
Monocarbonyl Analogs of Curcumin Based on the Pseudopelletierine Scaffold: Synthesis and Anti-Inflammatory Activity

 , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
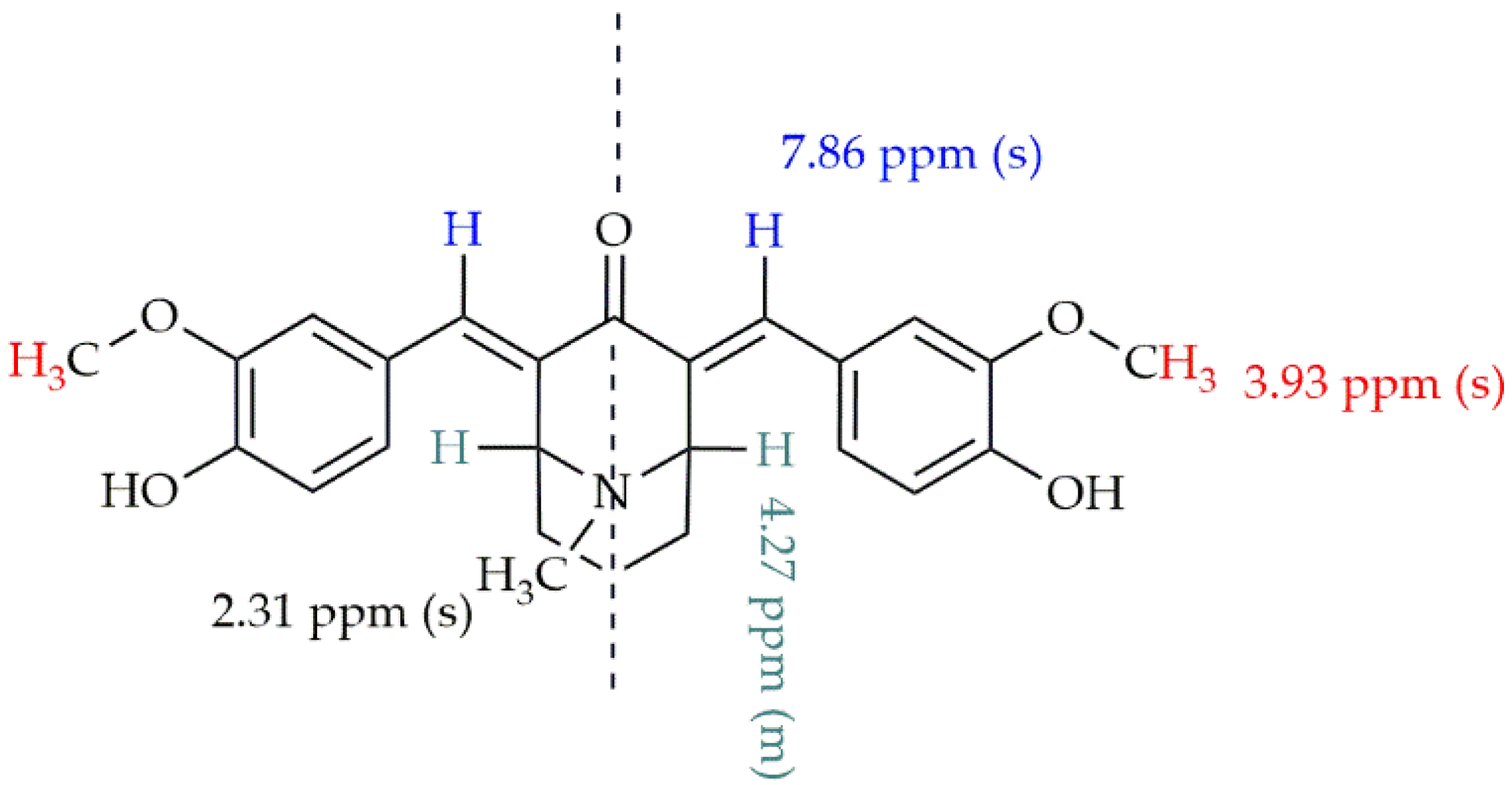
2.1. Synthesis of the Monocarbonyl Analogs of CUR and Their Characteristics
2.2. Limited Cytotoxicity of Monocarbonyl Analogs of CUR, CUR, and Naproxen
2.3. Monocarbonyl Analogs of CUR, CUR, and Naproxen Solutions Possess Low Endotoxin Contamination
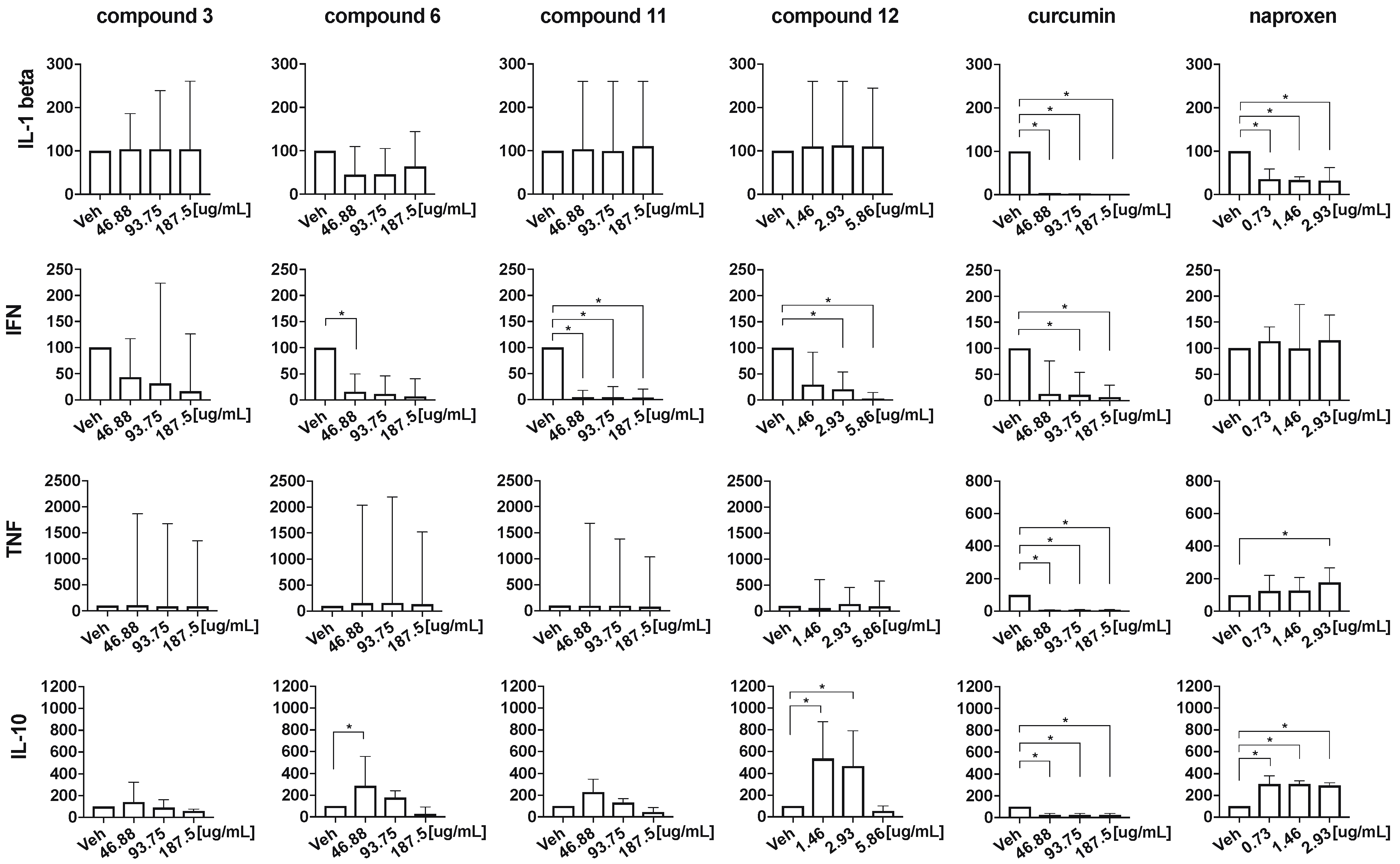
2.4. Anti-Inflammatory Properties of the Monocarbonyl Analogs of CUR
3. Materials and Methods
3.1. Synthesis of the Monocarbonyl Analogs of CUR
3.2. Instruments
3.3. Water Solubility Test
3.4. X-ray Crystallography
3.5. Peripheral Blood Mononuclear Cells Isolation
3.6. Endotoxin Tests: Preparation of the Monocarbonyl Analogs of CUR, CUR, and Naproxen
3.7. MTT Assay
3.8. Endotoxin Measurements
3.9. Cell Stimulation and Supernatant Collection
3.10. Cytokine Assay
3.11. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Z.-B.; Luo, D.-D.; Xie, J.-H.; Xian, Y.-F.; Lai, Z.-Q.; Liu, Y.-H.; Liu, W.-H.; Chen, J.-N.; Lai, X.-P.; Lin, Z.-X.; et al. Curcumin’s Metabolites, Tetrahydrocurcumin and Octahydrocurcumin, Possess Superior Anti-Inflammatory Effects in Vivo Through Suppression of TAK1-NF-ΚB Pathway. Front. Pharmacol. 2018, 9, 1181. [Google Scholar] [CrossRef]
- Amalraj, A.; Pius, A.; Gopi, S.; Gopi, S. Biological Activities of Curcuminoids, Other Biomolecules from Turmeric and Their Derivatives—A Review. J. Trad. Compl. Med. 2017, 7, 205–233. [Google Scholar] [CrossRef] [Green Version]
- Sandur, S.K.; Pandey, M.K.; Sung, B.; Ahn, K.S.; Murakami, A.; Sethi, G.; Limtrakul, P.; Badmaev, V.; Aggarwal, B.B. Curcumin, Demethoxycurcumin, Bisdemethoxycurcumin, Tetrahydrocurcumin and Turmerones Differentially Regulate Anti-Inflammatory and Anti-Proliferative Responses through a ROS-Independent Mechanism. Carcinogenesis 2007, 28, 1765–1773. [Google Scholar] [CrossRef]
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A Provisional Biopharmaceutical Classification of the Top 200 Oral Drug Products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Pan, M.-H.; Cheng, A.-L.; Lin, L.-I.; Ho, Y.-S.; Hsieh, C.-Y.; Lin, J.-K. Stability of Curcumin in Buffer Solutions and Characterization of Its Degradation Products. J. Pharm. Biomed. Anal. 1997, 15, 1867–1876. [Google Scholar] [CrossRef]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a Free Radical Method to Evaluate Antioxidant Activity. LWT-Food Sci. Tech. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Feng, J.-Y.; Liu, Z.-Q. Phenolic and Enolic Hydroxyl Groups in Curcumin: Which Plays the Major Role in Scavenging Radicals? J. Agric. Food Chem. 2009, 57, 11041–11046. [Google Scholar] [CrossRef]
- Somparn, P.; Phisalaphong, C.; Nakornchai, S.; Unchern, S.; Morales, N.P. Comparative Antioxidant Activities of Curcumin and Its Demethoxy and Hydrogenated Derivatives. Biol. Pharm. Bull. 2007, 30, 74–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, Y.; Kawakishi, S.; Osawa, T. Involvement of the β-Diketone Moiety in the Antioxidative Mechanism of Tetrahydrocurcumin. Biochem. Pharm. 1996, 52, 519–525. [Google Scholar] [CrossRef]
- Kiuchi, F.; Goto, Y.; Sugimoto, N.; Akao, N.; Kondo, K.; Tsuda, Y. Studies on Crude Drugs Effective on Visceral Larva Migrans. Part XVI. Nematocidal Activity of Turmeric: Synergistic Action of Curcuminoids. Chem. Pharm. Bull. 1993, 41, 1640–1643. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Narain, U.; Tripathi, S.; Misra, K. Syntheses of Curcumin Bioconjugates and Study of Their Antibacterial Activities against β-Lactamase-Producing Microorganisms. Bioconjugate Chem. 2001, 12, 464–469. [Google Scholar] [CrossRef]
- Pal, C.; Bandyopadhyay, U. Redox-Active Antiparasitic Drugs. Antioxid. Redox Sign. 2012, 17, 555–582. [Google Scholar] [CrossRef]
- Pérez-Arriaga, L.; Mendoza-Magaña, M.L.; Cortés-Zárate, R.; Corona-Rivera, A.; Bobadilla-Morales, L.; Troyo-Sanromán, R.; Ramírez-Herrera, M.A. Cytotoxic Effect of Curcumin on Giardia Lamblia Trophozoites. Acta Trop. 2006, 98, 152–161. [Google Scholar] [CrossRef]
- Jeong, G.-S.; Oh, G.-S.; Pae, H.-O.; Jeong, S.-O.; Kim, Y.-C.; Shin, M.-K.; Seo, B.Y.; Han, S.Y.; Lee, H.S.; Jeong, J.-G.; et al. Comparative Effects of Curcuminoids on Endothelial Heme Oxygenase-1 Expression: Ortho-Methoxy Groups Are Essential to Enhance Heme Oxygenase Activity and Protection. Exp. Mol. Med. 2006, 38, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Menon, V.P.; Sudheer, A.R. Antioxidant and anti-inflammatory properties of curcumin. In The Molecular Targets and Therapeutic Uses of Curcumin in Health and Disease; Aggarwal, B.B., Surh, Y.-J., Shishodia, S., Eds.; Advances in experimental medicine and biology; Springer: Boston, MA, USA, 2007; Volume 595, pp. 105–125. ISBN 978-0-387-46400-8. [Google Scholar]
- Lee, S.-Y.; Cho, S.-S.; Li, Y.; Bae, C.-S.; Park, K.M.; Park, D.-H. Anti-Inflammatory Effect of Curcuma Longa and Allium Hookeri Co-Treatment via NF-ΚB and COX-2 Pathways. Sci. Rep. 2020, 10, 5718. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Russo, S.; Gerace, D.; Alonci, A.; Musolino, C. Anticancer Activity of Curcumin and Its Analogues: Preclinical and Clinical Studies. Cancer Investig. 2017, 35, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Ki, K.J.; Jae, Y.C.; Rhee, M.H.; Hong, S.; Kwon, M.; Kim, S.H.; Kang, S.Y. Neuroprotective Effect of Curcumin Is Mainly Mediated by Blockade of Microglial Cell Activation. Pharmazie 2007, 937–942. [Google Scholar] [CrossRef]
- Bairwa, K.; Grover, J.; Kania, M.; Jachak, S.M. Recent Developments in Chemistry and Biology of Curcumin Analogues. RSC Adv. 2014, 4, 13946. [Google Scholar] [CrossRef]
- Rossi, J.-F.; Lu, Z.Y.; Massart, C.; Levon, K. Dynamic Immune/Inflammation Precision Medicine: The Good and the Bad Inflammation in Infection and Cancer. Front. Immunol. 2021, 12, 595722. [Google Scholar] [CrossRef] [PubMed]
- Black, P.H. Stress and the Inflammatory Response: A Review of Neurogenic Inflammation. Brain Behav. Immun. 2002, 16, 622–653. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Alvestrand, A. Review Articles: Inflammation in End-stage Renal Disease: Sources, Consequences, and Therapy. Semin. Dial. 2002, 15, 329–337. [Google Scholar] [CrossRef]
- Jacob, A.; Wu, R.; Zhou, M.; Wang, P. Mechanism of the Anti-Inflammatory Effect of Curcumin: PPAR-γ Activation. PPAR Res. 2007, 2007, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, A.M.; Cui, X.; Wu, R.; Dong, W.; Zhou, M.; Hu, M.; Simms, H.H.; Wang, P. The Anti-Inflammatory Effect of Curcumin in an Experimental Model of Sepsis Is Mediated by up-Regulation of Peroxisome Proliferator-Activated Receptor-Γ*. Crit. Care Med. 2006, 34, 1874–1882. [Google Scholar] [CrossRef] [PubMed]
- Saja, K.; Babu, M.S.; Karunagaran, D.; Sudhakaran, P.R. Anti-Inflammatory Effect of Curcumin Involves Downregulation of MMP-9 in Blood Mononuclear Cells. Intern. Immunopharm. 2007, 7, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Ung, V.Y.L.; Foshaug, R.R.; MacFarlane, S.M.; Churchill, T.A.; Doyle, J.S.G.; Sydora, B.C.; Fedorak, R.N. Oral Administration of Curcumin Emulsified in Carboxymethyl Cellulose Has a Potent Anti-Inflammatory Effect in the IL-10 Gene-Deficient Mouse Model of IBD. Dig. Dis. Sci. 2010, 55, 1272–1277. [Google Scholar] [CrossRef]
- Funes, S.C.; Rios, M.; Fernández-Fierro, A.; Covián, C.; Bueno, S.M.; Riedel, C.A.; Mackern-Oberti, J.P.; Kalergis, A.M. Naturally Derived Heme-Oxygenase 1 Inducers and Their Therapeutic Application to Immune-Mediated Diseases. Front. Immunol. 2020, 11, 1467. [Google Scholar] [CrossRef]
- Yadav, B.; Taurin, S.; Rosengren, R.J.; Schumacher, M.; Diederich, M.; Somers-Edgar, T.J.; Larsen, L. Synthesis and Cytotoxic Potential of Heterocyclic Cyclohexanone Analogues of Curcumin. Bioorganic Med. Chem. 2010, 18, 6701–6707. [Google Scholar] [CrossRef] [PubMed]
- Revalde, J.L.; Li, Y.; Hawkins, B.C.; Rosengren, R.J.; Paxton, J.W. Heterocyclic Cyclohexanone Monocarbonyl Analogs of Curcumin Can Inhibit the Activity of ATP-Binding Cassette Transporters in Cancer Multidrug Resistance. Biochem. Pharm. 2015, 93, 305–317. [Google Scholar] [CrossRef]
- Huber, I.; Rozmer, Z.; Gyöngyi, Z.; Budán, F.; Horváth, P.; Kiss, E.; Perjési, P. Structure Activity Relationship Analysis of Antiproliferative Cyclic C5-Curcuminoids without DNA Binding: Design, Synthesis, Lipophilicity and Biological Activity. J. Mol. Struc. 2020, 1206, 127661. [Google Scholar] [CrossRef]
- Qiu, C.; Hu, Y.; Wu, K.; Yang, K.; Wang, N.; Ma, Y.; Zhu, H.; Zhang, Y.; Zhou, Y.; Chen, C.; et al. Synthesis and Biological Evaluation of Allylated Mono-Carbonyl Analogues of Curcumin (MACs) as Anti-Cancer Agents for Cholangiocarcinoma. Bioorganic Med. Chem. Lett. 2016, 26, 5971–5976. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Y.; Ren, L.; Huang, Y.; Cai, Y.; Weng, Q.; Shen, X.; Li, X.; Liang, G.; Wang, Y. Evaluation of a Curcumin Analog as an Anti-Cancer Agent Inducing ER Stress-Mediated Apoptosis in Non-Small Cell Lung Cancer Cells. BMC Cancer 2013, 13, 494. [Google Scholar] [CrossRef] [Green Version]
- Weng, Q.; Fu, L.; Chen, G.; Hui, J.; Song, J.; Feng, J.; Shi, D.; Cai, Y.; Ji, J.; Liang, G. Design, Synthesis, and Anticancer Evaluation of Long-Chain Alkoxylated Mono-Carbonyl Analogues of Curcumin. Eur. J. Med. Chem. 2015, 103, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Shao, L.; Wang, Y.; Zhao, C.; Chu, Y.; Xiao, J.; Zhao, Y.; Li, X.; Yang, S. Exploration and Synthesis of Curcumin Analogues with Improved Structural Stability Both in Vitro and in Vivo as Cytotoxic Agents. Bioorganic Med. Chem. 2009, 17, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liu, Z.; Liang, G. Promising Curcumin-Based Drug Design: Mono-Carbonyl Analogues of Curcumin (MACs). Curr. Pharm. Design 2013, 19, 2114–2135. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid Dispersions as Strategy to Improve Oral Bioavailability of Poor Water Soluble Drugs. Drug Disc. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef]
- Armstrong, D.W.; Han, S.M.; Han, Y.I. Separation of Optical Isomers of Scopolamine, Cocaine, Homatropine, and Atropine. Anal. Biochem. 1987, 167, 261–264. [Google Scholar] [CrossRef]
- Huang, J.-P.; Wang, Y.-J.; Tian, T.; Wang, L.; Yan, Y.; Huang, S.-X. Tropane Alkaloid Biosynthesis: A Centennial Review. Nat. Prod. Rep. 2021. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of Curcumin: Problems and Promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Manju, S.; Sreenivasan, K. Conjugation of Curcumin onto Hyaluronic Acid Enhances Its Aqueous Solubility and Stability. J. Coll. Int. Sci. 2011, 359, 318–325. [Google Scholar] [CrossRef]
- Chen, L.; Bai, G.; Yang, S.; Yang, R.; Zhao, G.; Xu, C.; Leung, W. Encapsulation of Curcumin in Recombinant Human H-Chain Ferritin Increases Its Water-Solubility and Stability. Food Res. Int. 2014, 62, 1147–1153. [Google Scholar] [CrossRef]
- Ghosh, M.; Singh, A.T.K.; Xu, W.; Sulchek, T.; Gordon, L.I.; Ryan, R.O. Curcumin Nanodisks: Formulation and Characterization. Nanomed. Nanotechn. Biol. Med. 2011, 7, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Yen, F.-L.; Wu, T.-H.; Tzeng, C.-W.; Lin, L.-T.; Lin, C.-C. Curcumin Nanoparticles Improve the Physicochemical Properties of Curcumin and Effectively Enhance Its Antioxidant and Antihepatoma Activities. J. Agric. Food Chem. 2010, 58, 7376–7382. [Google Scholar] [CrossRef]
- Mohanty, C.; Das, M.; Sahoo, S.K. Emerging Role of Nanocarriers to Increase the Solubility and Bioavailability of Curcumin. Expert Opin. Drug Deliv. 2012, 9, 1347–1364. [Google Scholar] [CrossRef]
- Bhawana; Basniwal, R.K.; Buttar, H.S.; Jain, V.K.; Jain, N. Curcumin Nanoparticles: Preparation, Characterization, and Antimicrobial Study. J. Agric. Food Chem. 2011, 59, 2056–2061. [Google Scholar] [CrossRef]
- Takahashi, M.; Uechi, S.; Takara, K.; Asikin, Y.; Wada, K. Evaluation of an Oral Carrier System in Rats: Bioavailability and Antioxidant Properties of Liposome-Encapsulated Curcumin. J. Agric. Food Chem. 2009, 57, 9141–9146. [Google Scholar] [CrossRef] [PubMed]
- Margaritova Zaharieva, M.; Dimitrov Kroumov, A.; Dimitrova, L.; Tsvetkova, I.; Trochopoulos, A.; Mihaylov Konstantinov, S.; Reinhold Berger, M.; Momchilova, M.; Yoncheva, K.; Miladinov Najdenski, H. Micellar Curcumin Improves the Antibacterial Activity of the Alkylphosphocholines Erufosine and Miltefosine against Pathogenic Staphyloccocus Aureus Strains. Biotechnol. Biotechnol. Equip. 2019, 33, 38–53. [Google Scholar] [CrossRef] [Green Version]
- Paramera, E.I.; Konteles, S.J.; Karathanos, V.T. Stability and Release Properties of Curcumin Encapsulated in Saccharomyces Cerevisiae, β-Cyclodextrin and Modified Starch. Food Chem. 2011, 125, 913–922. [Google Scholar] [CrossRef]
- Pan, K.; Zhong, Q.; Baek, S.J. Enhanced Dispersibility and Bioactivity of Curcumin by Encapsulation in Casein Nanocapsules. J. Agric. Food Chem. 2013, 61, 6036–6043. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, K.; Hamada, H. Enzymatic Synthesis and Anti-Allergic Activities of Curcumin Oligosaccharides. Biochem. Insights 2010, 3, BCI–S2768. [Google Scholar] [CrossRef]
- Gong, F.; Chen, D.; Teng, X.; Ge, J.; Ning, X.; Shen, Y.; Li, J.; Wang, S. Curcumin-Loaded Blood-Stable Polymeric Micelles for Enhancing Therapeutic Effect on Erythroleukemia. Mol. Pharm. 2017, 14, 2585–2594. [Google Scholar] [CrossRef]
- Choudhury, A.; Raja, S.; Mahapatra, S.; Nagabhushanam, K.; Majeed, M. Synthesis and Evaluation of the Anti-Oxidant Capacity of Curcumin Glucuronides, the Major Curcumin Metabolites. Antioxidants 2015, 4, 750–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joe, B.; Vijaykumar, M.; Lokesh, B.R. Biological Properties of Curcumin-Cellular and Molecular Mechanisms of Action. Crit. Rev. Food Sci. Nutr. 2004, 44, 97–111. [Google Scholar] [CrossRef]
- Huminiecki, L.; Horbańczuk, J.; Atanasov, A.G. The Functional Genomic Studies of Curcumin. Semin. Cancer Biol. 2017, 46, 107–118. [Google Scholar] [CrossRef]
- Carolina Alves, R.; Perosa Fernandes, R.; Fonseca-Santos, B.; Damiani Victorelli, F.; Chorilli, M. A Critical Review of the Properties and Analytical Methods for the Determination of Curcumin in Biological and Pharmaceutical Matrices. Crit. Rev. Anal. Chem. 2019, 49, 138–149. [Google Scholar] [CrossRef]
- Noureddin, S.A.; El-Shishtawy, R.M.; Al-Footy, K.O. Curcumin Analogues and Their Hybrid Molecules as Multifunctional Drugs. Eur. J. Med. Chem. 2019, 182, 111631. [Google Scholar] [CrossRef]
- Rahman, A.F.M.M.; Ali, R.; Jahng, Y.; Kadi, A.A. A Facile Solvent Free Claisen-Schmidt Reaction: Synthesis of α,A′-Bis-(Substituted-Benzylidene)Cycloalkanones and α,A′-Bis-(Substituted-Alkylidene)Cycloalkanones. Molecules 2012, 17, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Wolosewicz, K.; Podgorska, K.; Rutkowska, E.; Lazny, R. Synthesis of Dicarbonyl Curcumin Analogues Containing the Tropane Scaffold: Synthesis of Dicarbonyl Curcumin Analogues Containing the Tropane Scaffold. Eur. J. Org. Chem. 2019, 2019, 4662–4674. [Google Scholar] [CrossRef]
- Medley, J.W.; Movassaghi, M. Robinson’s Landmark Synthesis of Tropinone. Chem. Commun. 2013, 49, 10775. [Google Scholar] [CrossRef]
- Afewerki, S.; Wang, J.-X.; Liao, W.-W.; Córdova, A. The Chemical Synthesis and Applications of Tropane Alkaloids. In The Alkaloids: Chemistry and Biology; Elsevier: Amsterdam, The Netherlands, 2019; Volume 81, pp. 151–233. ISBN 978-0-12-817151-6. [Google Scholar]
- Kamarul Zaman, M.A.; Mohamad Azzeme, A. Plant Toxins: Alkaloids and Their Toxicities. GSC Biol. Pharm. Sci. 2018, 6, 021–029. [Google Scholar] [CrossRef] [Green Version]
- Velema, W.A.; Kietrys, A.M.; Kool, E.T. RNA Control by Photoreversible Acylation. J. Am. Chem. Soc. 2018, 140, 3491–3495. [Google Scholar] [CrossRef] [PubMed]
- Pace, R.D.; Regmi, Y. The Finkelstein Reaction: Quantitative Reaction Kinetics of an SN2 Reaction Using Nonaqueous Conductivity. J. Chem. Educ. 2006, 83, 1344. [Google Scholar] [CrossRef]
- Petrov, O.; Ivanova, Y.; Gerova, M. SOCl2/EtOH: Catalytic System for Synthesis of Chalcones. Cat. Comm. 2008, 9, 315–316. [Google Scholar] [CrossRef]
- Das, B.; Thirupathi, P.; Mahender, I.; Reddy, K.R. Convenient and Facile Cross-Aldol Condensation Catalyzed by Molecular Iodine: An Efficient Synthesis of α,A′-Bis(Substituted-Benzylidene) Cycloalkanones. J. Mol. Cat. A Chem. 2006, 247, 182–185. [Google Scholar] [CrossRef]
- Li, J.; Su, W.; Li, N. Copper Triflate–Catalyzed Cross-Aldol Condensation: A Facile Synthesis of α,A′-Bis(Substituted Benzylidene) Cycloalkanones. Synt. Comm. 2005, 35, 3037–3043. [Google Scholar] [CrossRef]
- Motiur Rahman, A.F.M.; Jeong, B.-S.; Kim, D.H.; Park, J.K.; Lee, E.S.; Jahng, Y. A Facile Synthesis of α,A′-Bis(Substituted-Benzylidene)-Cycloalkanones and Substituted-Benzylidene Heteroaromatics: Utility of NaOAc as a Catalyst for Aldol-Type Reaction. Tetrahedron 2007, 63, 2426–2431. [Google Scholar] [CrossRef]
- George, H.; Roth, H.J. Photoisomerisierung und Cyclo-1,2-Addition α, β-ungesättigter Cyclanone. Tetrahedron Lett. 1971, 12, 4057–4060. [Google Scholar] [CrossRef]
- Chen, Z.; Izenwasser, S.; Katz, J.L.; Zhu, N.; Klein, C.L.; Trudell, M.L. Synthesis and Dopamine Transporter Affinity of 2-(Methoxycarbonyl)-9-Methyl-3-Phenyl-9-Azabicyclo[3.3.1]Nonane Derivatives. J. Med. Chem. 1996, 39, 4744–4749. [Google Scholar] [CrossRef]
- Nodzewska, A.; Bokina, A.; Romanowska, K.; Lazny, R. Environmentally Benign Diastereoselective Synthesis of Granatane and Tropane Aldol Derivatives. RSC Adv. 2014, 4, 29668. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A Found Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawelski, D.; Walewska, A.; Ksiezak, S.; Sredzinski, D.; Radziwon, P.; Moniuszko, M.; Gandusekar, R.; Eljaszewicz, A.; Lazny, R.; Brzezinski, K.; et al. Monocarbonyl Analogs of Curcumin Based on the Pseudopelletierine Scaffold: Synthesis and Anti-Inflammatory Activity. Int. J. Mol. Sci. 2021, 22, 11384. https://doi.org/10.3390/ijms222111384
Pawelski D, Walewska A, Ksiezak S, Sredzinski D, Radziwon P, Moniuszko M, Gandusekar R, Eljaszewicz A, Lazny R, Brzezinski K, et al. Monocarbonyl Analogs of Curcumin Based on the Pseudopelletierine Scaffold: Synthesis and Anti-Inflammatory Activity. International Journal of Molecular Sciences. 2021; 22(21):11384. https://doi.org/10.3390/ijms222111384
Chicago/Turabian StylePawelski, Damian, Alicja Walewska, Sylwia Ksiezak, Dariusz Sredzinski, Piotr Radziwon, Marcin Moniuszko, Ramesh Gandusekar, Andrzej Eljaszewicz, Ryszard Lazny, Krzysztof Brzezinski, and et al. 2021. "Monocarbonyl Analogs of Curcumin Based on the Pseudopelletierine Scaffold: Synthesis and Anti-Inflammatory Activity" International Journal of Molecular Sciences 22, no. 21: 11384. https://doi.org/10.3390/ijms222111384
APA StylePawelski, D., Walewska, A., Ksiezak, S., Sredzinski, D., Radziwon, P., Moniuszko, M., Gandusekar, R., Eljaszewicz, A., Lazny, R., Brzezinski, K., & Plonska-Brzezinska, M. E. (2021). Monocarbonyl Analogs of Curcumin Based on the Pseudopelletierine Scaffold: Synthesis and Anti-Inflammatory Activity. International Journal of Molecular Sciences, 22(21), 11384. https://doi.org/10.3390/ijms222111384