
Nod-like Receptors: Critical Intracellular Sensors for Host Protection and Cell Death in Microbial and Parasitic Infections

Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inflammatory Diseases | Affected Organs | Dysregulated NLRs Family | References |
|---|---|---|---|
| Thyroiditis | Thyroid gland | Over-expression and activation of NLRC4, NLRP1, AIM2 and NLRP3 inflammasome | [25,26,27] |
| Type 1 Diabetes | Pancreas | Over-expression NLRP1, NOD1/2, CIITA and NLRP3 | [26,28,29] |
| Inflammatory bowel diseases (IBD: Ulcerative colitis and Crohn’s disease) | Gastrointestinal | NOD1, NOD2, NLRP3 and NLRP1 | [26,30,31,32,33] |
| Celiac diseases | Small intestine | Enhanced expression of NLRP3 and CIITA, NLRP6 | [26,29,34] |
| Autoimmune hepatitis | Liver | Hyperactivation of NLRP3 and deficiency of NLRX1 | [2,26,35,36] |
| Arthritis | Joints | Excessive expression of NLRP3, NLRP2, CIITA, NOD2, NLRC5 and NLRP12 (beneficial), and NLRP9 and NLRP11 | [25,26,37,38,39,40] |
| Systemic Lupus Erythematous (SLE) | Multiple organs such as Kidney, Lung and CNS | Over-expression of NOD2, NLRP3, SNPs in CIITA, NLRP1 and NLRX1 | [26,41,42,43,44] |
| Vitiligo | Skin | Increased expression/activation of NLRP1 and NLRP3 | [26,44,45,46] |
| Psoriasis | Epidermal layer (from the limbs to eyelids) | Enhanced expression of NOD2, PYCARD, CARD6, CARD14, NLRP3, NLRP1 and IFI16 | [47,48,49,50] |
| Multiple Sclerosis | CNS: brain, spinal cord and optic nerves. | Over-activation of NOD1, NOD2 and NLRP1. Mutation in CIITA, NLRP3 and regulatory role of NLRP12, NLRC3 and NLRX1 | [51,52,53,54] |
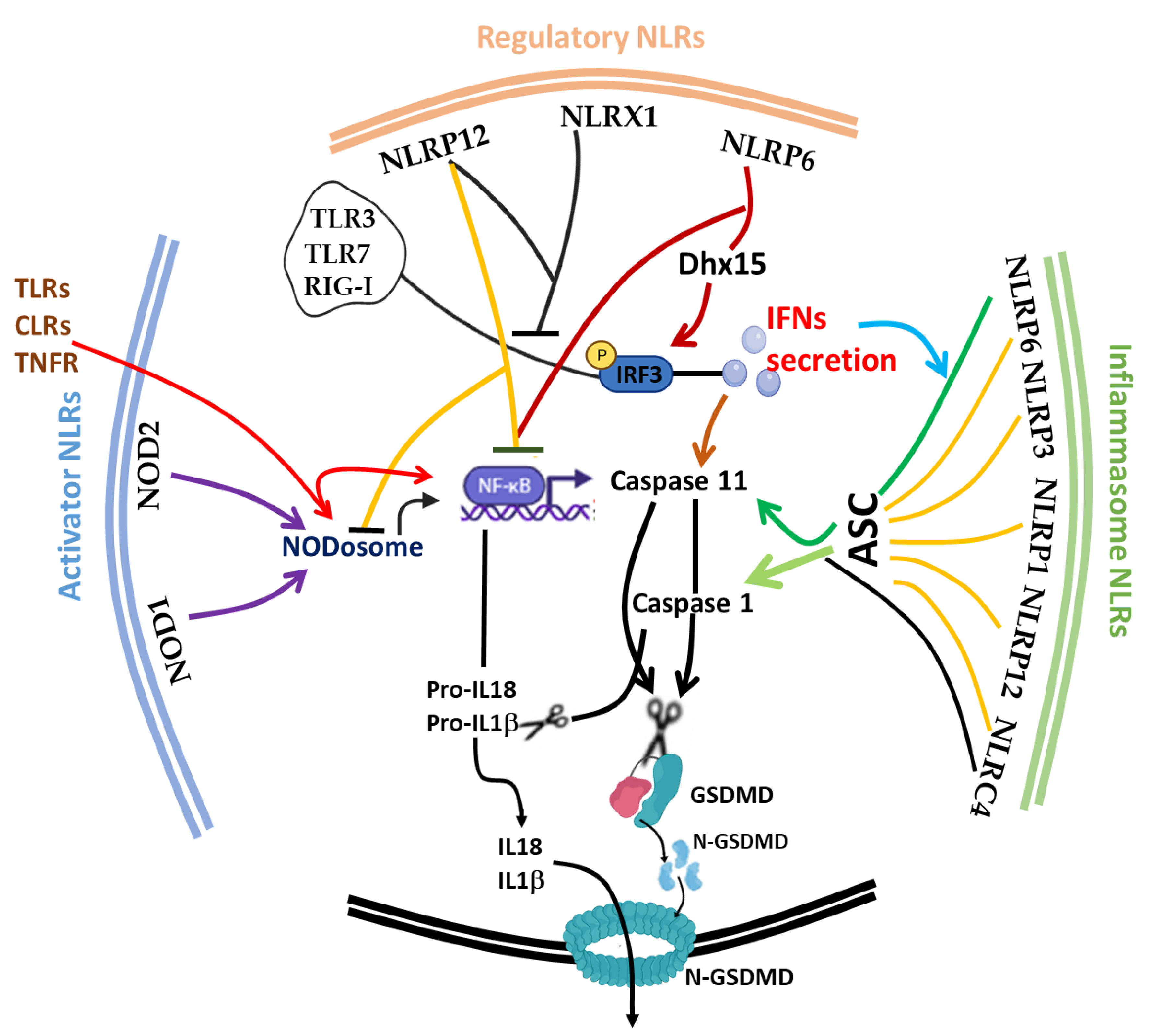
2. Structure, Function and Classification of NOD-like Receptors
- (i)
- Transcriptional trans-activators: members are CIITA and NLRC5, located at the promoter region of major histocompatibility complex (MHC) II and MHC-I, respectively [57,58]. CIITA, via its unique acidic domain (AD), is recruited to the MHC enhanceosome complex as a non-DNA binding activator to promote the transcriptional activation of MHC-II [57]. NLRC5, on the other hand, is conserved in vertebrates, with high expression in immune cells and mucosal epithelia. NLRC5 controls basal MHC I gene expression and is inducible by IFNγ stimulation to trans-activate the MHC-I gene in lymphoid and epithelial cells by reducing H3K27me3 in the MHC-I promoter [59,60,61]. The cis-regulatory elements of the promoter of the MHC-I gene interact with NLRC5 through a distinct transcriptional factor to recruit modifiers and initiate the MHC I enhanceosome transcriptional complex [60]. Nlrc5−/− mice exhibit impaired CTL responses, and NLRC5-null target cells are not efficiently cleared by CTLs, while the immunogenic melanoma was able to activate CD8+ T cells by restoring the expression of NLRC5 alongside with CD80 [56].
- (ii)
- Activators of NF-κB and MAPK pathways: The NLRs in this category are the first described NOD-like receptors; NOD1 (NLRC1) and NOD2 (NLRC2) recognize bacterial peptidoglycans components D-glutamyl-meso-diaminopimelic acid (iE-DAP) and muramyl dipeptide (MDP), respectively, to induce the production of proinflammatory cytokines (TNF, IL-6 and IL-1β) via NF-κB and MAPK pathways [9,62]. Peptidoglycans (PGN), a constituent of the bacterial cell wall, have been demonstrated to trigger NOD2 activity sufficiently [63]. A gain-of-function mutation in the NOD2 gene is associated with autoinflammatory condition Blau syndrome [64] and sarcoidosis [65], whereas its loss of function is linked to Crohn’s disease [66]. Intracellular ligands recognition through the LRR domain induced the formation of a protein complex at their CARD N-terminal with an adaptor protein RIP2 for the subsequent phosphorylation of NF-κB. Moreover, the downstream signal may also go through the activation of MAPKs, including the p38, extracellular signal-regulated protein kinase (ERK) and c-Jun N-terminal kinase (JNK) pathways [10].
- (iii)
- Inflammasome activators of the NLRs family: members of this family are involved in the inflammasome complex, i.e., an intracellular multi-protein complex that leads to the activation of caspase 1, required for the maturation of IL-1β and IL-18, as well as the amplification of NF-κB, JNK and p38 MAPK-signaling pathways [67,68]. These NLRs conduct robust secretion of proinflammatory cytokines and chemokines to the distressing site and also mediate pyroptotic cell death [57]. NLRC4 directly recruits pro-caspase 1 while intermediary cytosolic-resident adaptor apoptosis-associated speck-like protein containing a CARD (ASC) is required for PYD-carrying NLRs (such as NLRP3 and NLRP12) for the recruitment of pro-caspase 1. Aside from caspase 1, NALP1 has also been shown to participate in the activation of caspase 5 [69].
- (iv)
- Members of the NLR family are essentially involved in the negative regulation of the proinflammatory responses by limiting IL-1β secretion, NF-κB and type I IFN (IFN-I) signaling. These include NLRP2, NLRC3, NLRP4, NLRP6, NLRP7, NLRP10, NLRP12 and NLRX1 [57]. Although the majority of NLR members here exhibit both inflammasome activation and inhibitory functions under varying conditions, ASC is recruited for the exhibition of inflammasome function; meanwhile, different endogenous proteins are engaged for the inhibitory function. Information about the NOD-like receptors in this category is scant, and their therapeutic potentials are remarkable.
3. NOD1 and NOD2 in Sensing PAMP/DAMP and Inflammatory Responses
4. NLRP3 and NLRC4 Inflammasomes
5. NLRP6 and NLRP10 in Regulatory and Inflammatory Responses
6. NOD-Like Receptors in the Regulation of IFN-I and Proinflammatory Responses
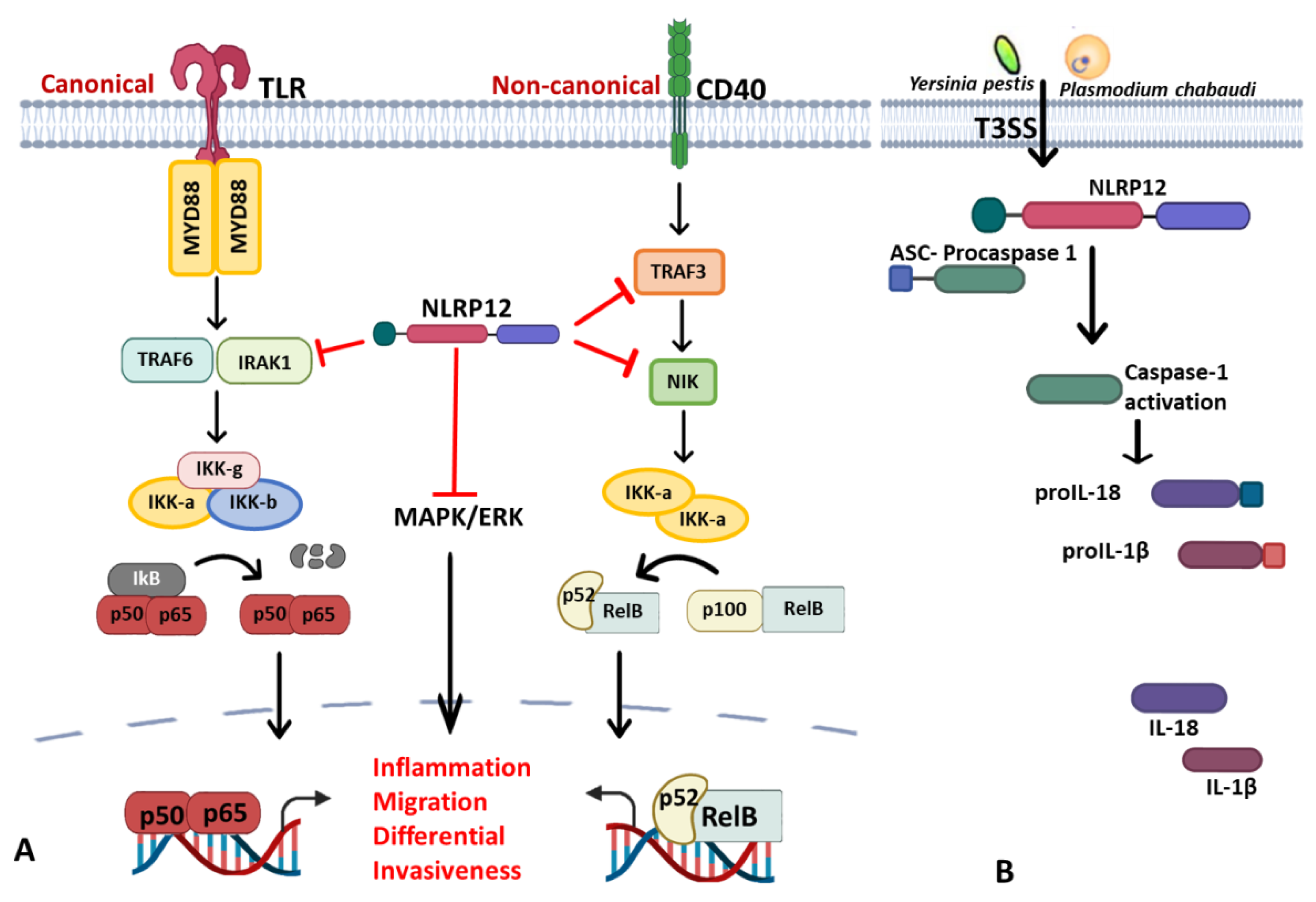
- (i)
- Regulator of canonical and noncanonical NF-κB and MAPK signaling pathways: NLRP12 is a negative regulator protein that inhibits canonical and noncanonical activations of NF-κB and ERK (Figure 2A). Nlrp12–/– mice show exaggerated NF-κB activation and ERK phosphorylation in colitis-associated colorectal cancer models [193], osteoclast differentiation [194] and in bone marrow-derived macrophages treated with Mycobacterium tuberculosis [190] and Salmonella LPS but not flagellin [195]. Canonical interference of NLRP12, as demonstrated by Zaki et al., is via the suppression of hyperphosphorylation of IRAK1 to limit of IκBα and ERK phosphorylation downstream of TLR-MyD88, thus reducing nuclear translocation of NF-κB and secretion of proinflammatory cytokines in macrophage stimulated with S. typhimurium [195]. Similarly, its interaction with NF-κB-inducing kinase (NIK) and TRAF3 was through its NOD and LRR domains, leading to proteasomal degradation of NIK in noncanonical NF-κB signaling [33,57,190] in microbial and parasitic (Leishmania major) infections [196]. Therefore, the constitutive elevation of NIK, processing of p100 to p52 and reduced degradation of TRAF3, was observed in Nlrp12–/– cells [33]. These studies recapitulate NLRP12 as a potential checkpoint for NF-κB signaling in murine macrophages and human THP-1 monocytic cells by negatively regulating both TLR and TNFR pathways [33]. In fact, NLRP12 also exhibits its inhibitory role by degrading NOD2 through the ubiquitin–proteasome pathway to raise host tolerance towards bacterial muramyl dipeptide (MDP) by sequestering heat-shock protein 90 (HSP90). Sequestration of HSP90 prevents the stabilization of NOD2/RIPK2 complex in response to MDP, thus repressing NOD2 signal transduction of NF-κB and subsequent activity of the JAK/STAT signaling pathway [197]. The physiological impact of NLRP12 regulation in the immune response is still elusive, for instance, the loss of NLRP12 in BMDC induces IL-6 and TNF upon M. tuberculosis or Klebsiella pneumonia without conferring resistance against these bacteria [198]. In hepatocellular carcinoma (HCC), however, NLRP12 downregulates the JNK-dependent inflammation and proliferation of hepatocytes and NLRP12 deficient mice were highly susceptible to diethyl nitrosamine (DEN)-induced HCC with increased inflammation, hepatocyte proliferation and tumor burden. In contrast, the upregulation of NLRP12 was reported in response to Porphyromonas gingivalis LPS in RAW264.7, and its depletion in the cell line corresponds to an increase in TNF production and iNOS expression [199].
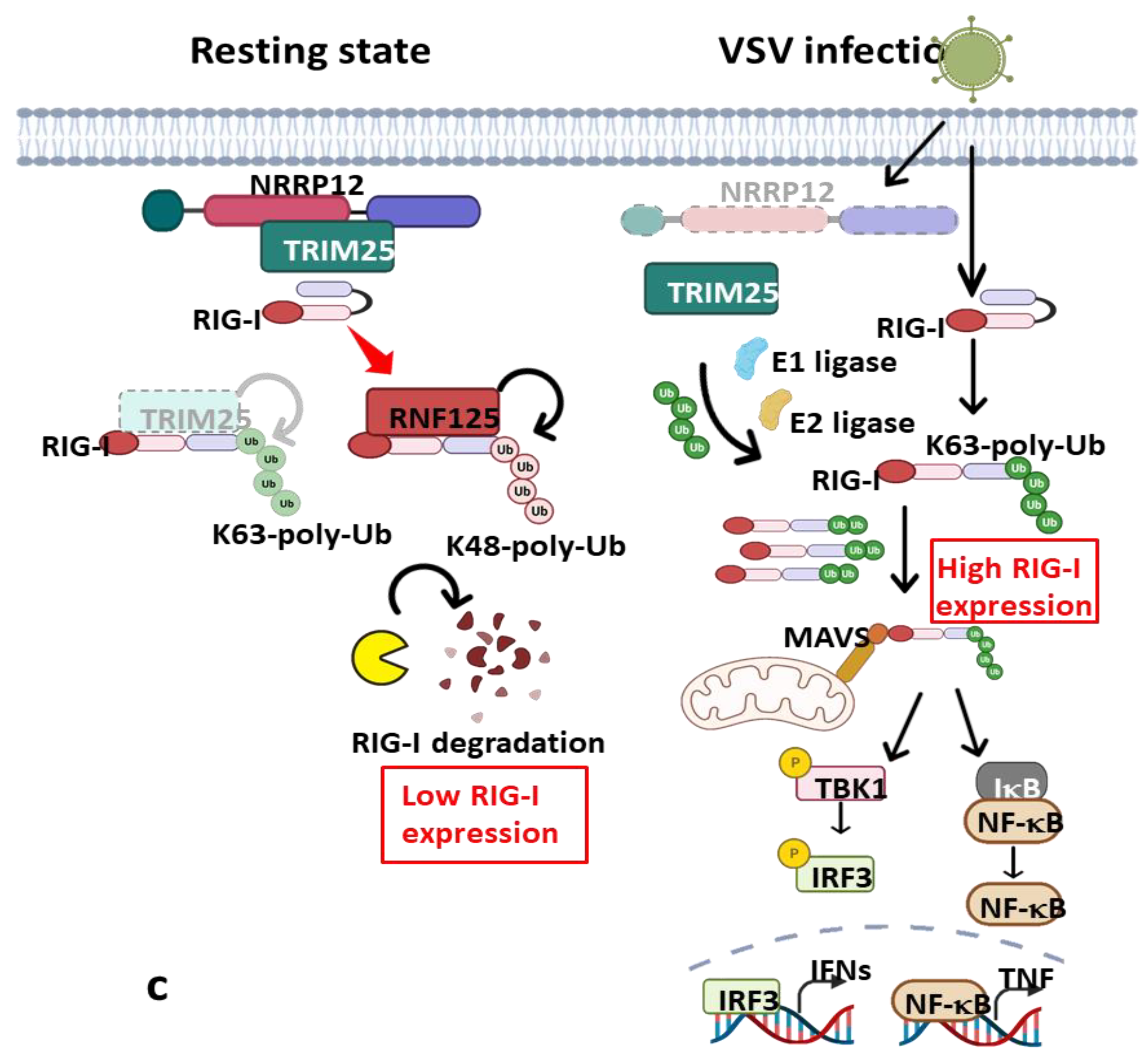
- (ii)
- A negative regulator of Type-I interferon and proinflammatory responses: This is another remarkable function of NLRP12 that was recently demonstrated by our research group; we reported an interference of NLRP12 on RIG-I-mediated IFN-I production during vesicular stomatitis virus (VSV) [200]. We found that VSV infection downregulates NLRP12 expression, and its deletion in BMDC provokes severe transcription and production of IFN-I (IFNα/β) and TNF that corresponds with reduced viral titer and relative genomic copy in Nlrp12–/– DCs upon infection. In the infected DCs, TRIM25, an E3 ligase required for Lys63-linked polyubiquitination and activation of RIG-I, mediates the downstream activation of MAVS (Figure 2C). MAVS associates with the adaptor protein TRAF3 and TRAF family member-associated NF-κB activator (TANK) to trigger the activation of TANK-binding kinase 1 (TBK1) and IκB kinase, leading to the activation of IRFs and production of IFN-I and TNF; thus, enhanced immune signaling cascades were observed in Nlrp12–/– DCs treated with VSV and 5′ppp dsRNA. However, the presence of NLRP12 relieved the binding of TRIM25 with RIG-I to suppress IFN-I production. Mechanistically, NLRP12 promotes RNF125-mediated degradation of RIG-I by associating with ubiquitin ligase TRIM25 to reduce K63-linked ubiquitination of the antiviral innate immune receptor RIG-I (Figure 2C). This will ultimately prevent RIG-I association with MAVS to checkmate the transcription and secretion of interferon and cytokine induction in response to RNA viruses. Domain mapping analysis showed that the NBD domain is presumably a critical target for TRIM25 interaction [200]. Nlrp12–/– mice are more resistant to VSV infection with lower viral loads in the brain and recover faster than WT mice with less neuronal loss in the ventral striatum and hypothalamus in in vivo study.
- (iii)
- NLRP12 inflammasome and its positive regulatory property in other inflammasomes: The foremost inflammasome functions of NLRP12 were reported in an overexpression system where NLRP12 co-expressed with ASC for caspase 1 and IL-1β production [201]. In primary cell and animal study, NLRP12 is involved in the caspase 1-mediating production of inflammatory cytokines (IL-18) and is crucial for the host defense against Yersinia pestis infection; thereby, deficiency of NLRP12 causes the susceptibility to Yersinia pestis infection as it occurred in the IL-18 deficient mice [202]. The actual ligand sensed by NLRP12 in Yersinia for its activation is not known, but it was noted that ligand generation requires a complex type III secretion system (T3SS) (Figure 2B).The inflammasome assemblage of NLRP12 was demonstrated in the dendritic cells from spleen and bone marrow treated with Plasmodium chabaudi, where NLRP12 was collaboratively required for ASC-dependent caspase 1 for the systemic production of IL-1β and pyroptosis [203]. Similarly, collaboration of NLRP12 with other inflammasomes was reported in pyroptosis mediating ganglion cell death of acute glaucoma, NLRP12 collaborates with NLRP3 and NLRC4 to elicit pyroptotic processes and IL-1β maturation through caspase 1 activation [204]. Not only that, simultaneous expression of the NLRP3, NLRP12 and IFI16 inflammasomes in cornea infection induced by virulent HSV-1 strains is ascribed to the enhanced caspase 1, IL-1β and IL18 alongside with co-expression of dense specks of the adapter molecule ASC [205]. However, a contrary report was obtained during Brucella abortus infection that portends NLRP12 as an anti-inflammatory regulator that inhibits not only NF-κB and MAPK signaling but also caspase 1 activation in BMDMs, and its absence conferred the host resistance in murine brucellosis [206]. All these indicate that the function of NLRP12 is stimuli-dependent, and its collaboration with other NLRs may be partially ascribed to dearth of specific ligands to be sensed. However, evidence-based reports have described it as a critical checkpoint in innate immunity in microbial and parasitic infections by regulating innate immune signaling cascades negatively or positively. Since its function varies with pathogens, it is pertinent to investigate the role of NLRP12 in other pathogens.
7. NOD-Like Receptors in the Regulation of Pyroptosis Cell Death
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fekete, T.; Bencze, D.; Bíró, E.; Benkő, S.; Pázmándi, K. Focusing on the Cell Type Specific Regulatory Actions of NLRX1. Int. J. Mol. Sci. 2021, 22, 1316. [Google Scholar] [CrossRef]
- Nagai-Singer, M.A.; Morrison, H.A.; Allen, I.C. NLRX1 Is a Multifaceted and Enigmatic Regulator of Immune System Function. Front. Immunol. 2019, 10, 2419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway, C.A.; Medzhitov, R. Innate Immune Recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowda, D.C.; Wu, X. Parasite Recognition and Signaling Mechanisms in Innate Immune Responses to Malaria. Front. Immunol. 2018, 9, 3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matzinger, P. Tolerance, Danger, and the Extended Family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.-M.; Hoffmann, J.A. The Dorsoventral Regulatory Gene Cassette Spätzle/Toll/Cactus Controls the Potent Antifungal Response in Drosophila Adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Janeway, C.A. Pillars Article: Approaching the Asymptote? Evolution and revolution in immunology. Cold spring harb symp quant biol. 1989, 54: 1–13. J. Immunol. 2013, 191, 4475–4487. [Google Scholar] [CrossRef] [PubMed]
- Bosch, T.C.G.; Augustin, R.; Anton-Erxleben, F.; Fraune, S.; Hemmrich, G.; Zill, H.; Rosenstiel, P.; Jacobs, G.; Schreiber, S.; Leippe, M.; et al. Uncovering the Evolutionary History of Innate Immunity: The Simple Metazoan Hydra Uses Epithelial Cells for Host Defence. Dev. Comp. Immunol. 2009, 33, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Shaw, M.H.; Kim, Y.-G.; Nuñez, G. NOD-Like Receptors: Role in Innate Immunity and Inflammatory Disease. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 365–398. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Ting, J.P.-Y.; Davis, B.K. Caterpiller: A Novel Gene Family Important in Immunity, Cell Death, and Diseases. Annu. Rev. Immunol. 2005, 23, 387–414. [Google Scholar] [CrossRef]
- Motta, V.; Soares, F.; Sun, T.; Philpott, D.J. NOD-Like Receptors: Versatile Cytosolic Sentinels. Physiol. Rev. 2015, 95, 149–178. [Google Scholar] [CrossRef] [Green Version]
- Fritz, J.H.; Kufer, T.A. Editorial: NLR-Protein Functions in Immunity. Front. Immunol. 2015, 6, 306. [Google Scholar] [CrossRef] [Green Version]
- Liwinski, T.; Zheng, D.; Elinav, E. The Microbiome and Cytosolic Innate Immune Receptors. Immunol. Rev. 2020, 297, 207–224. [Google Scholar] [CrossRef]
- Fang, C.; Wei, X.; Wei, Y. Mitochondrial DNA in the Regulation of Innate Immune Responses. Protein Cell 2016, 7, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Philpott, D.J.; Yamaoka, S.; Israël, A.; Sansonetti, P.J. Invasive Shigella Flexneri Activates NF-Kappa B through a Lipopolysaccharide-Dependent Innate Intracellular Response and Leads to IL-8 Expression in Epithelial Cells. J. Immunol. 2000, 165, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Lupfer, C.; Kanneganti, T.-D. Unsolved Mysteries in NLR Biology. Front. Immunol. 2013, 4, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.; Paudel, S.; Jin, L.; Ghimire, L.; Taylor, C.M.; Wakamatsu, N.; Bhattarai, D.; Jeyaseelan, S. NLRP6 Modulates Neutrophil Homeostasis in Bacterial Pneumonia-Derived Sepsis. Mucosal Immunol. 2021, 14, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, L.A.M.; Travassos, L.H. The Interplay between NLRs and Autophagy in Immunity and Inflammation. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Janowski, A.M.; Kolb, R.; Zhang, W.; Sutterwala, F.S. Beneficial and Detrimental Roles of NLRs in Carcinogenesis. Front. Immunol. 2013, 4, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, G.K.; Gutierrez, F.R.S.; Guedes, P.M.M.; Horta, C.V.; Cunha, L.D.; Mineo, T.W.P.; Santiago-Silva, J.; Kobayashi, K.S.; Flavell, R.A.; Silva, J.S.; et al. Cutting Edge: Nucleotide-Binding Oligomerization Domain 1-Dependent Responses Account for Murine Resistance against Trypanosoma Cruzi Infection. J. Immunol. 2010, 184, 1148–1152. [Google Scholar] [CrossRef] [Green Version]
- Kuss-Duerkop, S.K.; Keestra-Gounder, A.M. NOD1 and NOD2 Activation by Diverse Stimuli: A Possible Role for Sensing Pathogen-Induced Endoplasmic Reticulum Stress. Infect. Immun. 2020, 88, e00898-19. [Google Scholar] [CrossRef]
- Mandal, S.; Rajarammohan, S.; Kaur, J. ROS Accumulation and Associated Cell Death Mediates Susceptibility to Alternaria Brassicae in Arabidopsis Accessions. Physiol. Mol. Plant Pathol. 2019, 107, 51–59. [Google Scholar] [CrossRef]
- Saur, I.M.L.; Panstruga, R.; Schulze-Lefert, P. NOD-like Receptor-Mediated Plant Immunity: From Structure to Cell Death. Nat. Rev. Immunol. 2021, 21, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wu, Y.; Hou, Y.; Liu, Y.; Liu, T.; Zhang, H.; Fan, C.; Guan, H.; Li, Y.; Shan, Z.; et al. Cytokine Secretion and Pyroptosis of Thyroid Follicular Cells Mediated by Enhanced NLRP3, NLRP1, NLRC4, and AIM2 Inflammasomes Are Associated With Autoimmune Thyroiditis. Front. Immunol. 2018, 9, 1197. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cao, S.-Q.; Lin, Z.-M.; He, S.-J.; Zuo, J.-P. NOD-like Receptors in Autoimmune Diseases. Acta Pharmacol. Sin. 2021, 1–15. [Google Scholar] [CrossRef]
- Cunha, L.L.; Marcello, M.A.; Ward, L.S. The Role of the Inflammatory Microenvironment in Thyroid Carcinogenesis. Endocr. -Relat. Cancer 2014, 21, R85–R103. [Google Scholar] [CrossRef] [Green Version]
- Magitta, N.F.; Bøe Wolff, A.S.; Johansson, S.; Skinningsrud, B.; Lie, B.A.; Myhr, K.-M.; Undlien, D.E.; Joner, G.; Njølstad, P.R.; Kvien, T.K.; et al. A Coding Polymorphism in NALP1 Confers Risk for Autoimmune Addison’s Disease and Type 1 Diabetes. Genes Immun. 2009, 10, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Pontillo, A.; Brandao, L.; Guimaraes, R.; Segat, L.; Araujo, J.; Crovella, S. Two SNPs in NLRP3 Gene Are Involved in the Predisposition to Type-1 Diabetes and Celiac Disease in a Pediatric Population from Northeast Brazil. Autoimmunity 2010, 43, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.K.; Philipson, C.; Hontecillas, R.; Eden, K.; Bassaganya-Riera, J.; Allen, I.C. Emerging Significance of NLRs in Inflammatory Bowel Disease. Inflamm. Bowel. Dis. 2014, 20, 2412–2432. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, X. NLRP3 Inflammasome in Inflammatory Bowel Disease: Friend or Foe? Dig. Dis. Sci. 2017, 62, 2211–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wilson, J.E.; Koenigsknecht, M.J.; Chou, W.-C.; Montgomery, S.A.; Truax, A.D.; Brickey, W.J.; Packey, C.D.; Maharshak, N.; Matsushima, G.K.; et al. NLRP12 Attenuates Colon Inflammation by Maintaining Colonic Microbial Diversity and Promoting Protective Commensal Bacterial Growth. Nat. Immunol. 2017, 18, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Wilson, J.E.; Schneider, M.; Lich, J.D.; Roberts, R.A.; Arthur, J.C.; Woodford, R.-M.T.; Davis, B.K.; Uronis, J.M.; Herfarth, H.H.; et al. NLRP12 Suppresses Colon Inflammation and Tumorigenesis through the Negative Regulation of Noncanonical NF-ΚB Signaling. Immunity 2012, 36, 742–754. [Google Scholar] [CrossRef] [Green Version]
- Elinav, E.; Henao-Mejia, J.; Flavell, R.A. Integrative Inflammasome Activity in the Regulation of Intestinal Mucosal Immune Responses. Mucosal Immunol. 2013, 6, 4–13. [Google Scholar] [CrossRef]
- Xu, T.; Du, Y.; Fang, X.-B.; Chen, H.; Zhou, D.-D.; Wang, Y.; Zhang, L. New Insights into Nod-like Receptors (NLRs) in Liver Diseases. Int. J. Physiol. Pathophysiol. Pharmacol. 2018, 10, 1–16. [Google Scholar]
- Luan, J.; Zhang, X.; Wang, S.; Li, Y.; Fan, J.; Chen, W.; Zai, W.; Wang, S.; Wang, Y.; Chen, M.; et al. NOD-Like Receptor Protein 3 Inflammasome-Dependent IL-1β Accelerated ConA-Induced Hepatitis. Front. Immunol. 2018, 9, 758. [Google Scholar] [CrossRef]
- Guo, C.; Fu, R.; Wang, S.; Huang, Y.; Li, X.; Zhou, M.; Zhao, J.; Yang, N. NLRP3 Inflammasome Activation Contributes to the Pathogenesis of Rheumatoid Arthritis. Clin. Exp. Immunol. 2018, 194, 231–243. [Google Scholar] [CrossRef] [Green Version]
- Tadaki, H.; Saitsu, H.; Nishimura-Tadaki, A.; Imagawa, T.; Kikuchi, M.; Hara, R.; Kaneko, U.; Kishi, T.; Miyamae, T.; Miyake, N.; et al. De Novo 19q13.42 Duplications Involving NLRP Gene Cluster in a Patient with Systemic-Onset Juvenile Idiopathic Arthritis. J. Hum. Genet. 2011, 56, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Prado, D.S.; Veras, F.P.; Ferreira, R.G.; Damasceno, L.E.A.; Melo, P.H.; Zamboni, D.S.; Cunha, T.M.; Cunha, F.Q.; Alves-Filho, J.C. NLRP12 Controls Arthritis Severity by Acting as a Checkpoint Inhibitor of Th17 Cell Differentiation. FASEB J. 2020, 34, 10907–10919. [Google Scholar] [CrossRef]
- Eike, M.C.; Skinningsrud, B.; Ronninger, M.; Stormyr, A.; Kvien, T.K.; Joner, G.; Njølstad, P.R.; Førre, Ø.; Flatø, B.; Alfredsson, L.; et al. CIITA Gene Variants Are Associated with Rheumatoid Arthritis in Scandinavian Populations. Genes Immun. 2012, 13, 431–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickering, R.J.; Booty, L.M. NLR in EXile: Emerging Roles of NLRX1 in Immunity and Human Disease. Immunology 2021, 162, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, S.; Wang, M.-R.; Wang, T.-T.; Li, B.; Zhu, J.-M. Potential Roles of Nucleotide-Binding Oligomerization Domain 2 in the Pathogenesis of Systemic Lupus Erythematosus. Rheumatol. Int. 2014, 34, 1339–1344. [Google Scholar] [CrossRef] [PubMed]
- Chidambaram, A.C.; Ramamoorthy, J.; Krishnamurthy, S.; Manivannan, P.; Karunakar, P. Dysregulated Immune System Secondary to Novel Heterozygous Mutation of CIITA Gene Presenting with Recurrent Infections and Systemic Lupus Erythematosus. Authorea Prepr. 2020. Unpublished work. [Google Scholar]
- Pontillo, A.; Girardelli, M.; Kamada, A.; Pancotto, J.; Donadi, E.; Crovella, S.; Sandrin-Garcia, P. Polimorphisms in Inflammasome Genes Are Involved in the Predisposition to Systemic Lupus Erythematosus. Autoimmunity 2012, 45, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kang, P.; Zhang, W.; Jian, Z.; Zhang, Q.; Yi, X.; Guo, S.; Guo, W.; Shi, Q.; Li, B.; et al. Activated NLR Family Pyrin Domain Containing 3 (NLRP3) Inflammasome in Keratinocytes Promotes Cutaneous T-Cell Response in Patients with Vitiligo. J. Allergy Clin. Immunol. 2020, 145, 632–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.; Zhou, F. Inflammasomes in Common Immune-Related Skin Diseases. Front. Immunol. 2020, 11, 882. [Google Scholar] [CrossRef] [PubMed]
- Ciążyńska, M.; Olejniczak-Staruch, I.; Sobolewska-Sztychny, D.; Narbutt, J.; Skibińska, M.; Lesiak, A. The Role of NLRP1, NLRP3, and AIM2 Inflammasomes in Psoriasis: Review. Int. J. Mol. Sci. 2021, 22, 5898. [Google Scholar] [CrossRef]
- Yu, P.; Hao, S.; Zheng, H.; Zhao, X.; Li, Y. Association of NLRP1 and NLRP3 Polymorphisms with Psoriasis Vulgaris Risk in the Chinese Han Population. Biomed. Res. Int. 2018, 2018, 4714836. [Google Scholar] [CrossRef] [Green Version]
- Tervaniemi, M.H.; Katayama, S.; Skoog, T.; Siitonen, H.A.; Vuola, J.; Nuutila, K.; Sormunen, R.; Johnsson, A.; Linnarsson, S.; Suomela, S.; et al. NOD-like Receptor Signaling and Inflammasome-Related Pathways Are Highlighted in Psoriatic Epidermis. Sci. Rep. 2016, 6, 22745. [Google Scholar] [CrossRef]
- Zhong, F.L.; Mamaï, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverényi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 2016, 167, 187–202.e17. [Google Scholar] [CrossRef] [Green Version]
- Maver, A.; Lavtar, P.; Ristić, S.; Stopinšek, S.; Simčič, S.; Hočevar, K.; Sepčić, J.; Drulović, J.; Pekmezović, T.; Novaković, I.; et al. Identification of Rare Genetic Variation of NLRP1 Gene in Familial Multiple Sclerosis. Sci. Rep. 2017, 7, 3715. [Google Scholar] [CrossRef]
- Malhotra, S.; Río, J.; Urcelay, E.; Nurtdinov, R.; Bustamante, M.F.; Fernández, O.; Oliver, B.; Zettl, U.; Brassat, D.; Killestein, J.; et al. NLRP3 Inflammasome Is Associated with the Response to IFN-β in Patients with Multiple Sclerosis. Brain 2015, 138, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, R.H. The role of NOD-like receptors (NLRs) in the pathogenesis of metabolic diseases. Postepy Biochem. 2017, 63, 205–209. [Google Scholar] [PubMed]
- Dhaiban, S.; Al-Ani, M.; Elemam, N.M.; Al-Aawad, M.H.; Al-Rawi, Z.; Maghazachi, A.A. Role of Immune Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Science 2020, 3, 12. [Google Scholar] [CrossRef]
- Ting, J.P.-Y.; Lovering, R.C.; Alnemri, E.S.; Bertin, J.; Boss, J.M.; Davis, B.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A.; et al. The NLR Gene Family: An Official Nomenclature. Immunity 2008, 28, 285–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell Discov. 2020, 6, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Tuncer, S.; Fiorillo, M.T.; Sorrentino, R. The Multifaceted Nature of NLRP12. J. Leukoc. Biol. 2014, 96, 991–1000. [Google Scholar] [CrossRef]
- León Machado, J.A.; Steimle, V. The MHC Class II Transactivator CIITA: Not (Quite) the Odd-One-Out Anymore among NLR Proteins. Int. J. Mol. Sci. 2021, 22, 1074. [Google Scholar] [CrossRef]
- Rodriguez, G.M.; Bobbala, D.; Serrano, D.; Mayhue, M.; Champagne, A.; Saucier, C.; Steimle, V.; Kufer, T.A.; Menendez, A.; Ramanathan, S.; et al. NLRC5 Elicits Antitumor Immunity by Enhancing Processing and Presentation of Tumor Antigens to CD8+ T Lymphocytes. OncoImmunology 2016, 5, e1151593. [Google Scholar] [CrossRef] [Green Version]
- Meissner, T.B.; Li, A.; Biswas, A.; Lee, K.-H.; Liu, Y.-J.; Bayir, E.; Iliopoulos, D.; van den Elsen, P.J.; Kobayashi, K.S. NLR Family Member NLRC5 Is a Transcriptional Regulator of MHC Class I Genes. Proc. Natl. Acad. Sci. USA 2010, 107, 13794–13799. [Google Scholar] [CrossRef] [Green Version]
- Robbins, G.R.; Truax, A.D.; Davis, B.K.; Zhang, L.; Brickey, W.J.; Ting, J.P.-Y. Regulation of Class I Major Histocompatibility Complex (MHC) by Nucleotide-Binding Domain, Leucine-Rich Repeat-Containing (NLR) Proteins. J. Biol. Chem. 2012, 287, 24294–24303. [Google Scholar] [CrossRef] [Green Version]
- Philpott, D.J.; Sorbara, M.T.; Robertson, S.J.; Croitoru, K.; Girardin, S.E. NOD Proteins: Regulators of Inflammation in Health and Disease. Nat. Rev. Immunol. 2014, 14, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Caruso, R.; Warner, N.; Inohara, N.; Núñez, G. NOD1 and NOD2: Signaling, Host Defense, and Inflammatory Disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugan, J.; Griffiths, E.; Snow, P.; Rosenzweig, H.; Lee, E.; Brown, B.; Carr, D.W.; Rose, C.; Rosenbaum, J.; Davey, M.P. Blau Syndrome–Associated Nod2 Mutation Alters Expression of Full-Length NOD2 and Limits Responses to Muramyl Dipeptide in Knock-in Mice. J. Immunol. 2015, 194, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Davoudi, S.; Navarro-Gomez, D.; Shen, L.; Ung, C.; Ren, A.; Sullivan, L.; Kwong, M.; Janessian, M.; Comander, J.; Gai, X.; et al. NOD2 Genetic Variants and Sarcoidosis-Associated Uveitis. Am. J. Ophthalmol. Case Rep. 2016, 3, 39–42. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, J.E.; Warner, N.; Staples, J.; Crowley, E.; Gosalia, N.; Murchie, R.; Van Hout, C.; Fiedler, K.; Welch, G.; King, A.K.; et al. Mutation Spectrum of NOD2 Reveals Recessive Inheritance as a Main Driver of Early Onset Crohn’s Disease. Sci. Rep. 2021, 11, 5595. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Zhang, H.; Wang, C.; Jiao, F.; Xu, H.; Wang, X.; Luan, W.; Ma, F.; Ni, L.; Tang, X.; et al. Activation of ROS/MAPKs/NF-ΚB/NLRP3 and Inhibition of Efferocytosis in Osteoclast-Mediated Diabetic Osteoporosis. FASEB J. 2019, 33, 12515–12527. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-ΚB Pathway for the Therapy of Diseases: Mechanism and Clinical Study. Sig. Transduct. Target Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef]
- The Caspase-1 Inflammasome & Its Role in Autoinflammatory Diseases. Available online: https://www.rndsystems.com/resources/articles/caspase-1-inflammasome-its-role-autoinflammatory-diseases (accessed on 24 June 2021).
- Kaparakis, M.; Turnbull, L.; Carneiro, L.; Firth, S.; Coleman, H.A.; Parkington, H.C.; Le Bourhis, L.; Karrar, A.; Viala, J.; Mak, J.; et al. Bacterial Membrane Vesicles Deliver Peptidoglycan to NOD1 in Epithelial Cells. Cell Microbiol. 2010, 12, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Marijke Keestra-Gounder, A.; Tsolis, R.M. NOD1 and NOD2: Beyond Peptidoglycan Sensing. Trends Immunol. 2017, 38, 758–767. [Google Scholar] [CrossRef]
- Chamaillard, M.; Hashimoto, M.; Horie, Y.; Masumoto, J.; Qiu, S.; Saab, L.; Ogura, Y.; Kawasaki, A.; Fukase, K.; Kusumoto, S.; et al. An Essential Role for NOD1 in Host Recognition of Bacterial Peptidoglycan Containing Diaminopimelic Acid. Nat. Immunol. 2003, 4, 702–707. [Google Scholar] [CrossRef]
- Johnston, E.L.; Heras, B.; Kufer, T.A.; Kaparakis-Liaskos, M. Detection of Bacterial Membrane Vesicles by NOD-Like Receptors. Int. J. Mol. Sci. 2021, 22, 1005. [Google Scholar] [CrossRef]
- Pellegrini, E.; Desfosses, A.; Wallmann, A.; Schulze, W.M.; Rehbein, K.; Mas, P.; Signor, L.; Gaudon, S.; Zenkeviciute, G.; Hons, M.; et al. RIP2 Filament Formation Is Required for NOD2 Dependent NF-ΚB Signalling. Nat. Commun. 2018, 9, 4043. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Torres, R.J.; Chamaillard, M. The Ubiquitin Code of NODs Signaling Pathways in Health and Disease. Front. Immunol. 2019, 10, 2648. [Google Scholar] [CrossRef] [Green Version]
- Zangara, M.T.; Johnston, I.; Johnson, E.E.; McDonald, C. Mediators of Metabolism: An Unconventional Role for NOD1 and NOD2. Int. J. Mol. Sci. 2021, 22, 1156. [Google Scholar] [CrossRef]
- Boyle, J.P.; Parkhouse, R.; Monie, T.P. Insights into the Molecular Basis of the NOD2 Signalling Pathway. Open Biol. 2014, 4, 140178. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zheng, Y.; Coyaud, É.; Zhang, C.; Selvabaskaran, A.; Yu, Y.; Xu, Z.; Weng, X.; Chen, J.S.; Meng, Y.; et al. Palmitoylation of NOD1 and NOD2 Is Required for Bacterial Sensing. Science 2019, 366, 460–467. [Google Scholar] [CrossRef]
- Heim, V.J.; Stafford, C.A.; Nachbur, U. NOD Signaling and Cell Death. Front. Cell Dev. Biol. 2019, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Frutuoso, M.S.; Hori, J.I.; Pereira, M.S.F.; Junior, D.S.L.; Sônego, F.; Kobayashi, K.S.; Flavell, R.A.; Cunha, F.Q.; Zamboni, D.S. The Pattern Recognition Receptors Nod1 and Nod2 Account for Neutrophil Recruitment to the Lungs of Mice Infected with Legionella Pneumophila. Microbes Infect. 2010, 12, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.S.; Chamaillard, M.; Ogura, Y.; Henegariu, O.; Inohara, N.; Nuñez, G.; Flavell, R.A. Nod2-Dependent Regulation of Innate and Adaptive Immunity in the Intestinal Tract. Science 2005, 307, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Hisamatsu, T.; Suzuki, M.; Reinecker, H.-C.; Nadeau, W.J.; McCormick, B.A.; Podolsky, D.K. CARD15/NOD2 Functions as an Antibacterial Factor in Human Intestinal Epithelial Cells. Gastroenterology 2003, 124, 993–1000. [Google Scholar] [CrossRef]
- Jones, M.; Cunningham, M.E.; Wing, P.; DeSilva, S.; Challa, R.; Sheri, A.; Padmanabhan, S.; Iyer, R.P.; Korba, B.E.; Afdhal, N.; et al. SB 9200, a Novel Agonist of Innate Immunity, Shows Potent Antiviral Activity against Resistant HCV Variants. J. Med. Virol. 2017, 89, 1620–1628. [Google Scholar] [CrossRef]
- Davoli-Ferreira, M.; Fonseca, D.M.; Mota, C.M.; Dias, M.S.; Lima-Junior, D.S.; da Silva, M.V.; Quirino, G.F.S.; Zamboni, D.S.; Silva, J.S.; Mineo, T.W.P. Nucleotide-Binding Oligomerization Domain-Containing Protein 2 Prompts Potent Inflammatory Stimuli during Neospora Caninum Infection. Sci. Rep. 2016, 6, 29289. [Google Scholar] [CrossRef] [Green Version]
- Negroni, A.; Pierdomenico, M.; Cucchiara, S.; Stronati, L. NOD2 and Inflammation: Current Insights. J. Inflamm. Res. 2018, 11, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Tschöpe, C.; Müller, I.; Xia, Y.; Savvatis, K.; Pappritz, K.; Pinkert, S.; Lassner, D.; Heimesaat, M.M.; Spillmann, F.; Miteva, K.; et al. NOD2 (Nucleotide-Binding Oligomerization Domain 2) Is a Major Pathogenic Mediator of Coxsackievirus B3-Induced Myocarditis. Circ. Heart Fail. 2017, 10, e003870. [Google Scholar] [CrossRef]
- Jamontt, J.; Petit, S.; Clark, N.; Parkinson, S.J.; Smith, P. Nucleotide-Binding Oligomerization Domain 2 Signaling Promotes Hyperresponsive Macrophages and Colitis in IL-10–Deficient Mice. J. Immunol. 2013, 190, 2948–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-G.; Udayanga, K.G.S.; Totsuka, N.; Weinberg, J.B.; Núñez, G.; Shibuya, A. Gut Dysbiosis Promotes M2 Macrophage Polarization and Allergic Airway Inflammation via Fungi-Induced PGE2. Cell Host Microbe 2014, 15, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-G.; Park, J.-H.; Shaw, M.H.; Franchi, L.; Inohara, N.; Núñez, G. The Cytosolic Sensors Nod1 and Nod2 Are Critical for Bacterial Recognition and Host Defense after Exposure to Toll-like Receptor Ligands. Immunity 2008, 28, 246–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.S.; Park, J.-K.; Jo, E.-K. Toll-like Receptors and NOD-like Receptors in Innate Immune Defense during Pathogenic Infection. J. Bacteriol. Virol. 2014, 44, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Asano, N.; Fichtner-Feigl, S.; Gorelick, P.L.; Tsuji, Y.; Matsumoto, Y.; Chiba, T.; Fuss, I.J.; Kitani, A.; Strober, W. NOD1 Contributes to Mouse Host Defense against Helicobacter Pylori via Induction of Type I IFN and Activation of the ISGF3 Signaling Pathway. J. Clin. Investig. 2010, 120, 1645–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, T.A.; Lau, J.F.; Parisien, J.-P.; Horvath, C.M. A Hybrid IRF9-STAT2 Protein Recapitulates Interferon-Stimulated Gene Expression and Antiviral Response. J. Biol. Chem. 2003, 278, 13033–13038. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.M.; Zhang, J.; Li, P.W.; Hu, Y.W.; Cao, L.; Ouyang, S.; Bi, Y.H.; Nie, P.; Chang, M.X. NOD1 Promotes Antiviral Signaling by Binding Viral RNA and Regulating the Interaction of MDA5 and MAVS. J. Immunol. 2020, 204, 2216–2231. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Koseki, T.; del Peso, L.; Hu, Y.; Yee, C.; Chen, S.; Carrio, R.; Merino, J.; Liu, D.; Ni, J.; et al. Nod1, an Apaf-1-like Activator of Caspase-9 and Nuclear Factor-KappaB. J. Biol. Chem. 1999, 274, 14560–14567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedl, M.; Abraham, C. Distinct Roles for Nod2 Protein and Autocrine Interleukin-1β in Muramyl Dipeptide-Induced Mitogen-Activated Protein Kinase Activation and Cytokine Secretion in Human Macrophages. J. Biol. Chem. 2011, 286, 26440–26449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, L.-C.; Ali, S.R.; McGillivray, S.; Tseng, P.-H.; Mariathasan, S.; Humke, E.W.; Eckmann, L.; Powell, J.J.; Nizet, V.; Dixit, V.M.; et al. A NOD2–NALP1 Complex Mediates Caspase-1-Dependent IL-1β Secretion in Response to Bacillus Anthracis Infection and Muramyl Dipeptide. Proc. Natl. Acad. Sci. USA 2008, 105, 7803–7808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.-K. An Update on the Regulatory Mechanisms of NLRP3 Inflammasome Activation. Cell Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef] [PubMed]
- Vanaja, S.K.; Rathinam, V.A.K.; Fitzgerald, K.A. Mechanisms of Inflammasome Activation: Recent Advances and Novel Insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Gritsenko, A.; Yu, S.; Martin-Sanchez, F.; Diaz-del-Olmo, I.; Nichols, E.-M.; Davis, D.M.; Brough, D.; Lopez-Castejon, G. Priming Is Dispensable for NLRP3 Inflammasome Activation in Human Monocytes In Vitro. Front. Immunol. 2020, 11, 2573. [Google Scholar] [CrossRef]
- Chen, S.-T.; Li, F.-J.; Hsu, T.-Y.; Liang, S.-M.; Yeh, Y.-C.; Liao, W.-Y.; Chou, T.-Y.; Chen, N.-J.; Hsiao, M.; Yang, W.-B.; et al. CLEC5A Is a Critical Receptor in Innate Immunity against Listeria Infection. Nat. Commun. 2017, 8, 299. [Google Scholar] [CrossRef] [Green Version]
- Swanson, K.V.; Junkins, R.D.; Kurkjian, C.J.; Holley-Guthrie, E.; Pendse, A.A.; El Morabiti, R.; Petrucelli, A.; Barber, G.N.; Benedict, C.A.; Ting, J.P.-Y. A Noncanonical Function of CGAMP in Inflammasome Priming and Activation. J. Exp. Med. 2017, 214, 3611–3626. [Google Scholar] [CrossRef] [Green Version]
- Malireddi, R.K.S.; Kanneganti, T.-D. Role of Type I Interferons in Inflammasome Activation, Cell Death, and Disease during Microbial Infection. Front. Cell Infect. Microbiol. 2013, 3, 77. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical Inflammasome Signaling Elicits Gasdermin D-Dependent Neutrophil Extracellular Traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef] [Green Version]
- de Carvalho, R.V.H.; Andrade, W.A.; Lima-Junior, D.S.; Dilucca, M.; de Oliveira, C.V.; Wang, K.; Nogueira, P.M.; Rugani, J.N.; Soares, R.P.; Beverley, S.M.; et al. Leishmania Lipophosphoglycan Triggers Caspase-11 and the Non-Canonical Activation of the NLRP3 Inflammasome. Cell Rep. 2019, 26, 429–437.e5. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, K. Inflammasome-associated Cell Death: Pyroptosis, Apoptosis, and Physiological Implications. Microbiol. Immunol. 2020, 64, 252–269. [Google Scholar] [CrossRef]
- Chu, L.H.; Indramohan, M.; Ratsimandresy, R.A.; Gangopadhyay, A.; Morris, E.P.; Monack, D.M.; Dorfleutner, A.; Stehlik, C. The Oxidized Phospholipid OxPAPC Protects from Septic Shock by Targeting the Non-Canonical Inflammasome in Macrophages. Nat. Commun. 2018, 9, 996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent Advances in the Mechanisms of NLRP3 Inflammasome Activation and Its Inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 Scaffolding Function and MLKL Regulate NLRP3 Inflammasome Activation Downstream of TLR3. Nat. Commun. 2015, 6, 7515. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, C.; Russo, H.M.; Sanadi, C.E.; Martin, B.N.; Li, X.; Kaiser, W.J.; Mocarski, E.S.; Dubyak, G.R. Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling. J. Biol. Chem. 2015, 290, 20167–20184. [Google Scholar] [CrossRef] [Green Version]
- Gurung, P.; Kanneganti, T.-D. Novel Roles for Caspase-8 in IL-1β and Inflammasome Regulation. Am. J. Pathol. 2015, 185, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.-B.; Yang, S.-H.; Toth, B.; Kovalenko, A.; Wallach, D. Caspase-8 Blocks Kinase RIPK3-Mediated Activation of the NLRP3 Inflammasome. Immunity 2013, 38, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, S.; Rathinam, V.A.K.; Bossaller, L.; Army, K.; Kaiser, W.J.; Mocarski, E.S.; Dillon, C.P.; Green, D.R.; Mayadas, T.N.; Levitz, S.M.; et al. Caspase-8 Modulates Dectin-1 and Complement Receptor 3–Driven IL-1β Production in Response to β-Glucans and the Fungal Pathogen, Candida Albicans. J. Immunol. 2014, 193, 2519–2530. [Google Scholar] [CrossRef] [Green Version]
- Allam, R.; Lawlor, K.E.; Yu, E.C.-W.; Mildenhall, A.L.; Moujalled, D.M.; Lewis, R.S.; Ke, F.; Mason, K.D.; White, M.J.; Stacey, K.J.; et al. Mitochondrial Apoptosis Is Dispensable for NLRP3 Inflammasome Activation but Non-Apoptotic Caspase-8 Is Required for Inflammasome Priming. EMBO Rep. 2014, 15, 982–990. [Google Scholar] [CrossRef] [Green Version]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.S.; Walle, L.V.; Opdenbosch, N.V.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.-D. FADD and Caspase-8 Mediate Priming and Activation of the Canonical and Noncanonical Nlrp3 Inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [Green Version]
- Sokolovska, A.; Becker, C.E.; Ip, W.K.E.; Rathinam, V.A.K.; Brudner, M.; Paquette, N.; Tanne, A.; Vanaja, S.K.; Moore, K.J.; Fitzgerald, K.A.; et al. Activation of Caspase-1 by the NLRP3 Inflammasome Regulates the NADPH Oxidase NOX2 to Control Phagosome Function. Nat. Immunol. 2013, 14, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Niu, J.; Wu, S.; Chen, M.; Xu, K.; Guo, Q.; Lu, A.; Zhao, L.; Sun, B.; Meng, G. Hyperactivation of the NLRP3 Inflammasome Protects Mice against Influenza A Virus Infection via IL-1β Mediated Neutrophil Recruitment. Cytokine 2019, 120, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Wu, S.; Cai, J.; Yang, Y.; Ren, X.; Feng, Y.; Chen, L.; Qin, B.; Xu, C.; Yang, H.; et al. The H7N9 Influenza A Virus Infection Results in Lethal Inflammation in the Mammalian Host via the NLRP3-Caspase-1 Inflammasome. Sci. Rep. 2017, 7, 7625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Song, J.-W.; Huang, H.-H.; Fan, X.; Huang, L.; Deng, J.-N.; Tu, B.; Wang, K.; Li, J.; Zhou, M.-J.; et al. NLRP3 Inflammasome Induces CD4+ T Cell Loss in Chronically HIV-1–Infected Patients. Available online: https://www.jci.org/articles/view/138861/pdf (accessed on 6 August 2021).
- Alhallaf, R.; Agha, Z.; Miller, C.M.; Robertson, A.A.B.; Sotillo, J.; Croese, J.; Cooper, M.A.; Masters, S.L.; Kupz, A.; Smith, N.C.; et al. The NLRP3 Inflammasome Suppresses Protective Immunity to Gastrointestinal Helminth Infection. Cell Rep. 2018, 23, 1085–1098. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, X.-C.; Gong, P.-T.; Zhang, N.; Zhang, X.; Li, S.; Li, X.; Liu, S.-X.; Zhang, X.-X.; Li, W.; et al. ROS-Mediated NLRP3 Inflammasome Activation Participates in the Response against Neospora Caninum Infection. Parasites Vectors 2020, 13, 449. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, R.V.H.; Lima-Junior, D.S.; da Silva, M.V.G.; Dilucca, M.; Rodrigues, T.S.; Horta, C.V.; Silva, A.L.N.; da Silva, P.F.; Frantz, F.G.; Lorenzon, L.B.; et al. Leishmania RNA Virus Exacerbates Leishmaniasis by Subverting Innate Immunity via TLR3-Mediated NLRP3 Inflammasome Inhibition. Nat. Commun. 2019, 10, 5273. [Google Scholar] [CrossRef] [Green Version]
- Shio, M.T.; Christian, J.G.; Jung, J.Y.; Chang, K.-P.; Olivier, M. PKC/ROS-Mediated NLRP3 Inflammasome Activation Is Attenuated by Leishmania Zinc-Metalloprotease during Infection. PLoS Negl. Trop. Dis. 2015, 9, e0003868. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, V.M.; Matteucci, K.C.; Buzzo, C.L.; Miollo, B.H.; Ferrante, D.; Torrecilhas, A.C.; Rodrigues, M.M.; Alvarez, J.M.; Bortoluci, K.R. NLRP3 Controls Trypanosoma Cruzi Infection through a Caspase-1-Dependent IL-1R-Independent NO Production. PLoS Negl. Trop. Dis. 2013, 7, e2469. [Google Scholar] [CrossRef] [Green Version]
- Paroli, A.F.; Gonzalez, P.V.; Díaz Luján, C.M.; Onofrio, L.I.; Arocena, A.R.; Cano, R.C.; Carrera Silva, E.A.; Gea, S. NLRP3 Inflammasome and Caspase-1/11 Pathway Orchestrate Different Outcomes in the Host Protection Against Trypanosoma Cruzi Acute Infection. Front. Immunol. 2018, 9, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorfu, G.; Cirelli, K.M.; Melo, M.B.; Mayer-Barber, K.; Crown, D.; Koller, B.H.; Masters, S.; Sher, A.; Leppla, S.H.; Moayeri, M.; et al. Dual Role for Inflammasome Sensors NLRP1 and NLRP3 in Murine Resistance to Toxoplasma Gondii. mBio 2014, 5, e01117-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riestra, A.M.; Valderrama, J.A.; Patras, K.A.; Booth, S.D.; Quek, X.Y.; Tsai, C.-M.; Nizet, V. Trichomonas Vaginalis Induces NLRP3 Inflammasome Activation and Pyroptotic Cell Death in Human Macrophages. J. Innate Immun. 2019, 11, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, L.; Moreau, F.; Cornick, S.; Chadee, K. The NLRP3 Inflammasome Is a Pathogen Sensor for Invasive Entamoeba Histolytica via Activation of A5β1 Integrin at the Macrophage-Amebae Intercellular Junction. PLoS Pathog. 2015, 11, e1004887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, L.; König, A.; Koenig, P.-A.; Gresnigt, M.S.; Westman, J.; Drummond, R.A.; Lionakis, M.S.; Groß, O.; Ruland, J.; Naglik, J.R.; et al. The Fungal Peptide Toxin Candidalysin Activates the NLRP3 Inflammasome and Causes Cytolysis in Mononuclear Phagocytes. Nat. Commun. 2018, 9, 4260. [Google Scholar] [CrossRef] [PubMed]
- Roselletti, E.; Perito, S.; Gabrielli, E.; Mencacci, A.; Pericolini, E.; Sabbatini, S.; Cassone, A.; Vecchiarelli, A. NLRP3 Inflammasome Is a Key Player in Human Vulvovaginal Disease Caused by Candida Albicans. Sci. Rep. 2017, 7, 17877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.F.; Dasari, H.; Van Keulen, V.P.; Carmona, E.M. Canonical Stimulation of the NLRP3 Inflammasome by Fungal Antigens Links Innate and Adaptive B-Lymphocyte Responses by Modulating IL-1β and IgM Production. Front. Immunol. 2017, 8, 1504. [Google Scholar] [CrossRef]
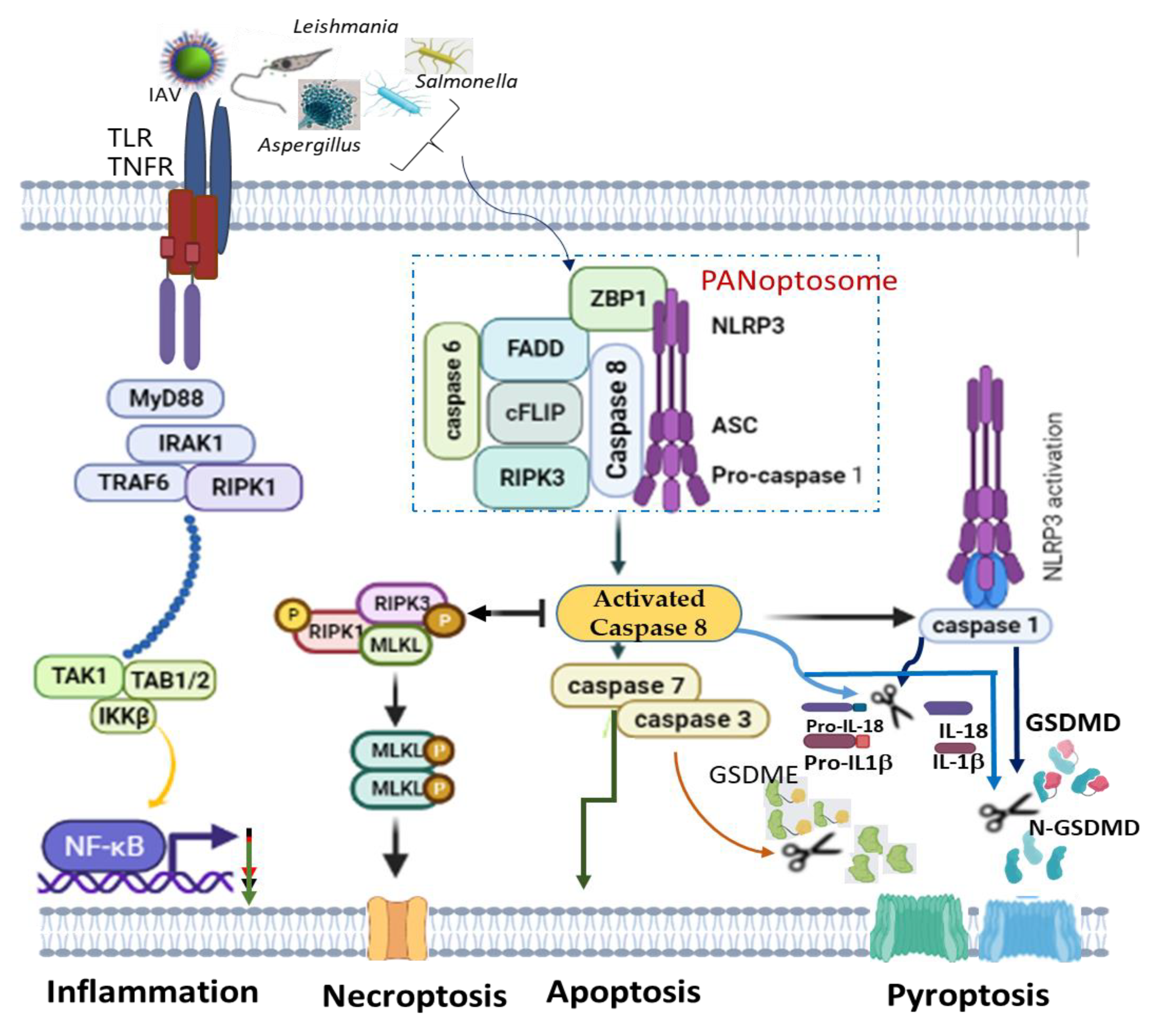
- Christgen, S.; Zheng, M.; Kesavardhana, S.; Karki, R.; Malireddi, R.K.S.; Banoth, B.; Place, D.E.; Briard, B.; Sharma, B.R.; Tuladhar, S.; et al. Identification of the PANoptosome: A Molecular Platform Triggering Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 2020, 10, 237. [Google Scholar] [CrossRef] [PubMed]
- Briard, B.; Malireddi, R.K.S.; Kanneganti, T.-D. Role of Inflammasomes/Pyroptosis and PANoptosis during Fungal Infection. PLoS Pathog. 2021, 17, e1009358. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Gurung, P.; Kesavardhana, S.; Samir, P.; Burton, A.; Mummareddy, H.; Vogel, P.; Pelletier, S.; Burgula, S.; Kanneganti, T.-D. Innate Immune Priming in the Absence of TAK1 Drives RIPK1 Kinase Activity-Independent Pyroptosis, Apoptosis, Necroptosis, and Inflammatory Disease. J. Exp. Med. 2020, 217, e20191644. [Google Scholar] [CrossRef]
- Lee, S.; Channappanavar, R.; Kanneganti, T.-D. Coronaviruses: Innate Immunity, Inflammasome Activation, Inflammatory Cell Death, and Cytokines. Trends Immunol. 2020, 41, 1083–1099. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) Is a Cytosolic DNA Sensor and an Activator of Innate Immune Response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Zheng, M.; Kanneganti, T. The Regulation of the ZBP1-NLRP3 Inflammasome and Its Implications in Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Immunol. Rev. 2020, 297, 26–38. [Google Scholar] [CrossRef]
- Zheng, M.; Kanneganti, T.-D. Newly Identified Function of Caspase-6 in ZBP1-Mediated Innate Immune Responses, NLRP3 Inflammasome Activation, PANoptosis, and Host Defense. J. Cell Immunol. 2020, 2, 341–347. [Google Scholar] [CrossRef]
- Zheng, M.; Williams, E.P.; Malireddi, R.K.S.; Karki, R.; Banoth, B.; Burton, A.; Webby, R.; Channappanavar, R.; Jonsson, C.B.; Kanneganti, T.-D. Impaired NLRP3 Inflammasome Activation/Pyroptosis Leads to Robust Inflammatory Cell Death via Caspase-8/RIPK3 during Coronavirus Infection. J. Biol. Chem. 2020, 295, 14040–14052. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhou, L.; Yuan, H.; Wu, S. Interconnections among Major Forms of Regulated Cell Death. Apoptosis 2020, 25, 616–624. [Google Scholar] [CrossRef]
- Zheng, Z.; Deng, W.; Bai, Y.; Miao, R.; Mei, S.; Zhang, Z.; Pan, Y.; Wang, Y.; Min, R.; Deng, F.; et al. The Lysosomal Rag-Ragulator Complex Licenses RIPK1– and Caspase-8–Mediated Pyroptosis by Yersinia. Science 2021, 372, eabg0269. [Google Scholar] [CrossRef]
- Duncan, J.A.; Canna, S.W. The NLRC4 Inflammasome. Immunol. Rev. 2018, 281, 115–123. [Google Scholar] [CrossRef]
- Gutierrez, O.; Pipaon, C.; Fernandez-Luna, J.L. Ipaf Is Upregulated by Tumor Necrosis Factor-Alpha in Human Leukemia Cells. FEBS Lett. 2004, 568, 79–82. [Google Scholar] [CrossRef] [Green Version]
- Sadasivam, S.; Gupta, S.; Radha, V.; Batta, K.; Kundu, T.K.; Swarup, G. Caspase-1 Activator Ipaf Is a P53-Inducible Gene Involved in Apoptosis. Oncogene 2005, 24, 627–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaram, B.; Kanneganti, T.-D. Advances in Understanding Activation and Function of the NLRC4 Inflammasome. Int. J. Mol. Sci. 2021, 22, 1048. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, D.P.A.; Cerqueira, D.M.; Pereira, M.S.F.; Castanheira, F.V.S.; Fernandes, T.D.; Manin, G.Z.; Cunha, L.D.; Zamboni, D.S. Inhibition of Caspase-1 or Gasdermin-D Enable Caspase-8 Activation in the Naip5/NLRC4/ASC Inflammasome. PLoS Pathog. 2017, 13, e1006502. [Google Scholar] [CrossRef] [Green Version]
- Guan, C.; Huang, X.; Yue, J.; Xiang, H.; Shaheen, S.; Jiang, Z.; Tao, Y.; Tu, J.; Liu, Z.; Yao, Y.; et al. SIRT3-Mediated Deacetylation of NLRC4 Promotes Inflammasome Activation. Theranostics 2021, 11, 3981–3995. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.-N.; Lu, Q.; Xu, H.; Liu, L.; Shao, F. The NLRC4 Inflammasome Receptors for Bacterial Flagellin and Type III Secretion Apparatus. Nature 2011, 477, 596–600. [Google Scholar] [CrossRef]
- Kofoed, E.M.; Vance, R.E. Innate Immune Recognition of Bacterial Ligands by NAIPs Determines Inflammasome Specificity. Nature 2011, 477, 592–595. [Google Scholar] [CrossRef]
- Rayamajhi, M.; Zak, D.E.; Chavarria-Smith, J.; Vance, R.E.; Miao, E.A. Cutting Edge: Mouse NAIP1 Detects the Type III Secretion System Needle Protein. J. Immunol. 2013, 191, 3986–3989. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, Y.; Shi, J.; Shao, F. Human NAIP and Mouse NAIP1 Recognize Bacterial Type III Secretion Needle Protein for Inflammasome Activation. Proc. Natl. Acad. Sci. USA 2013, 110, 14408–14413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortmann, J.; Brubaker, S.W.; Monack, D.M. Cutting Edge: Inflammasome Activation in Primary Human Macrophages Is Dependent on Flagellin. J. Immunol. 2015, 195, 815–819. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.W.; Groß, C.J.; Sotomayor, F.V.; Stacey, K.J.; Tschopp, J.; Sweet, M.J.; Schroder, K. The Neutrophil NLRC4 Inflammasome Selectively Promotes IL-1β Maturation without Pyroptosis during Acute Salmonella Challenge. Cell Rep. 2014, 8, 570–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, S.B.; Oh, C.; Maltez, V.I.; McGlaughon, B.D.; Verma, A.; Miao, E.A.; Aachoui, Y. Neutrophil Caspase-11 Is Essential to Defend against a Cytosol-Invasive Bacterium. Cell Rep. 2020, 32, 107967. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, A.; Böck, D.; Geiser, P.; Berthold, D.L.; Fattinger, S.A.; Furter, M.; Bouman, J.A.; Barthel-Scherrer, M.; Lang, C.M.; Bakkeren, E.; et al. Intestinal Epithelial NAIP/NLRC4 Restricts Systemic Dissemination of the Adapted Pathogen Salmonella Typhimurium Due to Site-Specific Bacterial PAMP Expression. Mucosal Immunol. 2020, 13, 530–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Liu, X.; Li, Y.; Zhao, J.; Liu, Z.; Hu, Z.; Wang, Y.; Yao, Y.; Miller, A.W.; Su, B.; et al. LRRK2 Promotes the Activation of NLRC4 Inflammasome during Salmonella Typhimurium Infection. J. Exp. Med. 2017, 214, 3051–3066. [Google Scholar] [CrossRef]
- He, M.; Chiang, H.-H.; Luo, H.; Zheng, Z.; Qiao, Q.; Wang, L.; Tan, M.; Ohkubo, R.; Mu, W.-C.; Zhao, S.; et al. An Acetylation Switch of the NLRP3 Inflammasome Regulates Aging-Associated Chronic Inflammation and Insulin Resistance. Cell Metab. 2020, 31, 580–591.e5. [Google Scholar] [CrossRef]
- Souza, C.O.S.; Ketelut-Carneiro, N.; Milanezi, C.M.; Faccioli, L.H.; Gardinassi, L.G.; Silva, J.S. NLRC4 Inhibits NLRP3 Inflammasome and Abrogates Effective Antifungal CD8+ T Cell Responses. iScience 2021, 24, 102548. [Google Scholar] [CrossRef] [PubMed]
- Grenier, J.M.; Wang, L.; Manji, G.A.; Huang, W.J.; Al-Garawi, A.; Kelly, R.; Carlson, A.; Merriam, S.; Lora, J.M.; Briskin, M.; et al. Functional Screening of Five PYPAF Family Members Identifies PYPAF5 as a Novel Regulator of NF-KappaB and Caspase-1. FEBS Lett. 2002, 530, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Venuprasad, K.; Theiss, A.L. NLRP6 in Host Defense and Intestinal Inflammation. Cell Rep. 2021, 35, 109043. [Google Scholar] [CrossRef]
- Wang, P.; Zhu, S.; Yang, L.; Cui, S.; Pan, W.; Jackson, R.; Zheng, Y.; Rongvaux, A.; Sun, Q.; Yang, G.; et al. Nlrp6 Regulates Intestinal Antiviral Innate Immunity. Science 2015, 350, 826–830. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.L.; Zhang, L.; Song, D.Z.; Yi, X.W.; Xu, W.Z.; Ye, L.; Huang, D.M. NLRP6 Suppresses the Inflammatory Response of Human Periodontal Ligament Cells by Inhibiting NF-ΚB and ERK Signal Pathways. Int. Endod. J. 2019, 52, 999–1009. [Google Scholar] [CrossRef]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 Inflammasome Regulates Colonic Microbial Ecology and Risk for Colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.Y.; Liu, M.; Wang, F.; Bertin, J.; Núñez, G. A Functional Role for Nlrp6 in Intestinal Inflammation and Tumorigenesis. J. Immunol. 2011, 186, 7187–7194. [Google Scholar] [CrossRef]
- Mukherjee, S.; Kumar, R.; Tsakem Lenou, E.; Basrur, V.; Kontoyiannis, D.L.; Ioakeimidis, F.; Mosialos, G.; Theiss, A.L.; Flavell, R.A.; Venuprasad, K. Deubiquitination of NLRP6 Inflammasome by Cyld Critically Regulates Intestinal Inflammation. Nat. Immunol. 2020, 21, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, L.; Paudel, S.; Jin, L.; Baral, P.; Cai, S.; Jeyaseelan, S. NLRP6 Negatively Regulates Pulmonary Host Defense in Gram-Positive Bacterial Infection through Modulating Neutrophil Recruitment and Function. PLoS Pathog. 2018, 14, e1007308. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Seregin, S.S.; Yang, D.; Fukase, K.; Chamaillard, M.; Alnemri, E.S.; Inohara, N.; Chen, G.Y.; Núñez, G. The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell 2018, 175, 1651–1664.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Wu, X.; Peng, L.; Chen, T.; Huang, Q.; Wang, Y.; Ye, C.; Peng, Y.; Hu, D.; Fang, R. The Critical Role of NLRP6 Inflammasome in Streptococcus Pneumoniae Infection In Vitro and In Vivo. Int. J. Mol. Sci. 2021, 22, 3876. [Google Scholar] [CrossRef] [PubMed]
- Sanches, R.C.O.; Souza, C.; Marinho, F.V.; Mambelli, F.S.; Morais, S.B.; Guimaraes, E.S.; Oliveira, S.C. NLRP6 Plays an Important Role in Early Hepatic Immunopathology Caused by Schistosoma Mansoni Infection. Front. Immunol. 2020, 11, 795. [Google Scholar] [CrossRef]
- Sateriale, A.; Gullicksrud, J.A.; Engiles, J.B.; McLeod, B.I.; Kugler, E.M.; Henao-Mejia, J.; Zhou, T.; Ring, A.M.; Brodsky, I.E.; Hunter, C.A.; et al. The Intestinal Parasite Cryptosporidium Is Controlled by an Enterocyte Intrinsic Inflammasome That Depends on NLRP6. Proc. Natl. Acad. Sci. USA 2021, 118, e2007807118. [Google Scholar] [CrossRef]
- Levy, M.; Shapiro, H.; Thaiss, C.A.; Elinav, E. NLRP6: A Multifaceted Innate Immune Sensor. Trends Immunol. 2017, 38, 248–260. [Google Scholar] [CrossRef]
- Anand, P.K.; Malireddi, R.K.S.; Lukens, J.R.; Vogel, P.; Bertin, J.; Lamkanfi, M.; Kanneganti, T.-D. NLRP6 Negatively Regulates Innate Immunity and Host Defence against Bacterial Pathogens. Nature 2012, 488, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Clay, G.M.; Valadares, D.G.; Graff, J.W.; Ulland, T.K.; Davis, R.E.; Scorza, B.M.; Zhanbolat, B.S.; Chen, Y.; Sutterwala, F.S.; Wilson, M.E. An Anti-Inflammatory Role for NLRP10 in Murine Cutaneous Leishmaniasis. J. Immunol. 2017, 199, 2823–2833. [Google Scholar] [CrossRef]
- Damm, A.; Lautz, K.; Kufer, T.A. Roles of NLRP10 in Innate and Adaptive Immunity. Microbes Infect. 2013, 15, 516–523. [Google Scholar] [CrossRef]
- Imamura, R.; Wang, Y.; Kinoshita, T.; Suzuki, M.; Noda, T.; Sagara, J.; Taniguchi, S.; Okamoto, H.; Suda, T. Anti-Inflammatory Activity of PYNOD and Its Mechanism in Humans and Mice. J. Immunol. 2010, 184, 5874–5884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenbarth, S.C.; Williams, A.; Colegio, O.R.; Meng, H.; Strowig, T.; Rongvaux, A.; Henao-Mejia, J.; Thaiss, C.A.; Joly, S.; Gonzalez, D.G.; et al. NLRP10 Is a NOD-like Receptor Essential to Initiate Adaptive Immunity by Dendritic Cells. Nature 2012, 484, 510–513. [Google Scholar] [CrossRef] [Green Version]
- Joly, S.; Eisenbarth, S.C.; Olivier, A.K.; Williams, A.; Kaplan, D.H.; Cassel, S.L.; Flavell, R.A.; Sutterwala, F.S. Nlrp10 Is Essential for Protective Anti-Fungal Adaptive Immunity against Candida Albicans. J. Immunol. 2012, 189, 4713–4717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebsamen, M.; Vazquez, J.; Tardivel, A.; Guarda, G.; Curran, J.; Tschopp, J. NLRX1/NOD5 Deficiency Does Not Affect MAVS Signalling. Cell Death Differ. 2011, 18, 1387. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Soares, F.; Tattoli, I.; Castanier, C.; Philpott, D.J.; Girardin, S.E. An N-Terminal Addressing Sequence Targets NLRX1 to the Mitochondrial Matrix. J. Cell Sci. 2009, 122, 3161–3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, B.L.; Ganesan, S.; Comstock, A.T.; Faris, A.N.; Hershenson, M.B.; Sajjan, U.S. Nod-Like Receptor X-1 Is Required for Rhinovirus-Induced Barrier Dysfunction in Airway Epithelial Cells. J. Virol. 2014, 88, 3705–3718. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.-H.; Liu, C.-Y.; Wu, S.-Y.; Chen, W.-Y.; Chang, T.-H.; Kan, H.-W.; Hsieh, S.-T.; Ting, J.P.-Y.; Wu-Hsieh, B.A. NLRX1 Facilitates Histoplasma Capsulatum-Induced LC3-Associated Phagocytosis for Cytokine Production in Macrophages. Front. Immunol. 2018, 9, 2761. [Google Scholar] [CrossRef]
- Singh, K.; Sripada, L.; Lipatova, A.; Roy, M.; Prajapati, P.; Gohel, D.; Bhatelia, K.; Chumakov, P.M.; Singh, R. NLRX1 Resides in Mitochondrial RNA Granules and Regulates Mitochondrial RNA Processing and Bioenergetic Adaptation. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1260–1276. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; König, R.; Deng, M.; Riess, M.; Mo, J.; Zhang, L.; Petrucelli, A.; Yoh, S.M.; Barefoot, B.; Samo, M.; et al. NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses. Cell Host Microbe 2016, 19, 515–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X.; Cui, J.; Wang, H.Y.; Zhu, L.; Matsueda, S.; Wang, Q.; Yang, X.; Hong, J.; Songyang, Z.; Chen, Z.J.; et al. NLRX1 Negatively Regulates TLR-Induced NF-ΚB Signaling by Targeting TRAF6 and IKK. Immunity 2011, 34, 843–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, I.C.; Moore, C.B.; Schneider, M.; Lei, Y.; Davis, B.K.; Scull, M.A.; Gris, D.; Roney, K.E.; Zimmermann, A.G.; Bowzard, J.B.; et al. NLRX1 Protein Attenuates Inflammatory Responses to Infection by Interfering with the RIG-I-MAVS and TRAF6-NF-ΚB Signaling Pathways. Immunity 2011, 34, 854–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaworska, J.; Coulombe, F.; Downey, J.; Tzelepis, F.; Shalaby, K.; Tattoli, I.; Berube, J.; Rousseau, S.; Martin, J.G.; Girardin, S.E.; et al. NLRX1 Prevents Mitochondrial Induced Apoptosis and Enhances Macrophage Antiviral Immunity by Interacting with Influenza Virus PB1-F2 Protein. Proc. Natl. Acad. Sci. USA 2014, 111, E2110–E2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Wen, H.; Yu, Y.; Taxman, D.J.; Zhang, L.; Widman, D.G.; Swanson, K.V.; Wen, K.-W.; Damania, B.; Moore, C.B.; et al. The Mitochondrial Proteins NLRX1 and TUFM Form a Complex That Regulates Type I Interferon and Autophagy. Immunity 2012, 36, 933–946. [Google Scholar] [CrossRef] [Green Version]
- Jin, T.; Huang, M.; Jiang, J.; Smith, P.; Xiao, T.S. Crystal Structure of Human NLRP12 PYD Domain and Implication in Homotypic Interaction. PLoS ONE 2018, 13, e0190547. [Google Scholar] [CrossRef] [Green Version]
- Tuladhar, S.; Kanneganti, T.-D. NLRP12 in Innate Immunity and Inflammation. Mol. Asp. Med. 2020, 76, 100887. [Google Scholar] [CrossRef]
- Williams, K.L.; Lich, J.D.; Duncan, J.A.; Reed, W.; Rallabhandi, P.; Moore, C.; Kurtz, S.; Coffield, V.M.; Accavitti-Loper, M.A.; Su, L.; et al. The CATERPILLER Protein Monarch-1 Is an Antagonist of Toll-like Receptor-, Tumor Necrosis Factor Alpha-, and Mycobacterium Tuberculosis-Induced pro-Inflammatory Signals. J. Biol. Chem. 2005, 280, 39914–39924. [Google Scholar] [CrossRef] [Green Version]
- Volpe, G.; Mauro, A.; Mellos, A. NLRP12- Associated Autoinflammatory Disorder: Case Report. Pediatric Rheumatol. 2014, 12, P262. [Google Scholar] [CrossRef] [Green Version]
- Zaki, M.H.; Vogel, P.; Subbarao Malireddi, R.K.; Body-Malapel, M.; Anand, P.K.; Bertin, J.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.-D. The NOD-like Receptor NLRP12 Attenuates Colon Inflammation and Tumorigenesis. Cancer Cell 2011, 20, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Krauss, J.L.; Zeng, R.; Hickman-Brecks, C.L.; Wilson, J.E.; Ting, J.P.-Y.; Novack, D.V. NLRP12 Provides a Critical Checkpoint for Osteoclast Differentiation. Proc. Natl. Acad. Sci. USA 2015, 112, 10455–10460. [Google Scholar] [CrossRef] [Green Version]
- Zaki, M.H.; Man, S.M.; Vogel, P.; Lamkanfi, M.; Kanneganti, T.-D. Salmonella Exploits NLRP12-Dependent Innate Immune Signaling to Suppress Host Defenses during Infection. Proc. Natl. Acad. Sci. USA 2014, 111, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Zamoshnikova, A.; Groß, C.J.; Schuster, S.; Chen, K.W.; Wilson, A.; Tacchini-Cottier, F.; Schroder, K. NLRP12 Is a Neutrophil-Specific, Negative Regulator of in Vitro Cell Migration but Does Not Modulate LPS- or Infection-Induced NF-ΚB or ERK Signalling. Immunobiology 2016, 221, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Normand, S.; Waldschmitt, N.; Neerincx, A.; Martinez-Torres, R.J.; Chauvin, C.; Couturier-Maillard, A.; Boulard, O.; Cobret, L.; Awad, F.; Huot, L.; et al. Proteasomal Degradation of NOD2 by NLRP12 in Monocytes Promotes Bacterial Tolerance and Colonization by Enteropathogens. Nat. Commun. 2018, 9, 5338. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; McElvania-TeKippe, E.; Wilson, J.E.; Lich, J.D.; Arthur, J.C.; Sullivan, J.T.; Braunstein, M.; Ting, J.P.Y. Characterization of NLRP12 during the In Vivo Host Immune Response to Klebsiella Pneumoniae and Mycobacterium Tuberculosis. PLoS ONE 2013, 8, e60842. [Google Scholar] [CrossRef]
- Pudla, M.; Srisaowakarn, C.; Utaisincharoen, P. NLRP12 Negatively Modulates Inducible Nitric Oxide Synthase (INOS) Expression and Tumor Necrosis Factor-α Production in Porphyromonas Gingivalis LPS-Treated Mouse Macrophage Cell Line (RAW264.7). Inflamm. Res. 2019, 68, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-T.; Chen, L.; Lin, D.S.-C.; Chen, S.-Y.; Tsao, Y.-P.; Guo, H.; Li, F.-J.; Tseng, W.-T.; Tam, J.W.; Chao, C.-W.; et al. NLRP12 Regulates Anti-Viral RIG-I Activation via Interaction with TRIM25. Cell Host Microbe 2019, 25, 602–616.e7. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Manji, G.A.; Grenier, J.M.; Al-Garawi, A.; Merriam, S.; Lora, J.M.; Geddes, B.J.; Briskin, M.; DiStefano, P.S.; Bertin, J. PYPAF7, a Novel Pyrin-Containing Apaf1-like Protein That Regulates Activation of NF-ΚB and Caspase-1-Dependent Cytokine Processing. J. Biol. Chem. 2002, 277, 29874–29880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vladimer, G.I.; Weng, D.; Paquette, S.W.M.; Vanaja, S.K.; Rathinam, V.A.K.; Aune, M.H.; Conlon, J.E.; Burbage, J.J.; Proulx, M.K.; Liu, Q.; et al. The NLRP12 Inflammasome Recognizes Yersinia Pestis. Immunity 2012, 37, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Ataide, M.A.; Andrade, W.A.; Zamboni, D.S.; Wang, D.; do Carmo Souza, M.; Franklin, B.S.; Elian, S.; Martins, F.S.; Pereira, D.; Reed, G.; et al. Malaria-Induced NLRP12/NLRP3-Dependent Caspase-1 Activation Mediates Inflammation and Hypersensitivity to Bacterial Superinfection. PLoS Pathog. 2014, 10, e1003885. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Deng, Y.; Gan, X.; Li, Y.; Huang, W.; Lu, L.; Wei, L.; Su, L.; Luo, J.; Zou, B.; et al. NLRP12 Collaborates with NLRP3 and NLRC4 to Promote Pyroptosis Inducing Ganglion Cell Death of Acute Glaucoma. Mol. Neurodegener. 2020, 15, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulon, P.-G.; Dhanushkodi, N.; Prakash, S.; Srivastava, R.; Roy, S.; Alomari, N.I.; Nguyen, A.M.; Warsi, W.R.; Ye, C.; Carlos-Cruz, E.A.; et al. NLRP3, NLRP12, and IFI16 Inflammasomes Induction and Caspase-1 Activation Triggered by Virulent HSV-1 Strains Are Associated With Severe Corneal Inflammatory Herpetic Disease. Front. Immunol. 2019, 10, 1631. [Google Scholar] [CrossRef] [Green Version]
- Silveira, T.N.; Gomes, M.T.R.; Oliveira, L.S.; Campos, P.C.; Machado, G.G.; Oliveira, S.C. NLRP12 Negatively Regulates Proinflammatory Cytokine Production and Host Defense against Brucella Abortus. Eur. J. Immunol. 2017, 47, 51–59. [Google Scholar] [CrossRef]
- Phulphagar, K.; Kühn, L.I.; Ebner, S.; Frauenstein, A.; Swietlik, J.J.; Rieckmann, J.; Meissner, F. Proteomics Reveals Distinct Mechanisms Regulating the Release of Cytokines and Alarmins during Pyroptosis. Cell Rep. 2021, 34, 108826. [Google Scholar] [CrossRef] [PubMed]
- Heilig, R.; Dick, M.S.; Sborgi, L.; Meunier, E.; Hiller, S.; Broz, P. The Gasdermin-D Pore Acts as a Conduit for IL-1β Secretion in Mice. Eur. J. Immunol. 2018, 48, 584–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Sun, B. Neutrophil Pyroptosis: New Perspectives on Sepsis. Cell. Mol. Life Sci. 2019, 76, 2031–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedlander, A.M. Macrophages Are Sensitive to Anthrax Lethal Toxin through an Acid-Dependent Process. J. Biol. Chem. 1986, 261, 7123–7126. [Google Scholar] [CrossRef]
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella Flexneri Induces Apoptosis in Infected Macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef]
- Black, R.A.; Kronheim, S.R.; Merriam, J.E.; March, C.J.; Hopp, T.P. A Pre-Aspartate-Specific Protease from Human Leukocytes That Cleaves Pro-Interleukin-1 β. J. Biol. Chem. 1989, 264, 5323–5326. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Bull, H.G.; Calaycay, J.R.; Chapman, K.T.; Howard, A.D.; Kostura, M.J.; Miller, D.K.; Molineaux, S.M.; Weidner, J.R.; Aunins, J. A Novel Heterodimeric Cysteine Protease Is Required for Interleukin-1 Beta Processing in Monocytes. Nature 1992, 356, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Cerretti, D.P.; Kozlosky, C.J.; Mosley, B.; Nelson, N.; Van Ness, K.; Greenstreet, T.A.; March, C.J.; Kronheim, S.R.; Druck, T.; Cannizzaro, L.A. Molecular Cloning of the Interleukin-1 Beta Converting Enzyme. Science 1992, 256, 97–100. [Google Scholar] [CrossRef]
- D’Souza, C.A.; Heitman, J. Dismantling the Cryptococcus Coat. Trends Microbiol. 2001, 9, 112–113. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and Diseases. Sig. Transduct. Target Ther. 2021, 6, 1–21. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of ProIL-Beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Saeki, N.; Kuwahara, Y.; Sasaki, H.; Satoh, H.; Shiroishi, T. Gasdermin (Gsdm) Localizing to Mouse Chromosome 11 Is Predominantly Expressed in Upper Gastrointestinal Tract but Significantly Suppressed in Human Gastric Cancer Cells. Mamm. Genome 2000, 11, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Wang, S.; Wang, J. Mechanism and Regulation of Pyroptosis-Mediated in Cancer Cell Death. Chem. -Biol. Interact. 2020, 323, 109052. [Google Scholar] [CrossRef]
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin Pores Permeabilize Mitochondria to Augment Caspase-3 Activation during Apoptosis and Inflammasome Activation. Nat. Commun. 2019, 10, 1689. [Google Scholar] [CrossRef]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 Induces Cleavage of Gasdermin D to Elicit Pyroptosis during Yersinia Infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897. [Google Scholar] [CrossRef] [Green Version]
- Burgener, S.S.; Leborgne, N.G.F.; Snipas, S.J.; Salvesen, G.S.; Bird, P.I.; Benarafa, C. Cathepsin G Inhibition by Serpinb1 and Serpinb6 Prevents Programmed Necrosis in Neutrophils and Monocytes and Reduces GSDMD-Driven Inflammation. Cell Rep. 2019, 27, 3646–3656.e5. [Google Scholar] [CrossRef] [Green Version]
- Sollberger, G.; Tilley, D.O.; Zychlinsky, A. Neutrophil Extracellular Traps: The Biology of Chromatin Externalization. Dev. Cell 2018, 44, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.K.; Vanaja, S.K.; Waggoner, L.; Sokolovska, A.; Becker, C.; Stuart, L.M.; Leong, J.M.; Fitzgerald, K.A. TRIF Licenses Caspase-11-Dependent NLRP3 Inflammasome Activation by Gram-Negative Bacteria. Cell 2012, 150, 606–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rühl, S.; Broz, P. Caspase-11 Activates a Canonical NLRP3 Inflammasome by Promoting K+ Efflux. Eur. J. Immunol. 2015, 45, 2927–2936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, J.K.Y.; Moriyama, M.; Iwasaki, A. Inflammasomes and Pyroptosis as Therapeutic Targets for COVID-19. J. Immunol. 2020, 205, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Chen, Q.; Li, X.; Zeng, Z.; Xiong, W.; Li, G.; Li, X.; Yang, J.; Xiang, B.; Yi, M. Pyroptosis: A New Paradigm of Cell Death for Fighting against Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 153. [Google Scholar] [CrossRef]
- Hachim, M.Y.; Khalil, B.A.; Elemam, N.M.; Maghazachi, A.A. Pyroptosis: The Missing Puzzle among Innate and Adaptive Immunity Crosstalk. J. Leukoc. Biol. 2020, 108, 323–338. [Google Scholar] [CrossRef]
- Guo, Y.; Li, L.; Xu, T.; Guo, X.; Wang, C.; Li, Y.; Yang, Y.; Yang, D.; Sun, B.; Zhao, X.; et al. HUWE1 Mediates Inflammasome Activation and Promotes Host Defense against Bacterial Infection. J. Clin. Investig. 2020, 130, 6301–6316. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-A.; Chiang, B.-L. Inflammasomes and Childhood Autoimmune Diseases: A Review of Current Knowledge. Clin. Rev. Allerg. Immunol. 2020, 61, 1–15. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, H.; Yao, X.; Zhang, D.; Zhou, Y.; Fu, B.; Wang, W.; Li, H.; Wang, Z.; Hu, Z.; et al. Pyroptotic Macrophages Stimulate the SARS-CoV-2-Associated Cytokine Storm. Cell Mol. Immunol. 2021, 18, 1305–1307. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed Cell Death as a Defence against Infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Tarayrah-Ibraheim, L.; Maurice, E.C.; Hadary, G.; Ben-Hur, S.; Kolpakova, A.; Braun, T.; Peleg, Y.; Yacobi-Sharon, K.; Arama, E. DNase II Mediates a Parthanatos-like Developmental Cell Death Pathway in Drosophila Primordial Germ Cells. Nat. Commun. 2021, 12, 2285. [Google Scholar] [CrossRef]
- Green, D.R. The Coming Decade of Cell Death Research: Five Riddles. Cell 2019, 177, 1094–1107. [Google Scholar] [CrossRef]
- Ashida, H.; Mimuro, H.; Ogawa, M.; Kobayashi, T.; Sanada, T.; Kim, M.; Sasakawa, C. Cell Death and Infection: A Double-Edged Sword for Host and Pathogen Survival. J. Cell Biol. 2011, 195, 931–942. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory Caspases Are Innate Immune Receptors for Intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Adler, B.; Adler, H. Type I Interferon Signaling and Macrophages: A Double-Edged Sword? Cell. Mol. Immunol. 2021, 1–2. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe COVID-19 Patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B.; Meyerholz, D.K.; Perlman, S. IFN-I Response Timing Relative to Virus Replication Determines MERS Coronavirus Infection Outcomes. J. Clin. Investig. 2019, 129, 3625–3639. [Google Scholar] [CrossRef]
- Peignier, A.; Parker, D. Impact of Type I Interferons on Susceptibility to Bacterial Pathogens. Trends Microbiol. 2021, 29, 823–835. [Google Scholar] [CrossRef]
- Li, Y.; Guo, X.; Hu, C.; Du, Y.; Guo, C.; Wang, D.; Zhao, W.; Huang, G.; Li, C.; Lu, Q.; et al. Type I IFN Operates Pyroptosis and Necroptosis during Multidrug-Resistant, A. Baumannii Infection. Cell Death Differ. 2018, 25, 1304–1318. [Google Scholar] [CrossRef]
- Mancuso, G.; Midiri, A.; Biondo, C.; Beninati, C.; Zummo, S.; Galbo, R.; Tomasello, F.; Gambuzza, M.; Macrì, G.; Ruggeri, A.; et al. Type I IFN Signaling Is Crucial for Host Resistance against Different Species of Pathogenic Bacteria. J. Immunol. 2007, 178, 3126–3133. [Google Scholar] [CrossRef]
- Lippmann, J.; Müller, H.C.; Naujoks, J.; Tabeling, C.; Shin, S.; Witzenrath, M.; Hellwig, K.; Kirschning, C.J.; Taylor, G.A.; Barchet, W.; et al. Dissection of a Type I Interferon Pathway in Controlling Bacterial Intracellular Infection in Mice. Cell. Microbiol. 2011, 13, 1668–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruangkiattikul, N.; Rys, D.; Abdissa, K.; Rohde, M.; Semmler, T.; Tegtmeyer, P.-K.; Kalinke, U.; Schwarz, C.; Lewin, A.; Goethe, R. Type I Interferon Induced by TLR2-TLR4-MyD88-TRIF-IRF3 Controls Mycobacterium Abscessus Subsp. Abscessus Persistence in Murine Macrophages via Nitric Oxide. Int. J. Med. Microbiol. 2019, 309, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Sebina, I.; Haque, A. Effects of Type I Interferons in Malaria. Immunology 2018, 155, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyedmousavi, S.; Davis, M.J.; Sugui, J.A.; Pinkhasov, T.; Moyer, S.; Salazar, A.M.; Chang, Y.C.; Kwon-Chung, K.J. Exogenous Stimulation of Type I Interferon Protects Mice with Chronic Granulomatous Disease from Aspergillosis through Early Recruitment of Host-Protective Neutrophils into the Lung. mBio 2018, 9, e00422-18. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, U.M.; Prantner, D.; Sikes, J.D.; Andrews, C.W.; Goodwin, A.M.; Nagarajan, S.; Darville, T. Type I Interferon Signaling Exacerbates Chlamydia Muridarum Genital Infection in a Murine Model. Infect. Immun. 2008, 76, 4642–4648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hussain, T.; Zhang, K.; Liao, Y.; Yao, J.; Song, Y.; Sabir, N.; Cheng, G.; Dong, H.; Li, M.; et al. Inhibition of Type I Interferon Signaling Abrogates Early Mycobacterium Bovis Infection. BMC Infect. Dis. 2019, 19, 1031. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Robinson, K.M.; McHugh, K.J.; Scheller, E.V.; Mandalapu, S.; Chen, C.; Di, Y.P.; Clay, M.E.; Enelow, R.I.; Dubin, P.J.; et al. Influenza-Induced Type I Interferon Enhances Susceptibility to Gram-Negative and Gram-Positive Bacterial Pneumonia in Mice. Am. J. Physiol. Lung. Cell Mol. Physiol. 2015, 309, L158–L167. [Google Scholar] [CrossRef] [PubMed]
- Henry, T.; Kirimanjeswara, G.S.; Ruby, T.; Jones, J.W.; Peng, K.; Perret, M.; Ho, L.; Sauer, J.-D.; Iwakura, Y.; Metzger, D.W.; et al. Type I IFN Signaling Constrains IL-17A/F Secretion by Gammadelta T Cells during Bacterial Infections. J. Immunol. 2010, 184, 3755–3767. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Zhang, X.; Ma, C.; Xu, W.; Gan, L.; Cui, J.; Yin, Y.; Wang, H. Nontypeable Haemophilus Influenzae DNA Stimulates Type I Interferon Expression via STING Signaling Pathway. Biochim. Biophys. Acta (BBA) -Mol. Cell Res. 2018, 1865, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Deriu, E.; Boxx, G.M.; He, X.; Pan, C.; Benavidez, S.D.; Cen, L.; Rozengurt, N.; Shi, W.; Cheng, G. Influenza Virus Affects Intestinal Microbiota and Secondary Salmonella Infection in the Gut through Type I Interferons. PLoS Pathog. 2016, 12, e1005572. [Google Scholar] [CrossRef] [PubMed]
- Riedelberger, M.; Penninger, P.; Tscherner, M.; Seifert, M.; Jenull, S.; Brunnhofer, C.; Scheidl, B.; Tsymala, I.; Bourgeois, C.; Petryshyn, A.; et al. Type I Interferon Response Dysregulates Host Iron Homeostasis and Enhances Candida Glabrata Infection. Cell Host Microbe 2020, 27, 454–466.e8. [Google Scholar] [CrossRef] [Green Version]
- Chessler, A.-D.C.; Caradonna, K.L.; Da’dara, A.; Burleigh, B.A. Type I Interferons Increase Host Susceptibility to Trypanosoma Cruzi Infection. Infect. Immun. 2011, 79, 2112–2119. [Google Scholar] [CrossRef] [Green Version]
- Pagliuso, A.; Tham, T.N.; Allemand, E.; Robertin, S.; Dupuy, B.; Bertrand, Q.; Bécavin, C.; Koutero, M.; Najburg, V.; Nahori, M.-A.; et al. An RNA-Binding Protein Secreted by a Bacterial Pathogen Modulates RIG-I Signaling. Cell Host Microbe 2019, 26, 823–835.e11. [Google Scholar] [CrossRef] [Green Version]
- Pitts, M.G.; Myers-Morales, T.; D’Orazio, S.E.F. Type I IFN Does Not Promote Susceptibility to Foodborne Listeria Monocytogenes. J. Immunol. 2016, 196, 3109–3116. [Google Scholar] [CrossRef] [Green Version]
- Hiruma, T.; Tsuyuzaki, H.; Uchida, K.; Trapnell, B.C.; Yamamura, Y.; Kusakabe, Y.; Totsu, T.; Suzuki, T.; Morita, S.; Doi, K.; et al. IFN-β Improves Sepsis-Related Alveolar Macrophage Dysfunction and Postseptic Acute Respiratory Distress Syndrome-Related Mortality. Am. J. Respir. Cell Mol. Biol. 2018, 59, 45–55. [Google Scholar] [CrossRef]
- Pylaeva, E.; Bordbari, S.; Spyra, I.; Decker, A.S.; Häussler, S.; Vybornov, V.; Lang, S.; Jablonska, J. Detrimental Effect of Type I IFNs during Acute Lung Infection With Pseudomonas Aeruginosa Is Mediated Through the Stimulation of Neutrophil NETosis. Front. Immunol. 2019, 10, 2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, N.; McComb, S.; Mulligan, R.; Dudani, R.; Krishnan, L.; Sad, S. Type I Interferon Induces Necroptosis in Macrophages during Infection with Salmonella Enterica Serovar Typhimurium. Nat. Immunol. 2012, 13, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Agbayani, G.; Wachholz, K.; Murphy, S.P.; Sad, S.; Krishnan, L. Type I Interferons Differentially Modulate Maternal Host Immunity to Infection by Listeria Monocytogenes and Salmonella Enterica Serovar Typhimurium during Pregnancy. Am. J. Reprod. Immunol. 2019, 81, e13068. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Lee-Lewis, H.; Hughes-Hanks, J.; Lewis, C.A.; Anderson, D.M. Opposing Roles for Interferon Regulatory Factor-3 (IRF-3) and Type I Interferon Signaling during Plague. PLoS Pathog. 2012, 8, e1002817. [Google Scholar] [CrossRef] [PubMed]
- Frantz, R.; Teubner, L.; Schultze, T.; La Pietra, L.; Müller, C.; Gwozdzinski, K.; Pillich, H.; Hain, T.; Weber-Gerlach, M.; Panagiotidis, G.-D.; et al. The SecRNome of Listeria Monocytogenes Harbors Small Noncoding RNAs That Are Potent Inducers of Beta Interferon. mBio 2019, 10, e01223-19. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Jiang, X.; Pfau, D.; Ling, Y.; Nathan, C.F. Type I Interferon Signaling Mediates Mycobacterium Tuberculosis-Induced Macrophage Death. J. Exp. Med. 2021, 218, e20200887. [Google Scholar] [CrossRef]
- Pothlichet, J.; Meunier, I.; Davis, B.K.; Ting, J.P.-Y.; Skamene, E.; von Messling, V.; Vidal, S.M. Type I IFN Triggers RIG-I/TLR3/NLRP3-Dependent Inflammasome Activation in Influenza A Virus Infected Cells. PLoS Pathog. 2013, 9, e1003256. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Ruby, T.; Belhocine, K.; Bouley, D.M.; Kayagaki, N.; Dixit, V.M.; Monack, D.M. Caspase-11 Increases Susceptibility to Salmonella Infection in the Absence of Caspase-1. Nature 2012, 490, 288–291. [Google Scholar] [CrossRef]
- Kumar, V. The Trinity of CGAS, TLR9, and ALRs Guardians of the Cellular Galaxy Against Host-Derived Self-DNA. Front. Immunol. 2021, 11, 3902. [Google Scholar] [CrossRef]
- Zhang, L.; Mo, J.; Swanson, K.V.; Wen, H.; Petrucelli, A.; Gregory, S.M.; Zhang, Z.; Schneider, M.; Jiang, Y.; Fitzgerald, K.A.; et al. NLRC3, a Member of the NLR Family of Proteins, Is a Negative Regulator of Innate Immune Signaling Induced by the DNA Sensor STING. Immunity 2014, 40, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Deng, M.; Petrucelli, A.S.; Zhu, C.; Mo, J.; Zhang, L.; Tam, J.W.; Ariel, P.; Zhao, B.; Zhang, S.; et al. Viral DNA Binding to NLRC3, an Inhibitory Nucleic Acid Sensor, Unleashes STING, a Cyclic Dinucleotide Receptor That Activates Type I Interferon. Immunity 2019, 50, 591–599.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]

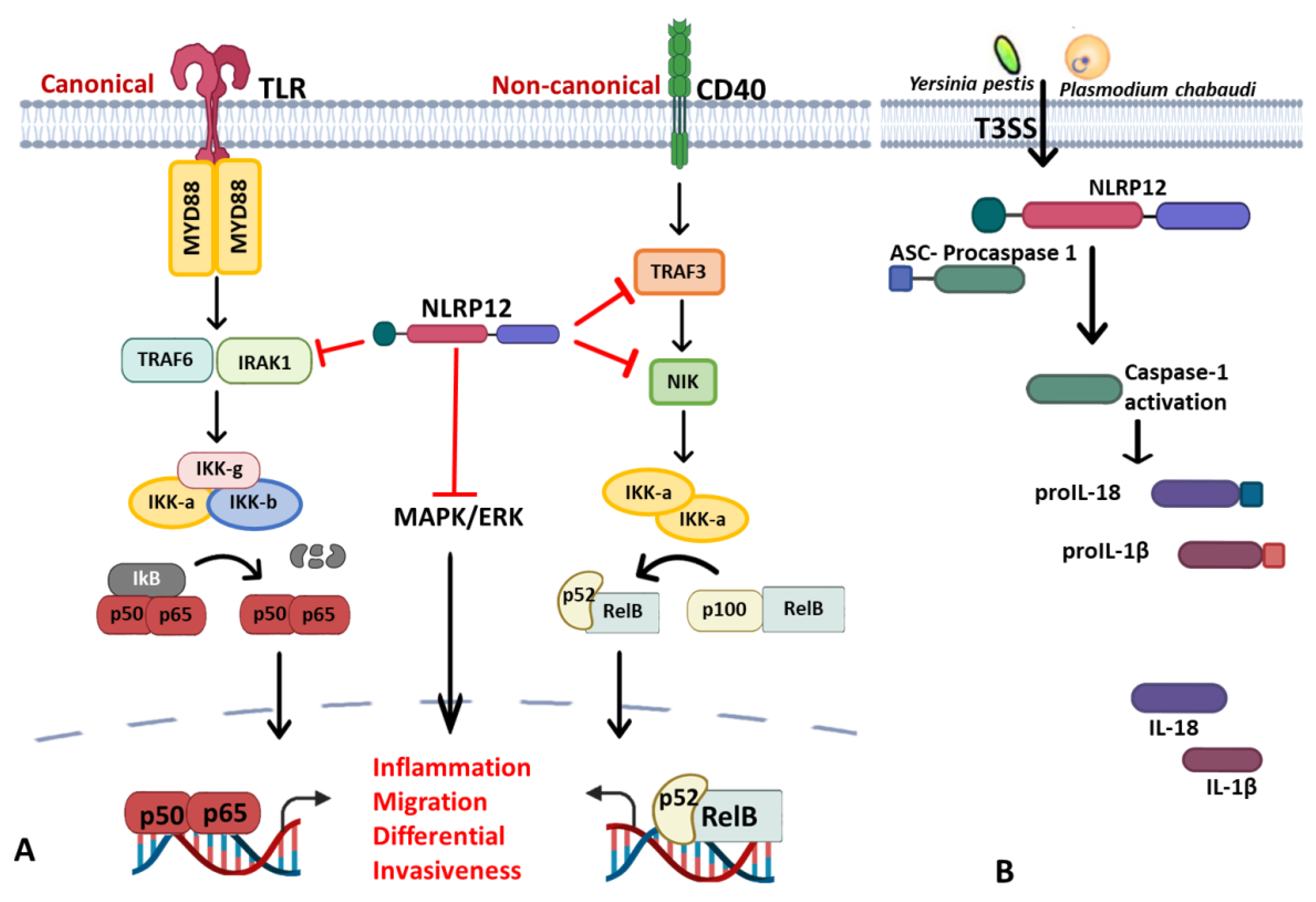


| NLRs Family | Sub-Family/Domain Architectures | Gene |
|---|---|---|
| Acidic transcription-carrying domain (NLRA) |  | CIITA (NLRA) |
| BIR- carrying domain (NLRB) |  | NLRB (NAIP) |
| CARD—carrying domain (NLRC) |  | NOD1, NLRC4 NOD2 |
| PYD-carrying domain |  | NLRP2-NLRP9 NLRP11-NLRP14 |
Additional domain (FIIND) | NLRP1 | |
No LRR | NLRP10 | |
| Unidentified domain | NLRs without PYD nor CARD | NLRC3 NLRC5 NLRX1 |
| NLR-related molecules |  | Apaf-1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babamale, A.O.; Chen, S.-T. Nod-like Receptors: Critical Intracellular Sensors for Host Protection and Cell Death in Microbial and Parasitic Infections. Int. J. Mol. Sci. 2021, 22, 11398. https://doi.org/10.3390/ijms222111398
Babamale AO, Chen S-T. Nod-like Receptors: Critical Intracellular Sensors for Host Protection and Cell Death in Microbial and Parasitic Infections. International Journal of Molecular Sciences. 2021; 22(21):11398. https://doi.org/10.3390/ijms222111398
Chicago/Turabian StyleBabamale, Abdulkareem Olarewaju, and Szu-Ting Chen. 2021. "Nod-like Receptors: Critical Intracellular Sensors for Host Protection and Cell Death in Microbial and Parasitic Infections" International Journal of Molecular Sciences 22, no. 21: 11398. https://doi.org/10.3390/ijms222111398
APA StyleBabamale, A. O., & Chen, S.-T. (2021). Nod-like Receptors: Critical Intracellular Sensors for Host Protection and Cell Death in Microbial and Parasitic Infections. International Journal of Molecular Sciences, 22(21), 11398. https://doi.org/10.3390/ijms222111398