
- Kessels, M.M.; Engqvist-Goldstein, A.E.; Drubin, D.G.; Qualmann, B. Mammalian Abp1, a signal-responsive F-actin-binding protein, links the actin cytoskeleton to endocytosis via the GTPase dynamin. J. Cell Biol. 2001, 153, 351–366. [Google Scholar] [CrossRef]
- Mooren, O.L.; Kotova, T.I.; Moore, A.J.; Schafer, D.A. Dynamin2 GTPase and cortactin remodel actin filaments. J. Biol. Chem. 2009, 284, 23995–24005. [Google Scholar] [CrossRef] [PubMed]
- Danowski, B.A.; Imanaka-Yoshida, K.; Sanger, J.M.; Sanger, J.W. Costameres are sites of force transmission to the substratum in adult rat cardiomyocytes. J. Cell Biol. 1992, 118, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M. Costameres: The Achilles’ heel of Herculean muscle. J. Biol. Chem. 2003, 278, 13591–13594. [Google Scholar] [CrossRef]
- Vassilopoulos, S.; Gentil, C.; Laine, J.; Buclez, P.O.; Franck, A.; Ferry, A.; Precigout, G.; Roth, R.; Heuser, J.E.; Brodsky, F.M.; et al. Actin scaffolding by clathrin heavy chain is required for skeletal muscle sarcomere organization. J. Cell Biol. 2014, 205, 377–393. [Google Scholar] [CrossRef]
- Brinas, L.; Vassilopoulos, S.; Bonne, G.; Guicheney, P.; Bitoun, M. Role of dynamin 2 in the disassembly of focal adhesions. J. Mol. Med. 2013, 91, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Ezratty, E.J.; Partridge, M.A.; Gundersen, G.G. Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat. Cell Biol. 2005, 7, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Drager, N.M.; Nachman, E.; Winterhoff, M.; Bruhmann, S.; Shah, P.; Katsinelos, T.; Boulant, S.; Teleman, A.A.; Faix, J.; Jahn, T.R. Bin1 directly remodels actin dynamics through its BAR domain. EMBO Rep. 2017, 18, 2051–2066. [Google Scholar] [CrossRef]
- Aspenstrom, P. BAR domain proteins regulate Rho GTPase signaling. Small GTPases 2014, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Cadot, B.; Gache, V.; Gomes, E.R. Moving and positioning the nucleus in skeletal muscle—One step at a time. Nucleus 2015, 6, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Shpetner, H.S.; Vallee, R.B. Identification of dynamin, a novel mechanochemical enzyme that mediates interactions between microtubules. Cell 1989, 59, 421–432. [Google Scholar] [CrossRef]
- Shpetner, H.S.; Vallee, R.B. Dynamin is a GTPase stimulated to high levels of activity by microtubules. Nature 1992, 355, 733–735. [Google Scholar] [CrossRef]
- Tanabe, K.; Takei, K. Dynamic instability of microtubules requires dynamin 2 and is impaired in a Charcot-Marie-Tooth mutant. J. Cell Biol. 2009, 185, 939–948. [Google Scholar] [CrossRef]
- Koutsopoulos, O.S.; Koch, C.; Tosch, V.; Bohm, J.; North, K.N.; Laporte, J. Mild functional differences of dynamin 2 mutations associated to centronuclear myopathy and charcot-marie-tooth peripheral neuropathy. PLoS ONE 2011, 6, e27498. [Google Scholar] [CrossRef]
- Agnetti, G.; Herrmann, H.; Cohen, S. New roles for desmin in the maintenance of muscle homeostasis. FEBS J. 2021, 1–16. [Google Scholar] [CrossRef]
- Roman, W.; Martins, J.P.; Carvalho, F.A.; Voituriez, R.; Abella, J.V.G.; Santos, N.C.; Cadot, B.; Way, M.; Gomes, E.R. Myofibril contraction and crosslinking drive nuclear movement to the periphery of skeletal muscle. Nat. Cell Biol. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Amoasii, L.; Bertazzi, D.L.; Tronchere, H.; Hnia, K.; Chicanne, G.; Rinaldi, B.; Cowling, B.S.; Ferry, A.; Klaholz, B.; Payrastre, B.; et al. Phosphatase-dead myotubularin ameliorates X-linked centronuclear myopathy phenotypes in mice. PLoS Genet. 2012, 8, e1002965. [Google Scholar] [CrossRef] [PubMed]
- Tasfaout, H.; Lionello, V.M.; Kretz, C.; Koebel, P.; Messaddeq, N.; Bitz, D.; Laporte, J.; Cowling, B.S. Single Intramuscular Injection of AAV-shRNA Reduces DNM2 and Prevents Myotubular Myopathy in Mice. Mol. Ther. 2018, 26, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Ralston, E.; Lu, Z.; Biscocho, N.; Soumaka, E.; Mavroidis, M.; Prats, C.; Lomo, T.; Capetanaki, Y.; Ploug, T. Blood vessels and desmin control the positioning of nuclei in skeletal muscle fibers. J. Cell Physiol. 2006, 209, 874–882. [Google Scholar] [CrossRef]
- Shah, S.B.; Davis, J.; Weisleder, N.; Kostavassili, I.; McCulloch, A.D.; Ralston, E.; Capetanaki, Y.; Lieber, R.L. Structural and functional roles of desmin in mouse skeletal muscle during passive deformation. Biophys. J. 2004, 86, 2993–3008. [Google Scholar] [CrossRef]
- Pierson, C.R.; Agrawal, P.B.; Blasko, J.; Beggs, A.H. Myofiber size correlates with MTM1 mutation type and outcome in X-linked myotubular myopathy. Neuromuscul. Disord. 2007, 17, 562–568. [Google Scholar] [CrossRef]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef]
- Fonseca, T.B.; Sanchez-Guerrero, A.; Milosevic, I.; Raimundo, N. Mitochondrial fission requires DRP1 but not dynamins. Nature 2019, 570, E34–E42. [Google Scholar] [CrossRef]
- Kamerkar, S.C.; Kraus, F.; Sharpe, A.J.; Pucadyil, T.J.; Ryan, M.T. Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat. Commun. 2018, 9, 5239. [Google Scholar] [CrossRef]
- Buono, S.; Ross, J.A.; Tasfaout, H.; Levy, Y.; Kretz, C.; Tayefeh, L.; Matson, J.; Guo, S.; Kessler, P.; Monia, B.P.; et al. Reducing dynamin 2 (DNM2) rescues DNM2-related dominant centronuclear myopathy. Proc. Natl. Acad. Sci. USA 2018, 115, 11066–11071. [Google Scholar] [CrossRef]
- Milner, D.J.; Mavroidis, M.; Weisleder, N.; Capetanaki, Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J. Cell Biol. 2000, 150, 1283–1298. [Google Scholar] [CrossRef]
- Kosaka, T.; Ikeda, K. Reversible blockage of membrane retrieval and endocytosis in the garland cell of the temperature-sensitive mutant of Drosophila melanogaster, shibirets1. J. Cell Biol. 1983, 97, 499–507. [Google Scholar] [CrossRef]
- van der Bliek, A.M.; Redelmeier, T.E.; Damke, H.; Tisdale, E.J.; Meyerowitz, E.M.; Schmid, S.L. Mutations in human dynamin block an intermediate stage in coated vesicle formation. J. Cell Biol. 1993, 122, 553–563. [Google Scholar] [CrossRef]
- Grabs, D.; Slepnev, V.I.; Songyang, Z.; David, C.; Lynch, M.; Cantley, L.C.; De Camilli, P. The SH3 domain of amphiphysin binds the proline-rich domain of dynamin at a single site that defines a new SH3 binding consensus sequence. J. Biol. Chem. 1997, 272, 13419–13425. [Google Scholar] [CrossRef]
- Lundmark, R.; Carlsson, S.R. Regulated membrane recruitment of dynamin-2 mediated by sorting nexin 9. J. Biol. Chem. 2004, 279, 42694–42702. [Google Scholar] [CrossRef]
- Zoncu, R.; Perera, R.M.; Sebastian, R.; Nakatsu, F.; Chen, H.; Balla, T.; Ayala, G.; Toomre, D.; De Camilli, P.V. Loss of endocytic clathrin-coated pits upon acute depletion of phosphatidylinositol 4,5-bisphosphate. Proc. Natl. Acad. Sci. USA 2007, 104, 3793–3798. [Google Scholar] [CrossRef] [PubMed]
- Bitoun, M.; Durieux, A.C.; Prudhon, B.; Bevilacqua, J.A.; Herledan, A.; Sakanyan, V.; Urtizberea, A.; Cartier, L.; Romero, N.B.; Guicheney, P. Dynamin 2 mutations associated with human diseases impair clathrin-mediated receptor endocytosis. Hum. Mutat. 2009, 30, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.W.; Lukiyanchuk, V.; Schmid, S.L. Common membrane trafficking defects of disease-associated dynamin 2 mutations. Traffic 2011, 12, 1620–1633. [Google Scholar] [CrossRef] [PubMed]
- Hartig, S.M.; Ishikura, S.; Hicklen, R.S.; Feng, Y.; Blanchard, E.G.; Voelker, K.A.; Pichot, C.S.; Grange, R.W.; Raphael, R.M.; Klip, A.; et al. The F-BAR protein CIP4 promotes GLUT4 endocytosis through bidirectional interactions with N-WASp and Dynamin-2. J. Cell Sci. 2009, 122, 2283–2291. [Google Scholar] [CrossRef] [PubMed]
- Kao, A.W.; Yang, C.; Pessin, J.E. Functional comparison of the role of dynamin 2 splice variants on GLUT-4 endocytosis in 3T3L1 adipocytes. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E825–E831. [Google Scholar] [CrossRef]
- Cao, C.; Backer, J.M.; Laporte, J.; Bedrick, E.J.; Wandinger-Ness, A. Sequential actions of myotubularin lipid phosphatases regulate endosomal PI(3)P and growth factor receptor trafficking. Mol. Biol. Cell 2008, 19, 3334–3346. [Google Scholar] [CrossRef] [PubMed]
- Leprince, C.; Le Scolan, E.; Meunier, B.; Fraisier, V.; Brandon, N.; De Gunzburg, J.; Camonis, J. Sorting nexin 4 and amphiphysin 2, a new partnership between endocytosis and intracellular trafficking. J. Cell Sci. 2003, 116, 1937–1948. [Google Scholar] [CrossRef] [PubMed]
- Posey, A.D., Jr.; Swanson, K.E.; Alvarez, M.G.; Krishnan, S.; Earley, J.U.; Band, H.; Pytel, P.; McNally, E.M.; Demonbreun, A.R. EHD1 mediates vesicle trafficking required for normal muscle growth and transverse tubule development. Dev. Biol. 2014, 387, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Pant, S.; Sharma, M.; Patel, K.; Caplan, S.; Carr, C.M.; Grant, B.D. AMPH-1/Amphiphysin/Bin1 functions with RME-1/Ehd1 in endocytic recycling. Nat. Cell. Biol. 2009, 11, 1399–1410. [Google Scholar] [CrossRef]
- Gonzalez-Jamett, A.M.; Momboisse, F.; Haro-Acuna, V.; Bevilacqua, J.A.; Caviedes, P.; Cardenas, A.M. Dynamin-2 function and dysfunction along the secretory pathway. Front. Endocrinol. 2013, 4, 126. [Google Scholar] [CrossRef] [PubMed]
- van Dam, E.M.; Stoorvogel, W. Dynamin-dependent transferrin receptor recycling by endosome-derived clathrin-coated vesicles. Mol. Biol. Cell 2002, 13, 169–182. [Google Scholar] [CrossRef]
- Castets, P.; Frank, S.; Sinnreich, M.; Ruegg, M.A. “Get the Balance Right”: Pathological Significance of Autophagy Perturbation in Neuromuscular Disorders. J. Neuromuscul. Dis. 2016, 3, 127–155. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Cebollero, E.; van der Vaart, A.; Reggiori, F. Understanding phosphatidylinositol-3-phosphate dynamics during autophagosome biogenesis. Autophagy 2012, 8, 1868–1870. [Google Scholar] [CrossRef] [PubMed]
- Cebollero, E.; van der Vaart, A.; Zhao, M.; Rieter, E.; Klionsky, D.J.; Helms, J.B.; Reggiori, F. Phosphatidylinositol-3-phosphate clearance plays a key role in autophagosome completion. Curr. Biol. 2012, 22, 1545–1553. [Google Scholar] [CrossRef] [PubMed]
- Vergne, I.; Deretic, V. The role of PI3P phosphatases in the regulation of autophagy. FEBS Lett. 2010, 584, 1313–1318. [Google Scholar] [CrossRef] [PubMed]
- Schulze, R.J.; Weller, S.G.; Schroeder, B.; Krueger, E.W.; Chi, S.; Casey, C.A.; McNiven, M.A. Lipid droplet breakdown requires dynamin 2 for vesiculation of autolysosomal tubules in hepatocytes. J. Cell Biol. 2013, 203, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, L. Development of Research into Autophagic Lysosome Reformation. Mol. Cells 2018, 41, 45–49. [Google Scholar] [CrossRef] [PubMed]
- McGrath, M.J.; Eramo, M.J.; Gurung, R.; Sriratana, A.; Gehrig, S.M.; Lynch, G.S.; Lourdes, S.R.; Koentgen, F.; Feeney, S.J.; Lazarou, M.; et al. Defective lysosome reformation during autophagy causes skeletal muscle disease. J. Clin. Investig. 2021, 131, 1–16. [Google Scholar] [CrossRef]
- East, D.A.; Campanella, M. Ca2+ in quality control: An unresolved riddle critical to autophagy and mitophagy. Autophagy 2013, 9, 1710–1719. [Google Scholar] [CrossRef]
- Pietri-Rouxel, F.; Gentil, C.; Vassilopoulos, S.; Baas, D.; Mouisel, E.; Ferry, A.; Vignaud, A.; Hourde, C.; Marty, I.; Schaeffer, L.; et al. DHPR alpha1S subunit controls skeletal muscle mass and morphogenesis. EMBO J. 2010, 29, 643–654. [Google Scholar] [CrossRef]
- Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Solovyova, A.; McWilliams, T.G.; Johnson, C.; Sahu, I.; Varghese, J.; Wood, N.; Wightman, M.; et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016, 166, 935–949. [Google Scholar] [CrossRef]
- Maani, N.; Sabha, N.; Rezai, K.; Ramani, A.; Groom, L.; Eltayeb, N.; Mavandadnejad, F.; Pang, A.; Russo, G.; Brudno, M.; et al. Tamoxifen therapy in a murine model of myotubular myopathy. Nat. Commun. 2018, 9, 4849. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, M.J. Emerging roles of the ubiquitin-proteasome system in the steroid receptor signaling. Arch. Pharm. Res. 2012, 35, 397–407. [Google Scholar] [CrossRef]
- Menconi, M.J.; Wei, W.; Yang, H.; Wray, C.J.; Hasselgren, P.O. Treatment of cultured myotubes with the calcium ionophore A23187 increases proteasome activity via a CaMK II-caspase-calpain-dependent mechanism. Surgery 2004, 136, 135–142. [Google Scholar] [CrossRef]
- Robb, S.A.; Sewry, C.A.; Dowling, J.J.; Feng, L.; Cullup, T.; Lillis, S.; Abbs, S.; Lees, M.M.; Laporte, J.; Manzur, A.Y.; et al. Impaired neuromuscular transmission and response to acetylcholinesterase inhibitors in centronuclear myopathies. Neuromuscul. Disord. 2011, 21, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Ambler, M.W.; Neave, C.; Singer, D.B. X-linked recessive myotubular myopathy: II. Muscle morphology and human myogenesis. Hum. Pathol. 1984, 15, 1107–1120. [Google Scholar] [CrossRef]
- Fidzianska, A.; Goebel, H.H. Aberrant arrested in maturation neuromuscular junctions in centronuclear myopathy. J. Neurol. Sci. 1994, 124, 83–88. [Google Scholar] [CrossRef]
- Liewluck, T.; Lovell, T.L.; Bite, A.V.; Engel, A.G. Sporadic centronuclear myopathy with muscle pseudohypertrophy, neutropenia, and necklace fibers due to a DNM2 mutation. Neuromuscul. Disord. 2010, 20, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.S.; Hsieh, T.L.; Liou, G.G.; Li, T.N.; Lin, H.C.; Chang, C.W.; Wu, H.Y.; Yao, C.K.; Liu, Y.W. Dynamin-2 Regulates Postsynaptic Cytoskeleton Organization and Neuromuscular Junction Development. Cell Rep. 2020, 33, 108310. [Google Scholar] [CrossRef]
- Noguchi, S.; Fujita, M.; Murayama, K.; Kurokawa, R.; Nishino, I. Gene expression analyses in X-linked myotubular myopathy. Neurology 2005, 65, 732–737. [Google Scholar] [CrossRef]
- Gartz Hanson, M.; Niswander, L.A. Rectification of muscle and nerve deficits in paralyzed ryanodine receptor type 1 mutant embryos. Dev. Biol. 2015, 404, 76–87. [Google Scholar] [CrossRef]
- Shakiryanova, D.; Klose, M.K.; Zhou, Y.; Gu, T.; Deitcher, D.L.; Atwood, H.L.; Hewes, R.S.; Levitan, E.S. Presynaptic ryanodine receptor-activated calmodulin kinase II increases vesicle mobility and potentiates neuropeptide release. J. Neurosci. 2007, 27, 7799–7806. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Marcelle, C. Muscle stem cells. Curr. Opin. Cell Biol. 2009, 21, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.F.; Fernandes, S.A.; Ribeiro Junior, A.F.; Keith Okamoto, O.; Vainzof, M. Muscle Satellite Cells: Exploring the Basic Biology to Rule Them. Stem Cells Int. 2016, 2016, 1078686. [Google Scholar] [CrossRef]
- Chazaud, B. Inflammation and Skeletal Muscle Regeneration: Leave It to the Macrophages! Trends Immunol. 2020, 41, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Tasfaout, H.; Cowling, B.S.; Laporte, J. Centronuclear myopathies under attack: A plethora of therapeutic targets. J. Neuromuscul. Dis. 2018, 5, 387–406. [Google Scholar] [CrossRef]
- Tasfaout, H.; Buono, S.; Guo, S.; Kretz, C.; Messaddeq, N.; Booten, S.; Greenlee, S.; Monia, B.P.; Cowling, B.S.; Laporte, J. Antisense oligonucleotide-mediated Dnm2 knockdown prevents and reverts myotubular myopathy in mice. Nat. Commun. 2017, 8, 15661. [Google Scholar] [CrossRef]
- Trochet, D.; Prudhon, B.; Beuvin, M.; Peccate, C.; Lorain, S.; Julien, L.; Benkhelifa-Ziyyat, S.; Rabai, A.; Mamchaoui, K.; Ferry, A.; et al. Allele-specific silencing therapy for Dynamin 2-related dominant centronuclear myopathy. EMBO Mol. Med. 2018, 10, 239–253. [Google Scholar] [CrossRef]
- Childers, M.K.; Joubert, R.; Poulard, K.; Moal, C.; Grange, R.W.; Doering, J.A.; Lawlor, M.W.; Rider, B.E.; Jamet, T.; Daniele, N.; et al. Gene therapy prolongs survival and restores function in murine and canine models of myotubular myopathy. Sci. Transl. Med. 2014, 6, 220ra210. [Google Scholar] [CrossRef]
- Buj-Bello, A.; Fougerousse, F.; Schwab, Y.; Messaddeq, N.; Spehner, D.; Pierson, C.R.; Durand, M.; Kretz, C.; Danos, O.; Douar, A.M.; et al. AAV-mediated intramuscular delivery of myotubularin corrects the myotubular myopathy phenotype in targeted murine muscle and suggests a function in plasma membrane homeostasis. Hum. Mol. Genet. 2008, 17, 2132–2143. [Google Scholar] [CrossRef]
- Mack, D.L.; Poulard, K.; Goddard, M.A.; Latournerie, V.; Snyder, J.M.; Grange, R.W.; Elverman, M.R.; Denard, J.; Veron, P.; Buscara, L.; et al. Systemic AAV8-Mediated Gene Therapy Drives Whole-Body Correction of Myotubular Myopathy in Dogs. Mol. Ther. 2017, 25, 839–854. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, M.W.; Armstrong, D.; Viola, M.G.; Widrick, J.J.; Meng, H.; Grange, R.W.; Childers, M.K.; Hsu, C.P.; O’Callaghan, M.; Pierson, C.R.; et al. Enzyme replacement therapy rescues weakness and improves muscle pathology in mice with X-linked myotubular myopathy. Hum. Mol. Genet. 2013, 22, 1525–1538. [Google Scholar] [CrossRef] [PubMed]
- Daniele, N.; Moal, C.; Julien, L.; Marinello, M.; Jamet, T.; Martin, S.; Vignaud, A.; Lawlor, M.W.; Buj-Bello, A. Intravenous Administration of a MTMR2-Encoding AAV Vector Ameliorates the Phenotype of Myotubular Myopathy in Mice. J. Neuropathol. Exp. Neurol. 2018, 77, 282–295. [Google Scholar] [CrossRef]
- Raess, M.A.; Cowling, B.S.; Bertazzi, D.L.; Kretz, C.; Rinaldi, B.; Xuereb, J.M.; Kessler, P.; Romero, N.B.; Payrastre, B.; Friant, S.; et al. Expression of the neuropathy-associated MTMR2 gene rescues MTM1-associated myopathy. Hum. Mol. Genet. 2017, 26, 3736–3748. [Google Scholar] [CrossRef]
- Rendu, J.; Brocard, J.; Denarier, E.; Monnier, N.; Pietri-Rouxel, F.; Beley, C.; Roux-Buisson, N.; Gilbert-Dussardier, B.; Perez, M.J.; Romero, N.; et al. Exon skipping as a therapeutic strategy applied to an RYR1 mutation with pseudo-exon inclusion causing a severe core myopathy. Hum. Gene Ther. 2013, 24, 702–713. [Google Scholar] [CrossRef]
- Lim, H.J.; Joo, S.; Oh, S.H.; Jackson, J.D.; Eckman, D.M.; Bledsoe, T.M.; Pierson, C.R.; Childers, M.K.; Atala, A.; Yoo, J.J. Syngeneic Myoblast Transplantation Improves Muscle Function in a Murine Model of X-Linked Myotubular Myopathy. Cell Transplant. 2015, 24, 1887–1900. [Google Scholar] [CrossRef]
- Lawlor, M.W.; Read, B.P.; Edelstein, R.; Yang, N.; Pierson, C.R.; Stein, M.J.; Wermer-Colan, A.; Buj-Bello, A.; Lachey, J.L.; Seehra, J.S.; et al. Inhibition of activin receptor type IIB increases strength and lifespan in myotubularin-deficient mice. Am. J. Pathol. 2011, 178, 784–793. [Google Scholar] [CrossRef]
- Volpatti, J.R.; Endo, Y.; Knox, J.; Groom, L.; Brennan, S.; Noche, R.; Zuercher, W.J.; Roy, P.; Dirksen, R.T.; Dowling, J.J. Identification of drug modifiers for RYR1-related myopathy using a multi-species discovery pipeline. Elife 2020, 9, e52946. [Google Scholar] [CrossRef]
- Todd, J.J.; Lawal, T.A.; Witherspoon, J.W.; Chrismer, I.C.; Razaqyar, M.S.; Punjabi, M.; Elliott, J.S.; Tounkara, F.; Kuo, A.; Shelton, M.O.; et al. Randomized controlled trial of N-acetylcysteine therapy for RYR1-related myopathies. Neurology 2020, 94, e1434–e1444. [Google Scholar] [CrossRef]
- Gayi, E.; Neff, L.A.; Massana Munoz, X.; Ismail, H.M.; Sierra, M.; Mercier, T.; Decosterd, L.A.; Laporte, J.; Cowling, B.S.; Dorchies, O.M.; et al. Tamoxifen prolongs survival and alleviates symptoms in mice with fatal X-linked myotubular myopathy. Nat. Commun. 2018, 9, 4848. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Mantegazza, R. Treatment of myasthenia gravis: Focus on pyridostigmine. Clin. Drug Investig. 2011, 31, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Elverman, M.; Goddard, M.A.; Mack, D.; Snyder, J.M.; Lawlor, M.W.; Meng, H.; Beggs, A.H.; Buj-Bello, A.; Poulard, K.; Marsh, A.P.; et al. Long-term effects of systemic gene therapy in a canine model of myotubular myopathy. Muscle Nerve 2017, 56, 943–953. [Google Scholar] [CrossRef]
- Shieh, P.B.; Bönnemann, C.G.; Müller-Felber, W.; Blaschek, A.; Dowling, J.J.; Kuntz, N.L.; Seferian, A.M. Re: “Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy” by Wilson and Flotte. Hum. Gene Ther. 2020, 31, 787. [Google Scholar] [CrossRef]
- Dowling, J.J.; Shieh, P.; Kuntz, N.; Bonnemann, C.; Muller-Felber, W.; Lawlor, M.; Servais, L.; Smith, B.; Noursalehi, M.; Rico, S.; et al. ASPIRO phase 1/2 gene therapy trial in X-linked motubular myopathy (XLMTM): Update on preliminary safety and efficacy findings. Neuromuscul. Disord. 2019, 29, S207. [Google Scholar] [CrossRef]
- Shieh, P.; Kuntz, N.; Dowling, J.J.; Müller-Felber, W.; Blaschek, A.; Bönnemann, C.; Foley, R.; Saade, D.; Seferian, A.; Servais, L.; et al. ASPIRO gene therapy trial in X-linked myotubular myopathy (XLMTM): Update on preliminary efficacy and safety findings. Present. WMS 2021 2021, 31, S47. [Google Scholar] [CrossRef]
- Laporte, J.; Liaubet, L.; Blondeau, F.; Tronchere, H.; Mandel, J.L.; Payrastre, B. Functional redundancy in the myotubularin family. Biochem. Biophys. Res. Commun. 2002, 291, 305–312. [Google Scholar] [CrossRef]
- Berger, A.; Maire, S.; Gaillard, M.C.; Sahel, J.A.; Hantraye, P.; Bemelmans, A.P. mRNA trans-splicing in gene therapy for genetic diseases. Wiley Interdiscip. Rev. RNA 2016, 7, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Trochet, D.; Prudhon, B.; Jollet, A.; Lorain, S.; Bitoun, M. Reprogramming the Dynamin 2 mRNA by Spliceosome-mediated RNA Trans-splicing. Mol. Ther. Nucleic Acids 2016, 5, e362. [Google Scholar] [CrossRef] [PubMed]
- Rybalka, E.; Timpani, C.A.; Debruin, D.A.; Bagaric, R.M.; Campelj, D.G.; Hayes, A. The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle. Cells 2020, 9, 2657. [Google Scholar] [CrossRef]
- Koch, C.; Buono, S.; Menuet, A.; Robe, A.; Djeddi, S.; Kretz, C.; Gomez-Oca, R.; Depla, M.; Monseur, A.; Thielemans, L.; et al. Myostatin: A Circulating Biomarker Correlating with Disease in Myotubular Myopathy Mice and Patients. Mol. Ther. Methods Clin. Dev. 2020, 17, 1178–1189. [Google Scholar] [CrossRef] [PubMed]
- Mariot, V.; Joubert, R.; Hourde, C.; Feasson, L.; Hanna, M.; Muntoni, F.; Maisonobe, T.; Servais, L.; Bogni, C.; Le Panse, R.; et al. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat. Commun. 2017, 8, 1859. [Google Scholar] [CrossRef]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Maryon, E.B.; Coronado, R.; Anderson, P. unc-68 encodes a ryanodine receptor involved in regulating C. elegans body-wall muscle contraction. J. Cell Biol. 1996, 134, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Dorchies, O.M.; Reutenauer-Patte, J.; Dahmane, E.; Ismail, H.M.; Petermann, O.; Patthey- Vuadens, O.; Comyn, S.A.; Gayi, E.; Piacenza, T.; Handa, R.J.; et al. The anticancer drug tamoxifen counteracts the pathology in a mouse model of duchenne muscular dystrophy. Am. J. Pathol. 2013, 182, 485–504. [Google Scholar] [CrossRef] [PubMed]
- Gayi, E.; Neff, L.A.; Ismail, H.M.; Ruegg, U.T.; Scapozza, L.; Dorchies, O.M. Repurposing the Selective Oestrogen Receptor Modulator Tamoxifen for the Treatment of Duchenne Muscular Dystrophy. Chimia 2018, 72, 238–240. [Google Scholar] [CrossRef]
- Nagy, S.; Hafner, P.; Schmidt, S.; Rubino-Nacht, D.; Schadelin, S.; Bieri, O.; Fischer, D. Tamoxifen in Duchenne muscular dystrophy (TAMDMD): Study protocol for a multicenter, randomized, placebo-controlled, double-blind phase 3 trial. Trials 2019, 20, 637. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| CNM Form/ Mutated Gene | Incidence/ Prevalence [9,21] | Severity/ Age of Onset | Clinical Presentations (Common Findings) [1,21] | Muscle Histology [21,22] | Altered Pathways | Ref(s) |
|---|---|---|---|---|---|---|
| XLMTM/ MTM1 | 17 per mln births */ 57% CNM patients | +++/ Neonatal,20% moderate late-onset form | Severe neonatal hypotonia, generalized muscle weakness. 25% male die in the first year of life. In late-onset cases, slowly progressive weakness. Moderate ptosis and ophthalmoplegia. Respiratory failure and swallowing difficulties. Others: dolichocephaly, possible hepatobiliary disease | Fiber hypotrophy, rounded fibers, centralized nuclei, type 1 fiber predominance. Pale peripheral halo lacking of oxidative activity. Female and late-onset cases: necklaces and internalized nuclei. | Abnormal triads Satellite cell deficiency NMJ dysfunction Increased levels of PI(3)P Epigenetic dysregulation Defective autophagy Defective endosome recycling | [3,26,29,30,31,32,33,34,35,36] |
| ADCNM/ DNM2 | 2 per mln births */ 12% CNM patients | + or ++/ Adulthood or adolescence, neonatal | Slowly progressive muscle weakness. Pediatric cases: generalized muscle weakness, hypotonia and breathing difficulties, improving over the time. Ophthalmoplegia and ptosis frequently present. Other: mild peripheral nerve involvement in some cases. | Hypotrophy of type 1 fibers, with some hypertrophic fibers, centralized or internalized nuclei, type 1 fiber predominance. Radiating sarcoplasmic strands, accumulation of oxidative activity around centralized nuclei. | Abnormal triad Defective autophagy NMJ dysfunction | [4,33,37,38,39] |
| ARCNM/ BIN1 | 1 per mln births */ 4% CNM patients | ++/ Childhood | Diffuse muscle weakness from slowly to rapidly progressive and facial weakness. Ophthalmoplegia and ptosis (some cases). | Fiber hypotrophy, rounded fibers, centralized and clustered nuclei, type 1 fiber predominance. Central accumulation of oxidative activity. | Abnormal triads Abnormal nuclear shape | [6,33,40,41] |
| ADCNM/ BIN1 | +/ Adulthood | Mildly progressive muscle weakness without facial involvement | Abnormal triads Defective autophagy | [5] | ||
| ARCNM/ RYR1 | 2 per mln births*/ 12% CNM patients | ++/ Neonatal, childhood | Proximal muscle weakness and hypotonia, improving over time. Ophthalmoplegia with or without ptosis. Possible breathing difficulties. | Fiber hypotrophy with size heterogenicity, predominantly internalized nuclei (some centralized), type 1 fiber predominancy. Cores depleted of oxidative activity with undefined boundaries. | Defective calcium homeostasis Epigenetic dysregulation Oxidative stress | [7,8,42,43,44] |
| Genotype and Specie | Skeletal Muscle Phenotypes | Altered Pathways in Muscle | Ref(s) | |
|---|---|---|---|---|
| Lifespan and Motor Phenotype | Muscle Histology | |||
| Mtm1−/y (Mtm1KO, Mtm1δ4) mouse | Short lifespan (6–8 weeks), decreased body weight and progressive and generalized severe myopathy (from 3 weeks) with breathing difficulties. | Fiber hypotrophy, centralized and internalized nuclei, pale peripheral halo in oxidative staining. | Abnormal triads and defective ECC Deficient autophagy and UPS Dysfunctional mitochondria Abnormal NMJ Satellite cell deficiency and defective regeneration Increased levels of PI(3)P Defective endosome recycling | [31,35,59,63,66,67,68,69,70,71,72,73] |
| Mtm1gt/y (gene trap) mouse | Short lifespan (6 weeks), decreased body weight and progressive and generalized severe myopathy (from 3 weeks). | Fiber hypotrophy, centralized and internalized nuclei. | Defective autophagy mTORC1 overactivation Dysfunctional mitochondria Increased levels of PI(3)P | [61] |
| Mtm1Δ5/y, Mtm1Δ7/y mouse | Short lifespan (6–7 weeks) decreased body weight and generalized severe myopathy (from 3wks). | Fiber hypotrophy, centralized nuclei. | Defective muscular postnatal development and muscle maturation | [62] |
| Mtm1R69C/y (Mtm1-KI) mouse | Reduced lifespan (median 66 weeks), non-progressive mild myopathy (from 8 weeks) and altered breathing. | Fiber hypotrophy, centralized nuclei. Central and peripheral accumulations of oxidative staining. | Abnormal triads NMJ dysfunction Satellite cell deficiency | [60,68,74] |
| mtm1 morphant zebrafish | Impaired motor function from 24 hpf. | Fiber hypotrophy, abnormal nuclei position and shape. | Abnormal triads and ECC Abnormal mitochondria (disrupted cristae) Increased levels of PI(3)P | [34] |
| mtm1-null (mtm1Δ8/Δ8) zebrafish | Reduced lifespan (7–9 dpf) and impaired motor function and phenotypic changes from 3 dpf. Enlarged, globular and fatty liver. | Not specified. | Abnormal triads | [63] |
| MTM1 N155K, Q384P dog | Reduced lifespan, generalized and progressive severe myopathy (from 2–3 month). | Fiber size variability, hypotrophy type 1 fibers, centralized nuclei, type 1 fiber predominance. Subsarcolemmal and central accumulations of oxidative staining. | Abnormal triads Increased levels of PI(3)P Defective autophagy with Q384P mutation Transcriptional dysregulation with N155K | [64,65,75] |
| Genotype and Specie | Skeletal Muscle Phenotypes | Altered Pathways in Muscle | Ref(s) | ||
|---|---|---|---|---|---|
| Lifespan and Motor Phenotypes | Muscle Histology | ||||
| Dnm2R465W/+ mouse (mild adult CNM form) | Homozygous: died at P1 (2% survived 3 weeks). Heterozygous: normal lifespan, normal body weight, progressive moderate myopathy, normal body weight. Progressive moderate myopathy (from 3 weeks). | Fiber hypotrophy, normal nuclei position, central accumulation of oxidative activity. | Defective ECC and calcium homeostasis Defective autophagy Defective actin organization and polymerization Defective GLUT4 trafficking Defective costamere Deficient satellite cells | [93,106,107,108,109,110,111,112,113,114] | |
| Dnm2S619L/+ (Dnm2SL/+) mouse (severe neonatal CNM form) | Homozygous: none survived to P2. Heterozygous: partial mortality from E18.5 to P10. Normal lifespan after P10, decreased body weight, generalized severe myopathy. | Fiber hypotrophy, normal nuclei position, central accumulation of oxidative activity. | Abnormal mitochondria (swollen and disrupted cristae) | [94] | |
| Transient expression of DNM2 | Mouse hDNM2-WT, R465W, S619L | Decreased muscle force 2- and 4-weeks post-injection (higher impact of mutants). | Fiber hypotrophy, centralized and internalized nuclei Central and peripheral accumulations of oxidative activity. | Abnormal triads Abnormal mitochondria (enlarge and disrupted cristae with S619L mutant) Abnormal NMJ | [97,98] |
| Zebrafish hDNM2-WT, S619L | Motor function impaired at 2dpf (higher impact mutant). | Fiber hypotrophy. Disorganized perinuclear material. | Abnormal triads and deficient ECC Swollen organelles Abnormal NMJ | [39,102] | |
| Zebrafish hDNM2-R522H | Motor function impaired Dose-dependent 24 dpf mortality. | Fiber size variability and increased central nuclei. | Abnormal NMJ | [104] | |
| Stable expression of DNM2 | Mouse Tg MCK-rat Dnm2 rat Dnm2-WT | Impaired motor function. | Fiber hypotrophy and centralized nuclei. Central accumulation of oxidative staining and radial strands. | Abnormal T-tubule | [99] |
| Drosophila Tg hDNM2-WT R465W, S619L, A618T | Defects in the eclosion. Defective locomotor activity. | Fiber hypotrophy. | Abnormal T-tubule | [77] | |
| Zebrafish Tg(hDNM2-EGFP) WT,R465W,S619L | Impaired motor function (highest impact for S619L) from 2dpf. | Not specified. | Abnormal triad Abnormal NMJ | [103] | |
| DNM2R465W/+ dog | Mildly progressive weakness. | Fiber size variability, central nuclei. Abnormal mitochondrial positioning: necklace fibers. | Not described yet | [105] | |
| Genotype and Specie | Skeletal Muscle Phenotypes | Altered Pathways in Muscle | Ref (s) | |
|---|---|---|---|---|
| Lifespan and Motor Phenotypes | Muscle Histology | |||
| Bin1−/− mouse (Bin1ex3-6 deletion) | Perinatal death at P0. | Skeletal muscle not examined in detail. | Non investigated | [132] |
| Bin1ex20−/− mouse (Bin1ex20 deletion) | Perinatal death at P0, feeding defect, no difference in body weight. | Centralized nuclei Central collapse of oxidative staining. | Abnormal triads | [84,131] |
| Bin1ex20hsa−/− mouse (Bin1ex20skm−/−, Bin1ex20 skeletal muscle deletion from E9) | Perinatal death at P0, feeding defect. | Not described. | Not described | [84,131] |
| Bin1ex20mck−/− mouse (Bin1ex20 skeletal muscle deletion from E17) | Normal lifespan, decreased body weight (from 4 months) and progressive moderate myopathy (from 8 weeks). | Fiber hypotrophy, normal nuclei position until 8 months. Central accumulation of oxidative staining. | Abnormal triads and defective ECC Defective autophagy | [135] |
| Bin1ex20hsa(i)−/− mouse (Bin1ex20skm(i)−/− deletion in adult skeletal muscle) | No impact on lifespan or body weight and normal motor function. | Muscle histology comparable to WT. | None | [131] |
| Bin1shRNA knock-down in adult muscle | Not described. | Not described. | Abnormal triads and defective ECC | [134] |
| Bin1ex11−/− mouse (splice switching from muscle-specific to ubiquitous isoform) | Normal lifespan and body weight and normal motor function. | Muscle histology comparable to WT. Slight, but significant, increased in mis-localized nuclei. | Satellite cell deficiency and defective muscle regeneration | [131] |
| Bin1ex11 transient skipping by U7-ex11 AS in mouse | Reduced muscle force. | Histology comparable with WT. | Abnormal T-tubule orientation | [122] |
| bin1morphant zebrafish | Defective motor function (from 17–26 hpf). | Mislocalized, rounded and grouped nuclei. | Abnormal triads and defective ECC | [136] |
| BIN1ex11 splice acceptor mutation in dog (decreased BIN1 expression) | Highly progressive myopathy. | Fiber hypotrophy, internalized nuclei. Central accumulation of oxidative staining and some radial strands. | Abnormal triads Defective autophagy | [40] |
| Genotype and Specie | Skeletal Muscle Phenotypes | Altered Pathways in Muscle | Ref (s) | |
|---|---|---|---|---|
| Lifespan and Motor Phenotypes | Muscle Histology | |||
| Ryr1TM/Indel mouse (compound heterozygous mutation) | Short lifespan (6-8 weeks), decreased body weight, progressive and severe myopathy. | Fiber hypotrophy, some centralized nuclei. No cores. | Defective ECC | [144] |
| Ryr1Q1970fsX16+A4329D mouse (compound heterozygous mutation) | Normal lifespan, decreased body weight and moderate myopathy (from 3–4 months)/ | Hypotrophy of type 2 fibers, nuclei position not described. Cores. | Abnormal triads and defective ECC Epigenetic dysregulation | [145] |
| skrm1/skrm1 mouse (Ryr1 null mutation, dyspedic mouse) | Perinatal death, respiratory failure. | Fiber hypotrophy. | Abnormal triads and defective ECC | [146,147] |
| Ryr1-Rec mouse (Ryr1 deletion in adult skeletal muscle) | Progressive myopathy, body weight reduction. | Fiber hypotrophy, normal nuclei position. Accumulation/depletion oxidative staining. “Dusty” cores. | Abnormal triads (multiple triads) and defective ECC Defective autophagy | [43] |
| ryr1bmi340 relatively relaxed zebrafish mutant | Decreased motor function (from 36 hpf) and lethality at 7–15 dpf. | Amorphous cores. | Abnormal triads and defective ECC Oxidative stress | [150,151] |
| ryr1a;ryr1b zebrafish double-mutant | Complete paralysis and lethality at 7 dpf. | Not described. | Defective ECC | [152] |
| Approach | Purpose | CNM Form/Model | Therapeutic Effect Observed | Ref(s) | Status |
|---|---|---|---|---|---|
| DNM2 reduction with ASO | Normalization/reduction of DNM2 | XLMTM/ Mtm1−/y mouse | Prevention and reversion of CNM phenotypes: lifespan prolongation and rescue of body weight, improvement/rescue of muscle mass, histology, force, motor function and histology. | [236] | Phase 1/2 clinical trial initiated in 2020 in MTM1 and DNM2 patients (UNITE-CNM: NCT04033159) |
| AD-CNM/Dnm2 R465W/+ mouse | Rescue of muscle mass and histology. | [191] | |||
| AD-CNM/Dnm2 S619L/+ mouse | Reversion force, motor function, fiber size and histology phenotypes. | [94] | |||
| AR-CNM/ Bin1ex20mck−/− mouse | Improvement of force, fiber size and histology phenotypes. | [135] | |||
| DNM2 reduction with AAV-shRNA | Normalization/reduction of DNM2 | XLMTM/ Mtm1−/y mouse | Improvement of muscle mass, force and rescue of histology. | [184] | Preclinical studies |
| AD-CNM/Dnm2 R465W/+ mouse | Rescue of muscle mass and histology. | [191] | |||
| Specific reduction of Dnm2-R465W transcript | AD-CNM/Dnm2 R465W/+ mouse | Rescue of muscle mass, force and histology. | [237] | Preclinical studies | |
| Acetylcholine esterase inhibitor (AChEI: Edrophonium, pyridostigmine) | NMJ transmission improvement | XLMTM/ Mtm1R69C/y mouse | Improvement of motor function including exercise intolerance and fatigability. | [68] | FDA-approved drug. Use in clinic for myopathies as symptomatic treatment. Alleviated fatigability and improved strength in MTM1 and DNM2 patients [39,224] |
| XLMTM/ mtm1 morphant zebrafish | Fast improvement in motor function. | [224] | |||
| AD-CNM/hDNM2-S619L zebrafish (transient expression) | Rescue of the motor function. | [39] | |||
| MTM1 gene replacement (AAV-MTM1) | MTM1 expression | XLMTM/ Mtm1−/y mouse | Prevention and reversion of CNM phenotypes: lifespan prolongation, rescue of body weight and histology, improvement in muscle mass, force, and motor function. | [238,239] | Phase 1/2 clinical trial initiated in 2017 in MTM1 patients (ASPIRO: NCT03199469). First results showing striking muscular improvements. On hold due to fatal serious adverse events |
| XLMTM/ XLMTM dog | Prolongation of survival, improve/rescue muscle mass, force, histology, and respiratory function | [238,240] | |||
| Myotubularin replacement (3E10Fv-MTM1) | MTM1 re-expression | XLMTM/ Mtm1−/y mouse | Intramuscular injection slightly improves muscle force. No amelioration of muscle histology. | [241] | Preclinical studies by Valerion Therapeutics |
| MTMR2 gene therapy (AAV-MTMR2) | MTMR2 expression | XLMTM/ Mtm1−/y mouse | Lifespan prolongation, improvement in body weight, muscle mass, force and histology. Better rescue with short isoform of MTMR2 (MTMR2-S). | [242,243] | Preclinical studies |
| BIN1 gene therapy (AAV-BIN1) | BIN1 expression | XLMTM/ Mtm1−/y mouse | Lifespan prolongation and rescue/improvement in muscle mass, force and histology. | [73] | Preclinical studies |
| Allele-specific RNA silencing (AAV-shRNA against Dnm2-R465W) | Reduction of mutated DNM2 | AD-CNM/Dnm2 R465W/+ mouse | Rescued in muscle mass, specific force and histology. | [237] | Preclinical studies |
| Exon skipping (ASO to skip pseudo-exon) | Increase RyR1 protein level | AR-CNM/ Fetal primary muscle cells carrying RyR1 patient mutation | Restore of calcium release from SR. | [244] | Cell studies |
| Cell transplantation | Muscle regeneration | XLMTM/ Mtm1R69C/y mouse | Improvement in muscle strength and mass. | [245] | Preclinical studies |
| Myostatin inhibition (ActRIIB-mFC) | Muscle growth signaling pathway | XLMTM/ Mtm1−/y mouse | Slight prolongation of lifespan, increase in muscle weight and transient slight improvement of muscle force. | [246] | Preclinical studies |
| XLMTM/ Mtm1R69C/y mouse | Increased gastrocnemius weight. No other improvements noted. | [74] | |||
| mTORC1 inhibition (AZD8055) | Autophagy activation | XLMTM/ Mtm1gt/y | Restoration of muscle mass. | [61] | Preclinical studies |
| PI3K inhibition (Wortmannin) | Decreased PI(3)P levels | XLMTM/ Mtm1−/y mouse | Lifespan prolongation and improvement of muscle histology. No improvement in body weight. | [63,70] | Preclinical studies |
| XLMTM/ mtm1 morphant zebrafish | Lifespan prolongation and improved motor function. | [63] | |||
| p38MAPK inhibition | Drug repurposing, mechanism of action to be investigated | AR-CNM/ ryr relatively relaxed zebrafish | Not therapeutic improvement of motor function, although positive chemical-genetic interactions. | [247] | Preclinical studies |
| Antioxidant N-acetylcysteine (NAC) | Drug repurposing, decreased oxidative stress | AR-CNM/ ryr relatively relaxed zebrafish | Restore motor function and improve histology. | [151] | Phase 1/2 clinical trial initiated in 2015 and completed in 2018. Neither elevated oxidative stress nor exercise intolerance were rescued [248] |
| Tamoxifen (Estrogen receptor modulator) | Drug repurposing, pathways to be investigated. | XLMTM/ Mtm1−/y mouse | Lifespan prolongation and delay of disease progression: improvement in muscle force, histology and motor function. No rescue of body weight. | [221,249] | Phase 1/2 clinical trial initiated at the end of 2020 in MTM1 patients (TAM4MTM: NCT04915846) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Oca, R.; Cowling, B.S.; Laporte, J. Common Pathogenic Mechanisms in Centronuclear and Myotubular Myopathies and Latest Treatment Advances. Int. J. Mol. Sci. 2021, 22, 11377. https://doi.org/10.3390/ijms222111377
Gómez-Oca R, Cowling BS, Laporte J. Common Pathogenic Mechanisms in Centronuclear and Myotubular Myopathies and Latest Treatment Advances. International Journal of Molecular Sciences. 2021; 22(21):11377. https://doi.org/10.3390/ijms222111377
Chicago/Turabian StyleGómez-Oca, Raquel, Belinda S. Cowling, and Jocelyn Laporte. 2021. "Common Pathogenic Mechanisms in Centronuclear and Myotubular Myopathies and Latest Treatment Advances" International Journal of Molecular Sciences 22, no. 21: 11377. https://doi.org/10.3390/ijms222111377
APA StyleGómez-Oca, R., Cowling, B. S., & Laporte, J. (2021). Common Pathogenic Mechanisms in Centronuclear and Myotubular Myopathies and Latest Treatment Advances. International Journal of Molecular Sciences, 22(21), 11377. https://doi.org/10.3390/ijms222111377