
Targetable Pathways for Alleviating Mitochondrial Dysfunction in Neurodegeneration of Metabolic and Non-Metabolic Diseases





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
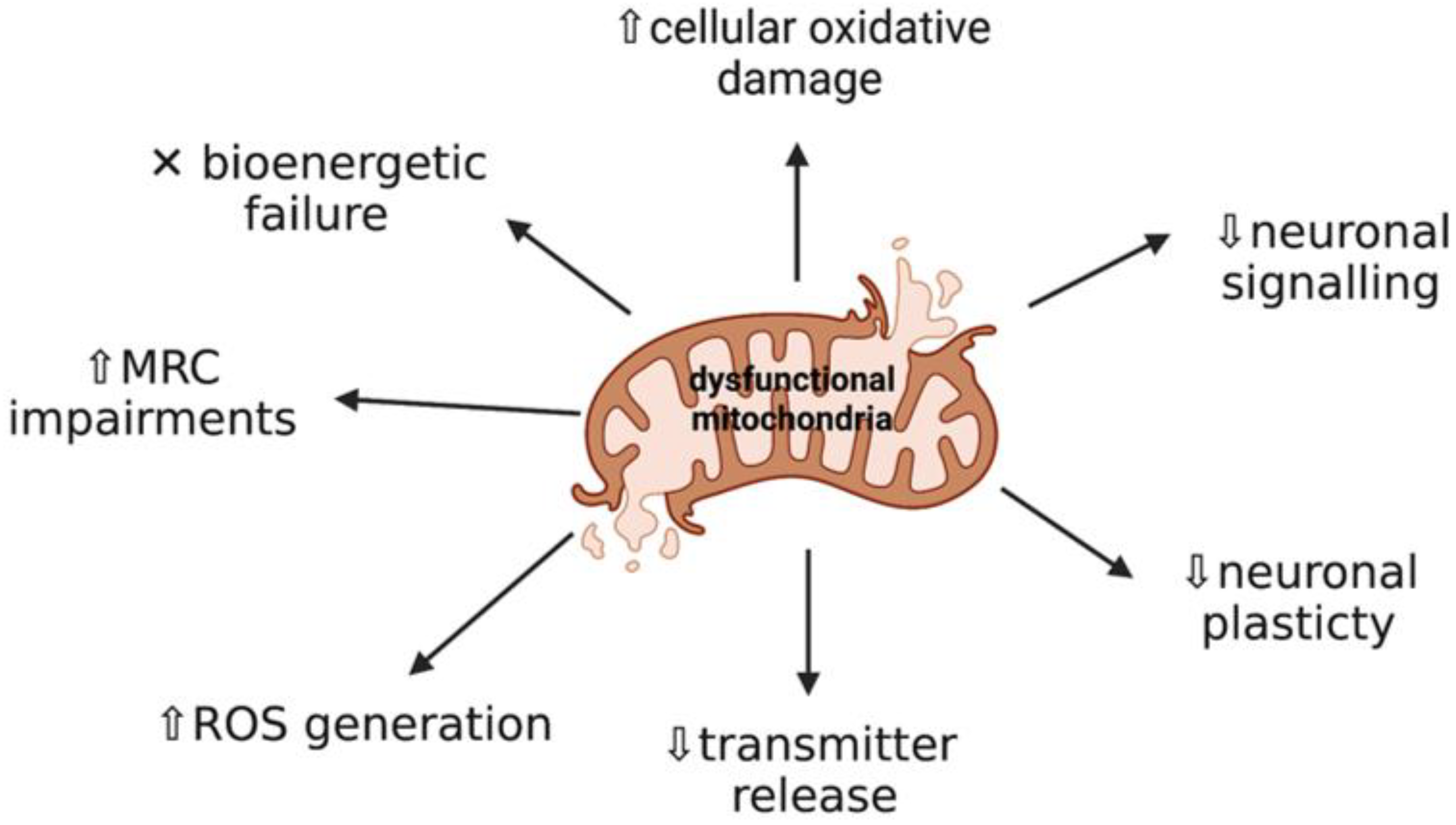
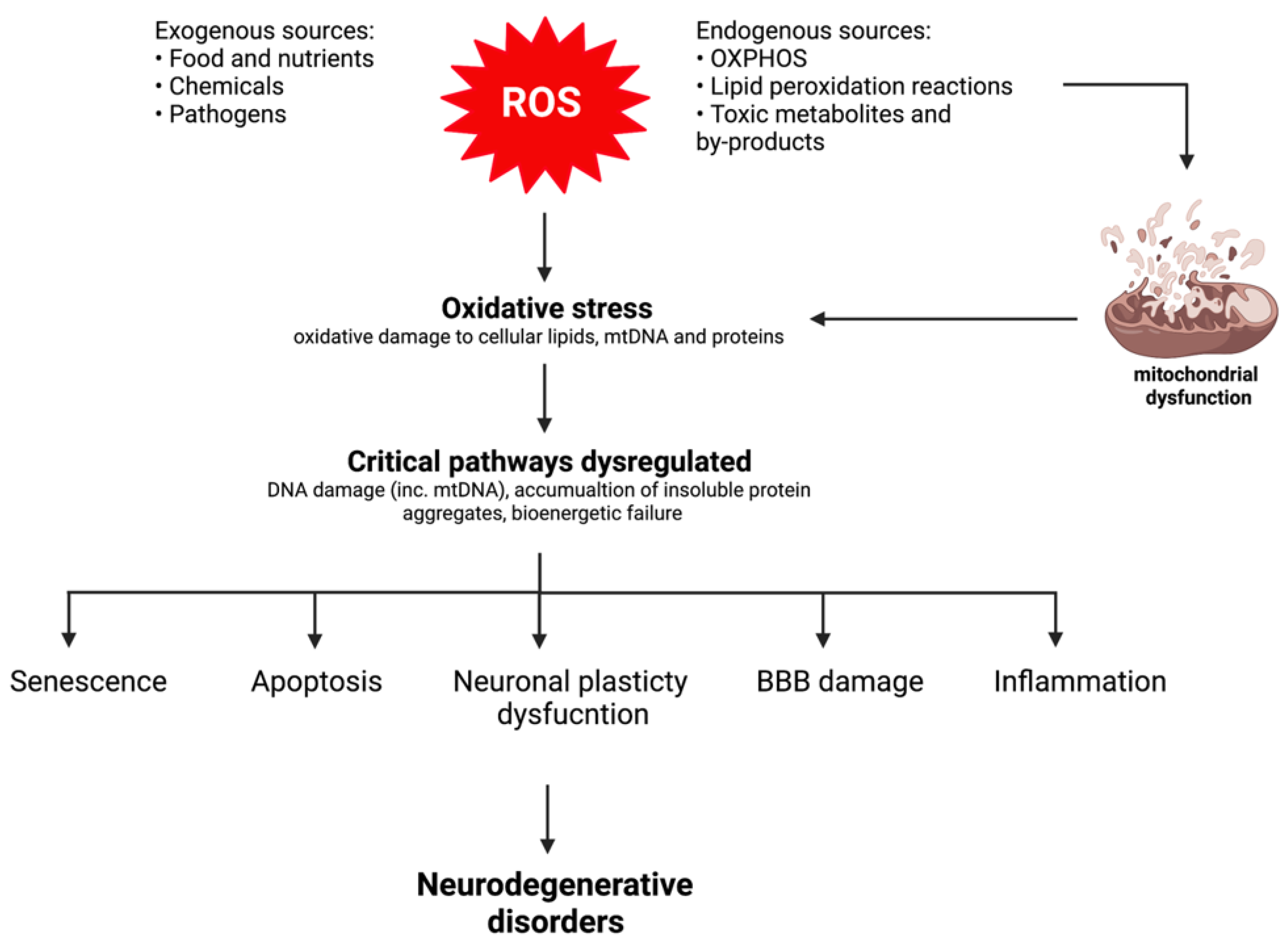
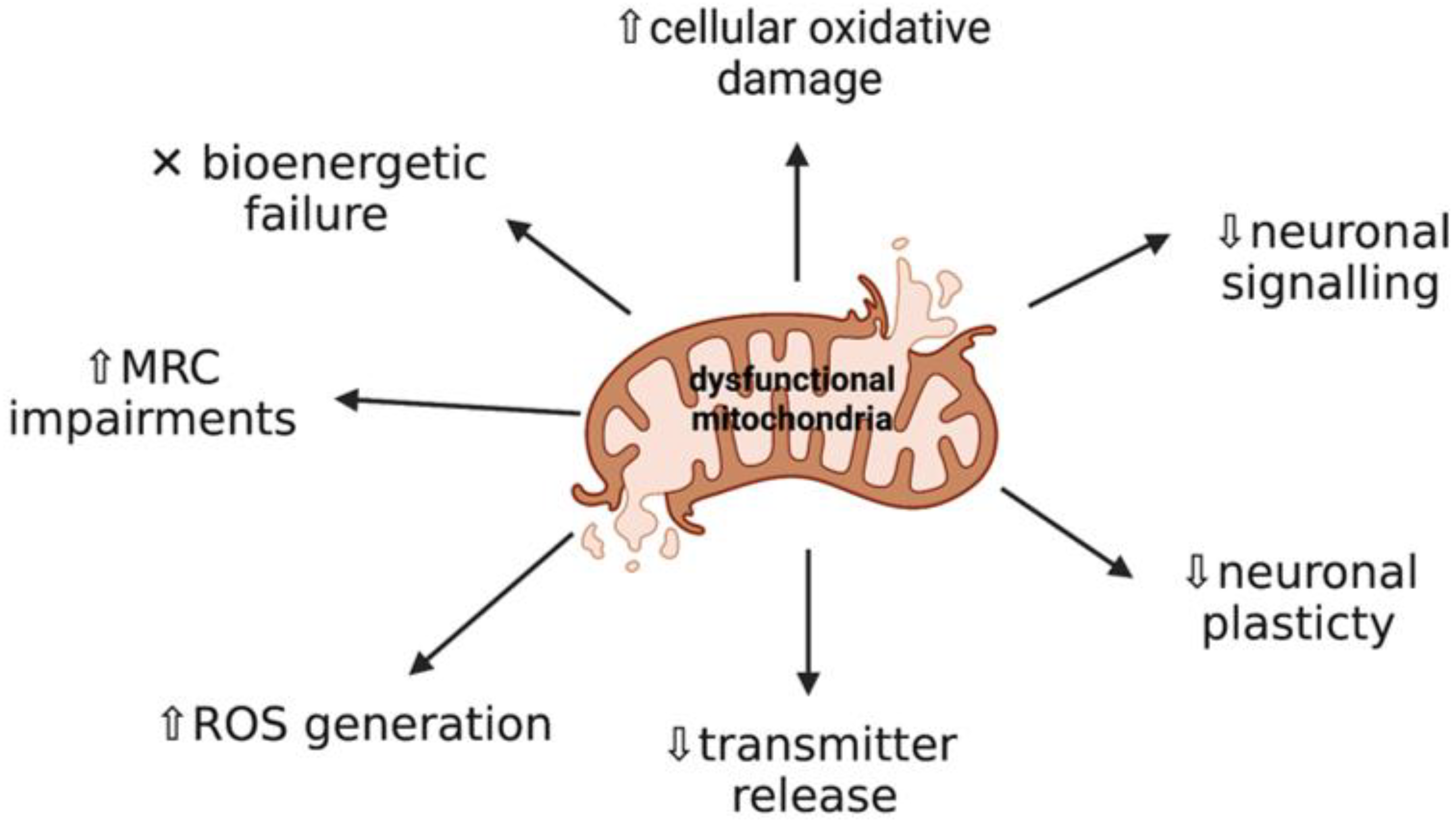
2. Mitochondrial Dysfunction and Oxidative Stress: Key Events in Neurodegeneration
3. Altered Signaling Pathways Leading to Mitochondrial Dysfunction in Neurological Disease
3.1. Protein Function in Health
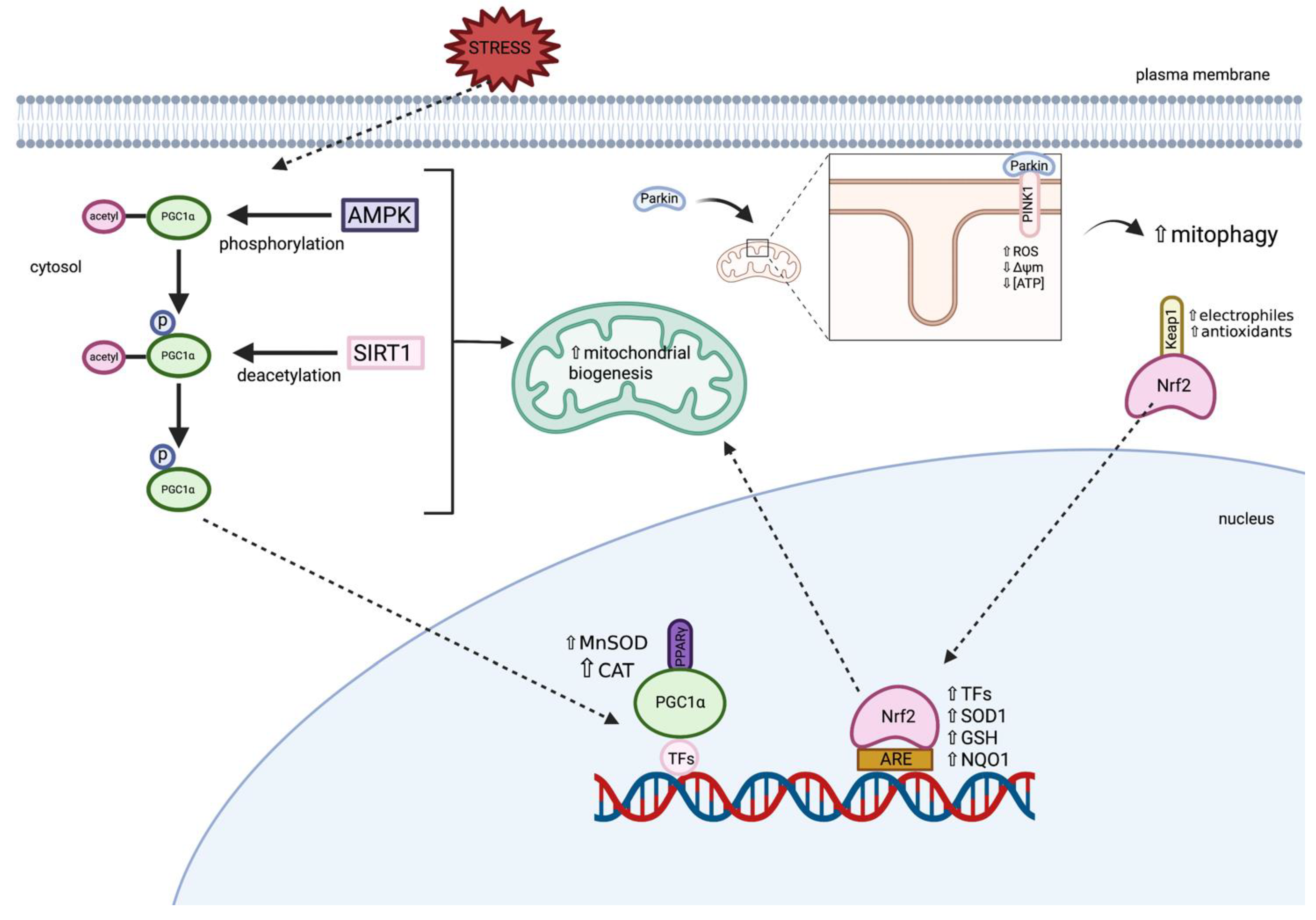
3.1.1. Parkin and PINK1
3.1.2. Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha (PGC1α)
3.1.3. Peroxisome Proliferator-Activated Receptor Gamma (PPARγ)
3.1.4. Nuclear Factor Erythroid 2-Related Factor (Nrf2)
3.2. Non-Communicable Diseases
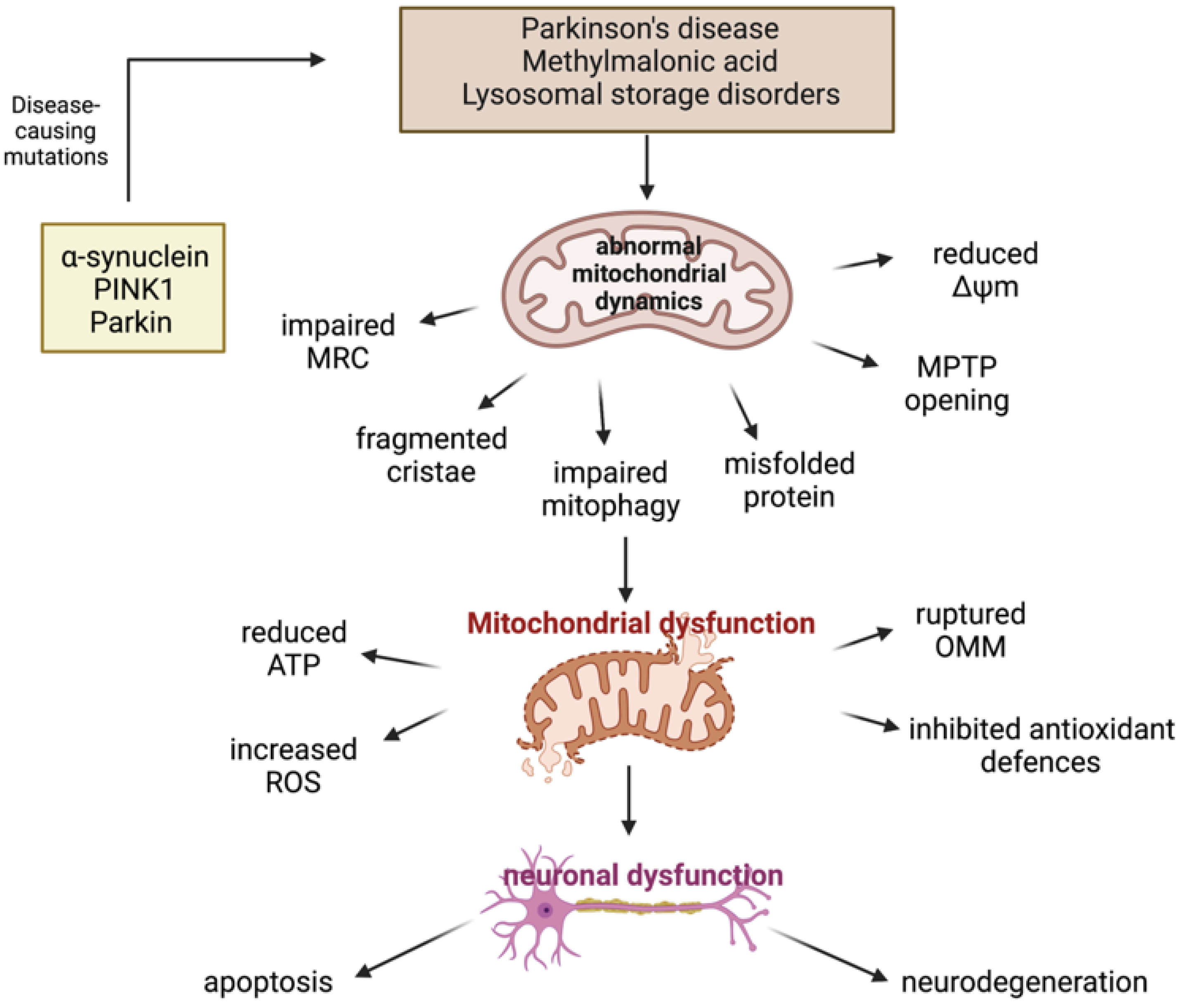
3.2.1. Parkinson’s Disease
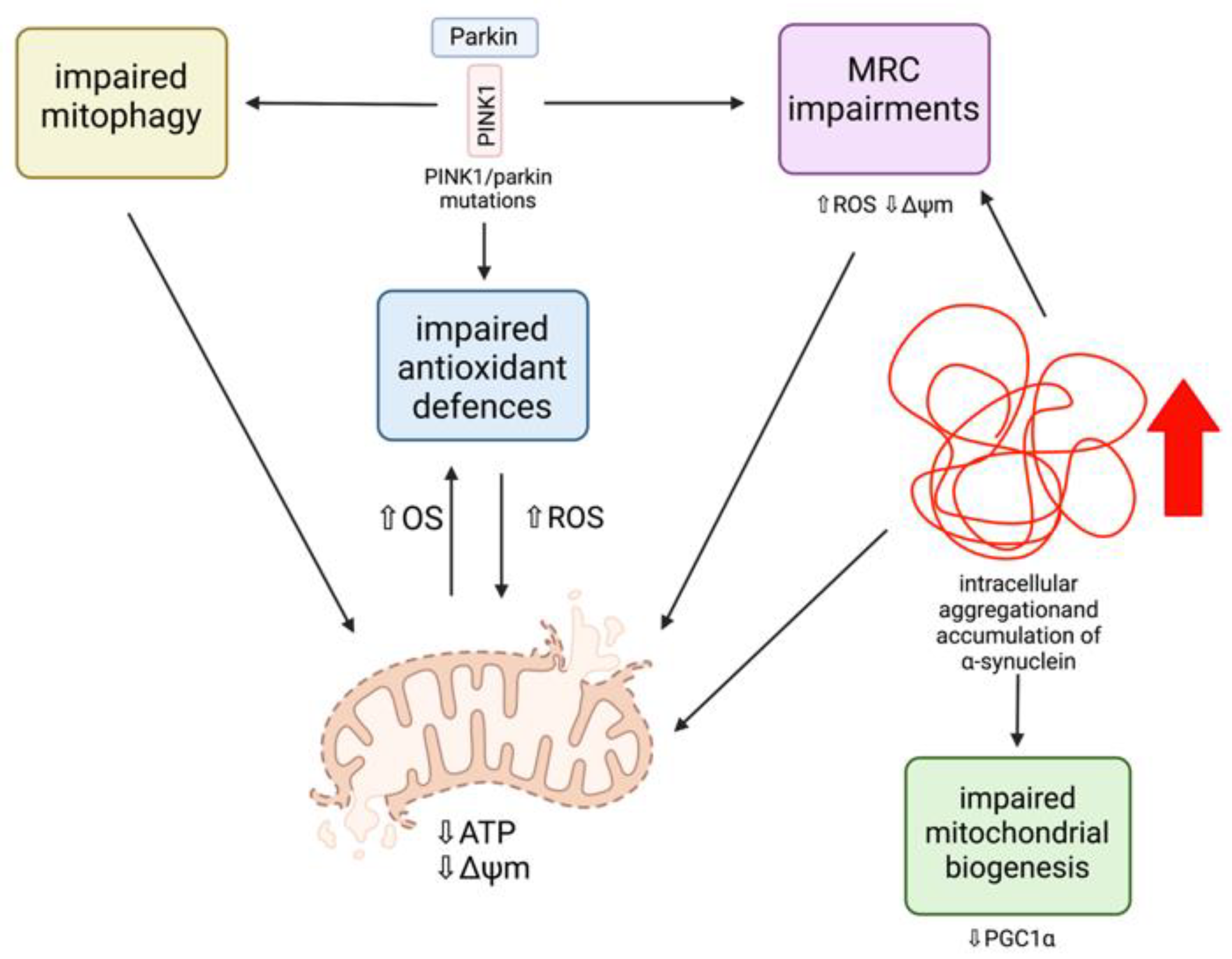
PINK1/Parkin Mutations and Mitochondrial Dysfunction
α-Synuclein and Mitochondrial Dysfunction
3.3. Inherited Metabolic Disorders
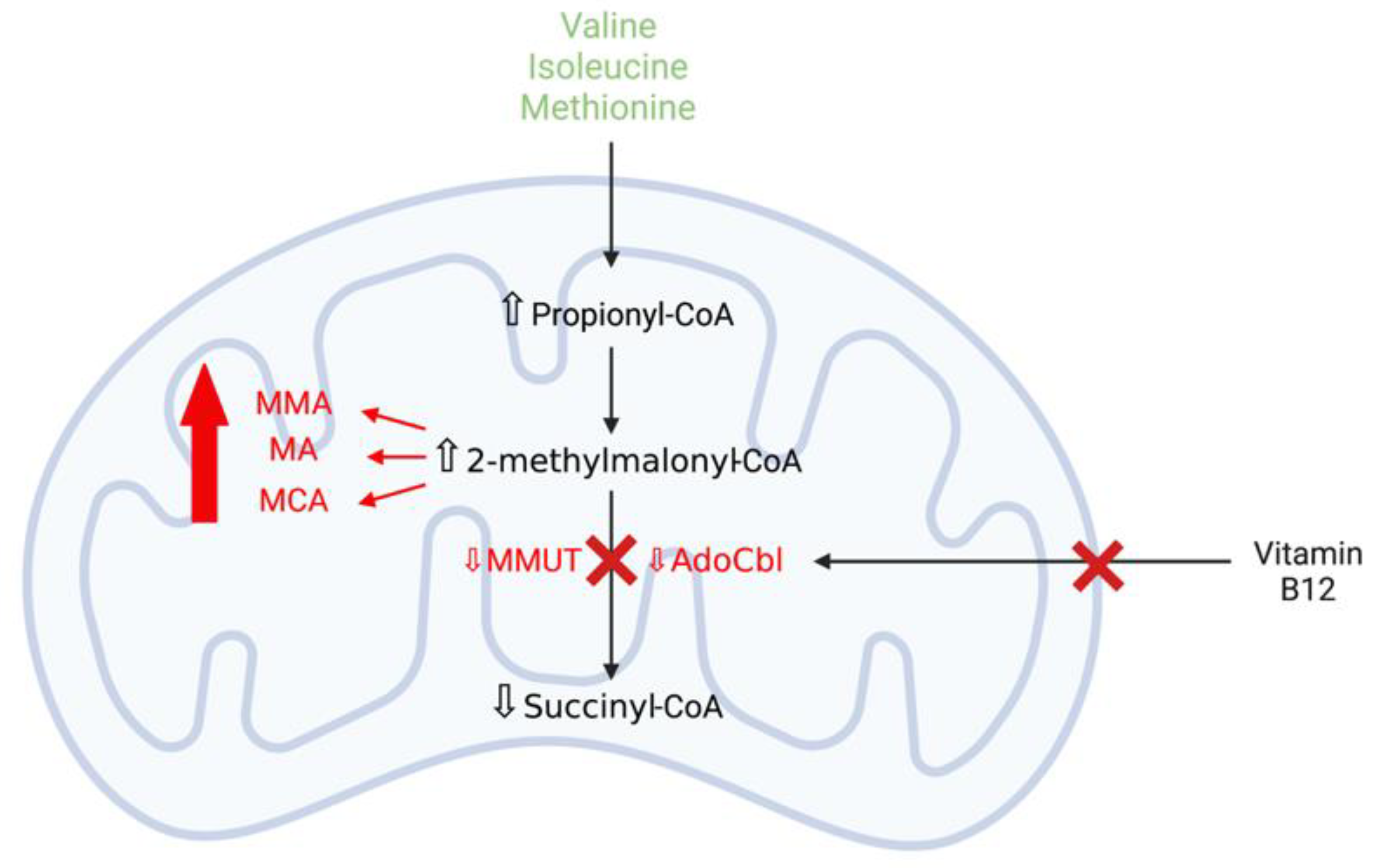
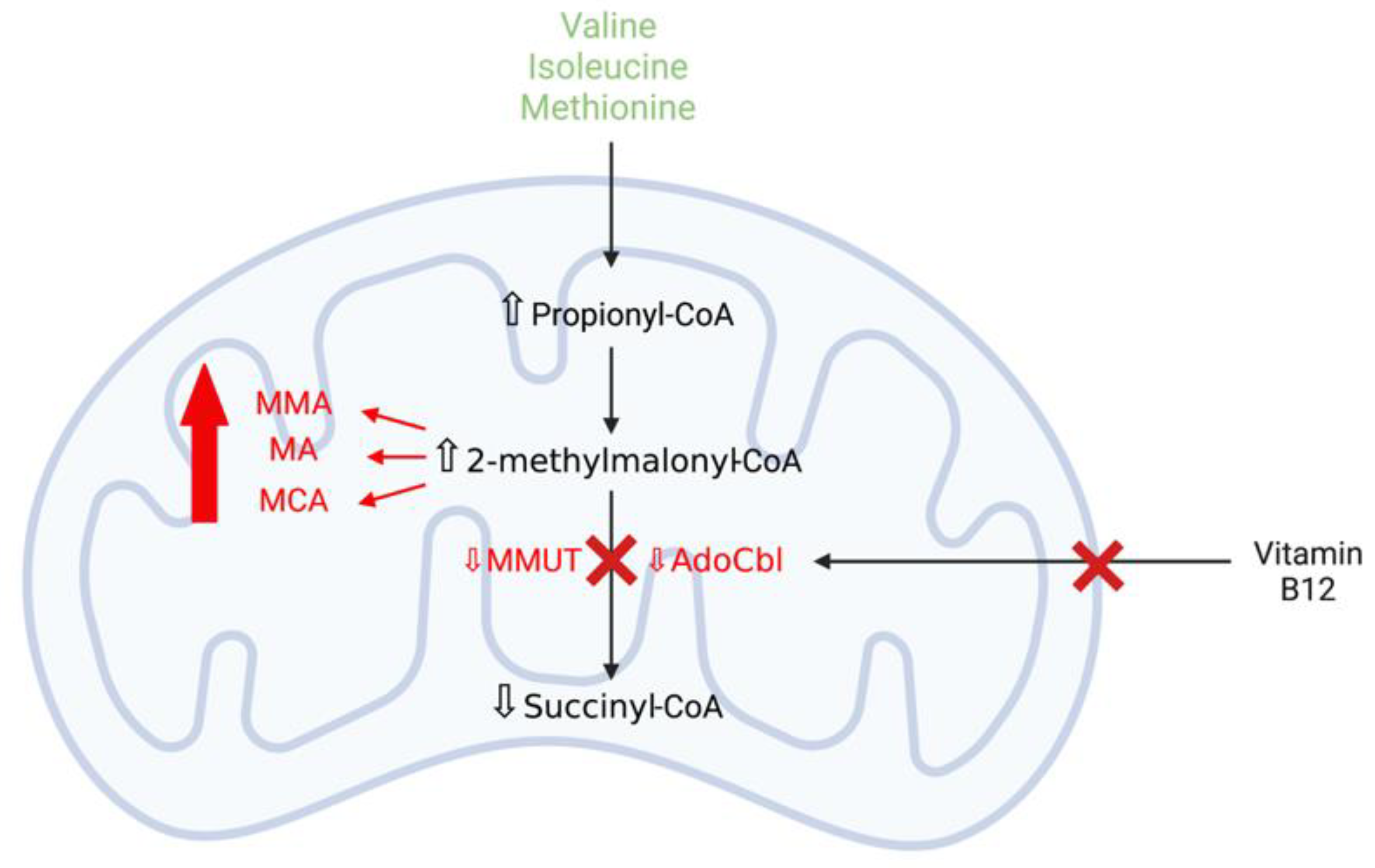
3.3.1. Methylmalonic Acidaemia
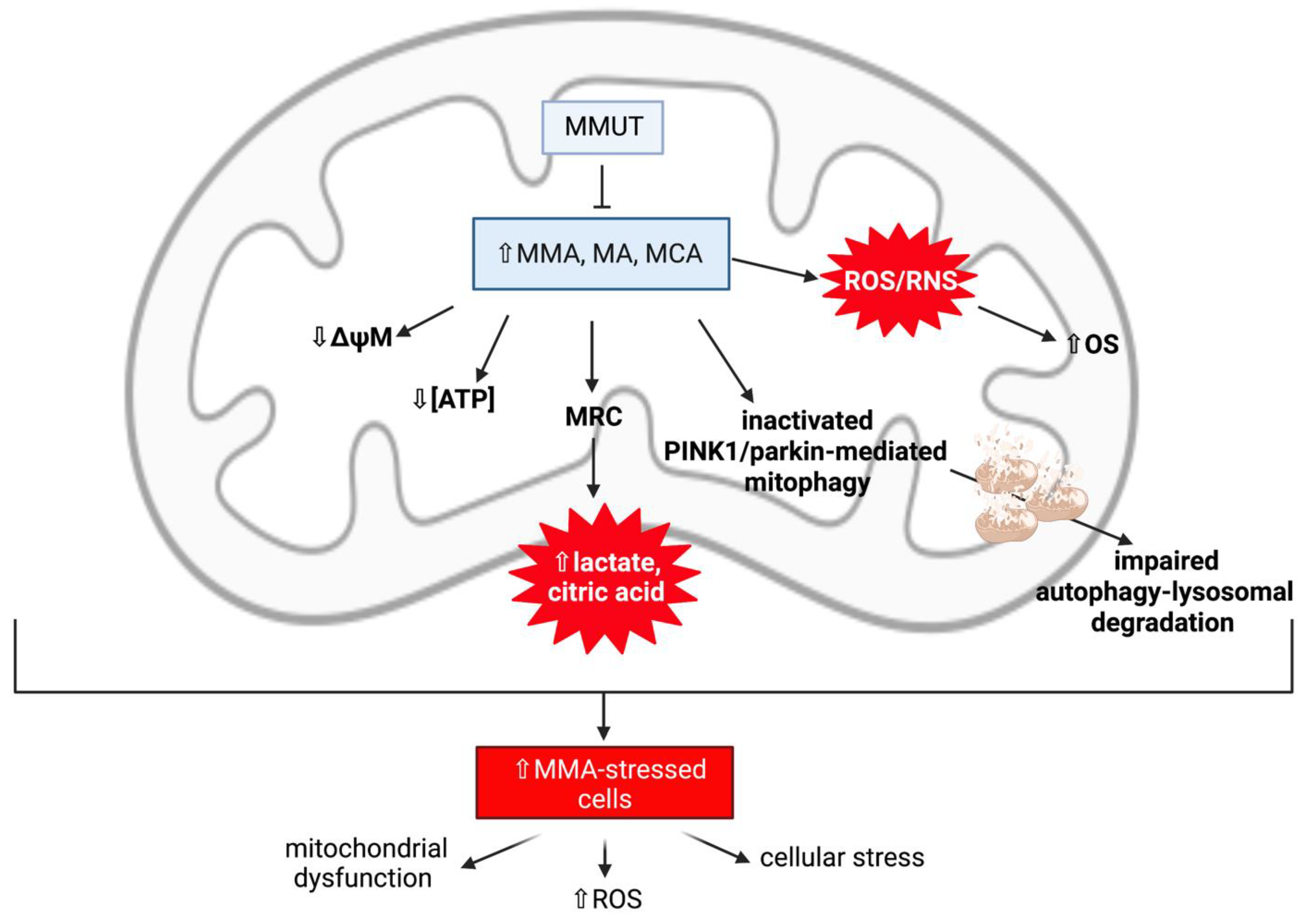
Mitochondrial Dysfunction and Oxidative Stress Contribute to Neurodegeneration in Methylmalonic Acidaemia
MMUT Deficiency and Mitochondrial Dysfunction
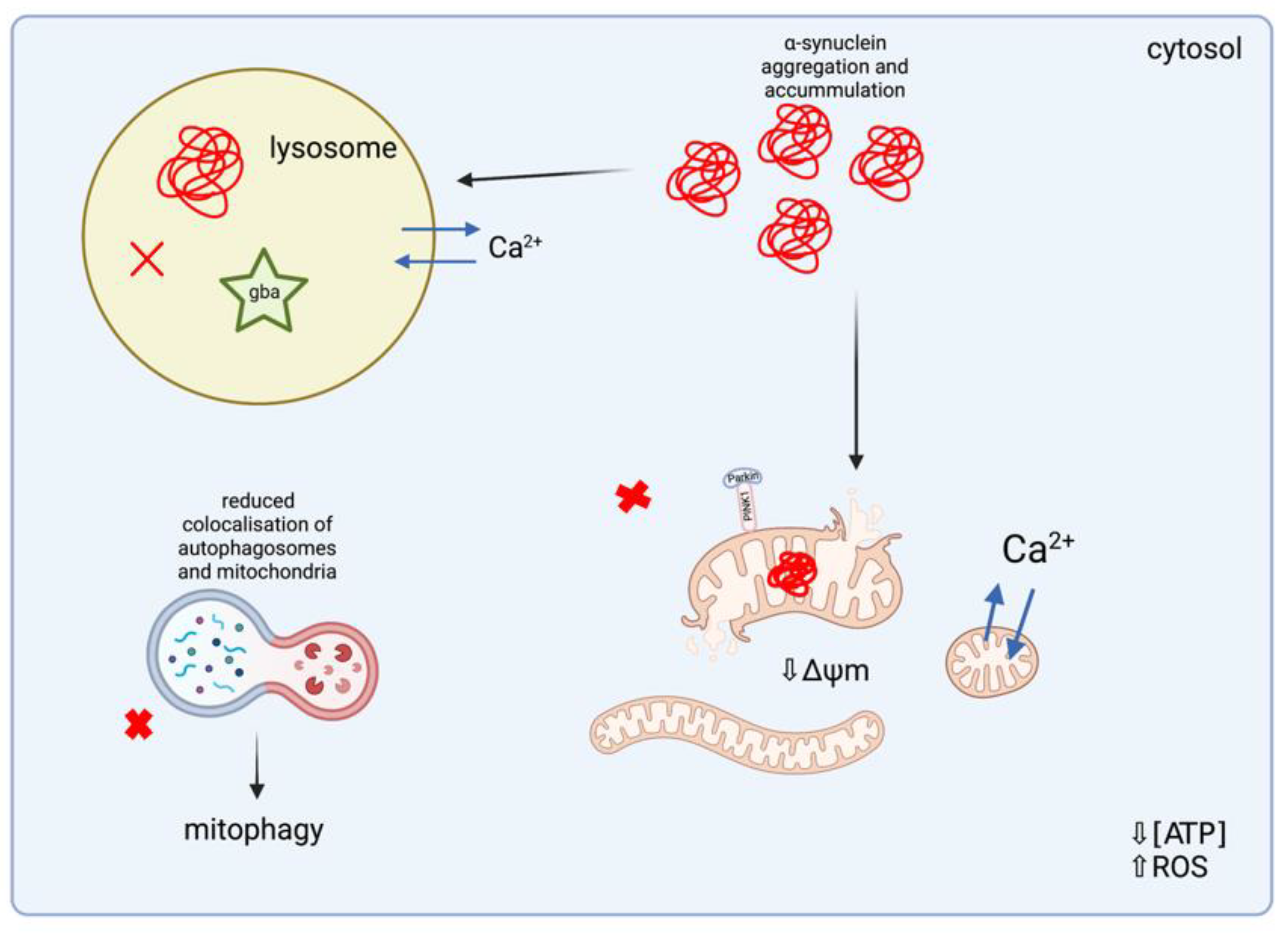
3.3.2. Lysosomal Storage Disorders
Mitochondrial Dysfunction in Lysosomal Storage Disorders
Oxidative Stress in Lysosomal Storage Disorders
Dysregulated Mitochondrial Biogenesis and Mitophagy in Lysosomal Storage Disorders
Mitochondrial Dysfunction and α-Synuclein Aggregation in Lysosomal Storage Disorders
4. Therapeutic Approaches
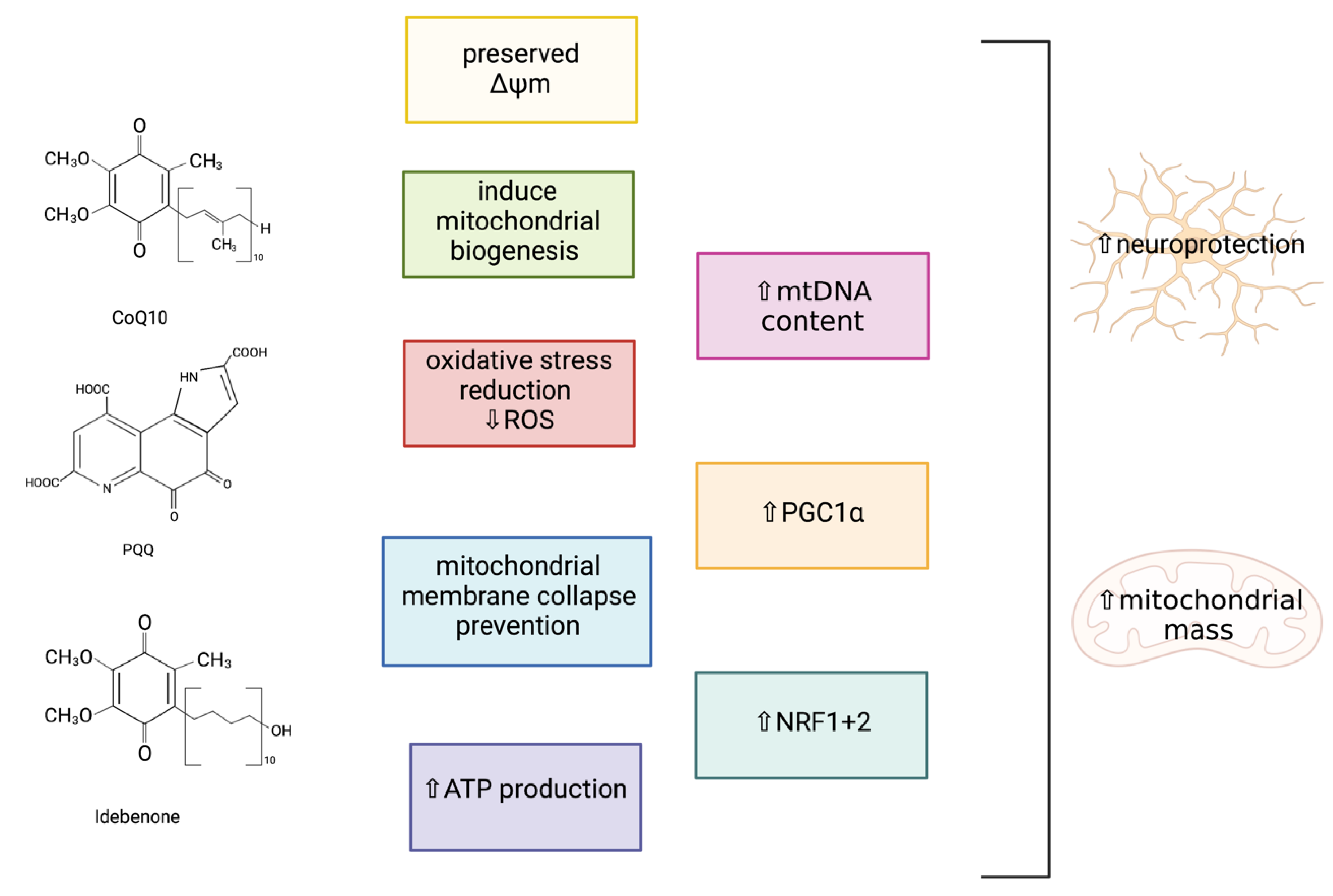
4.1. Targeting Dysfunctional Mitochondria to Induce Mitochondrial Biogenesis
4.1.1. Natural Polyphenols
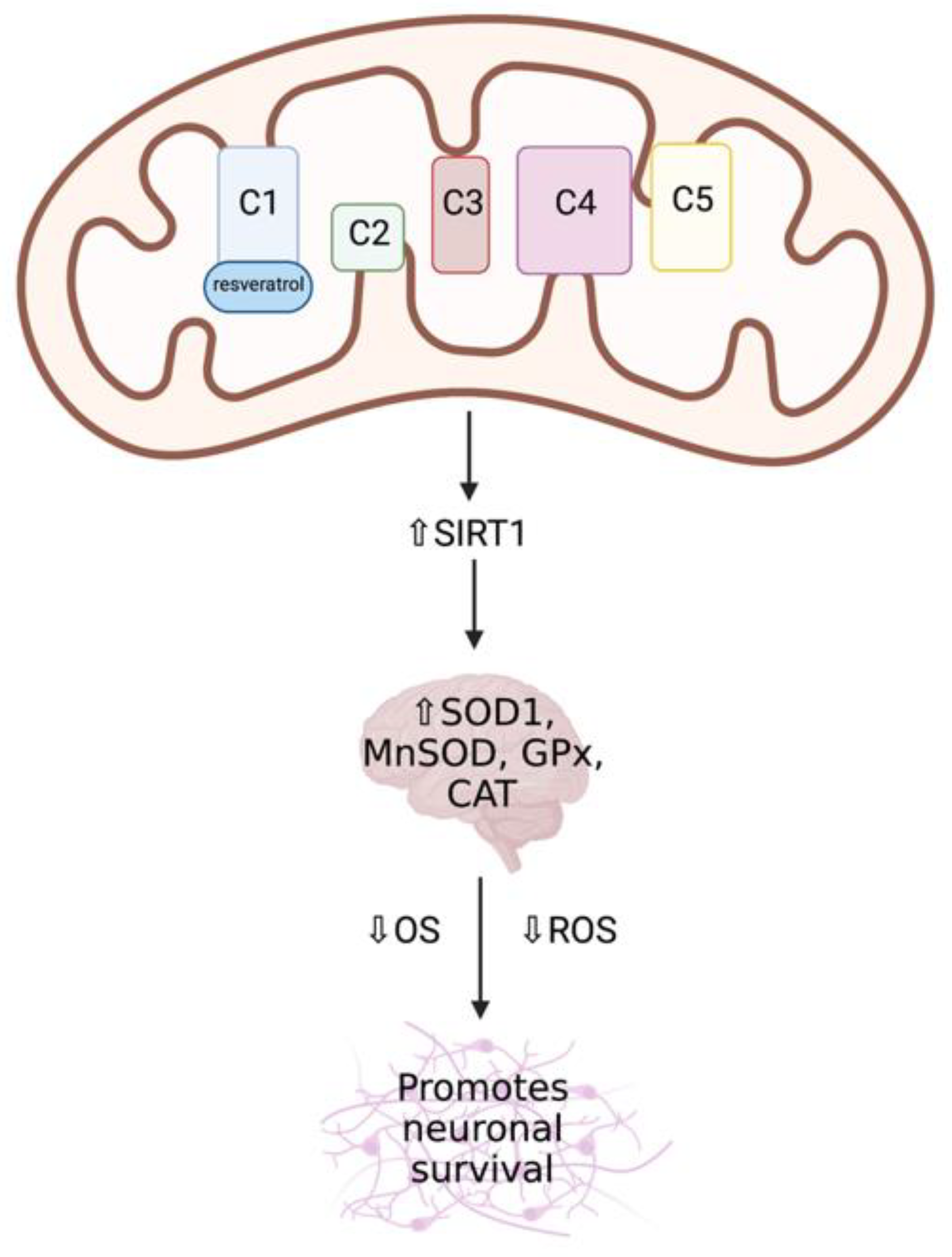
Resveratrol
Curcumin
4.1.2. Bioactive Quinones
Coenzyme Q10 (CoQ10)
Pyrroloquinoline Quinone (PQQ)
Idebenone
4.1.3. PPARγ Agonists
4.2. Targeting Nrf2-ARE Pathway
4.2.1. Chrysin
4.2.2. Fumaric Acid Esters
4.3. Targeting Impaired Mitophagy and/or Autophagy
4.3.1. Deubiquitinases
4.3.2. Trehalose
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AdoCbl | 5′-deoxyadenosylcobalamin |
| Akt | Protein kinase B |
| ARE | Antioxidant-responsive element |
| ADP | Adenosine diphosphate |
| AMPK | AMP-dependent kinase |
| ATP | Adenosine triphosphate |
| AZ | Alzheimer’s disease |
| BBB | Blood-brain-barrier |
| Bcl-2 | B-cell lymphoma 2 |
| C8 | Octanoic acid |
| C10 | Decanoic acid |
| cAMP | Cyclic adenosine monophosphate |
| CAT | Catalase |
| CCCP | Carbonyl cyanide-m-chlorophenyl-hydrazine |
| CNS | Central nervous system |
| CoQ10 | Coenzyme Q10 |
| Complex I | NADH: ubiquinone oxidoreductase |
| Complex II | Succinate dehydrogenase |
| Complex III | Ubiquinol-cytochrome c oxidoreductase |
| Complex IV | Cytochrome c oxidase |
| Complex V | ATP synthase |
| CREB | cAMP response element-binding protein |
| CS | Citrate synthase |
| CSF | Cerebrospinal fluid |
| CβE | Conduritol-β-epoxide |
| DMF | Dimethyl fumarate |
| DUB | Deubiquitinase |
| ETC | Electron transport chain |
| FAE | Fumaric acid esters |
| FCCP | 4-(trifluoromethoxy)phenylhydrazone |
| GCase | Glucocerebrosidase |
| GD | Gaucher disease |
| GPx | Glutathione peroxidase |
| GSH | Glutathione |
| H2O2 | Hydrogen peroxide |
| HNE | 4-hydroxyl-2-nonenal |
| HO-1 | Heme oxygenase-1 |
| IMD | Inherited metabolic disorder |
| iPSC | Induced pluripotent stem cell |
| KD | Ketogenic diet |
| Keap1 | Kelch-like ECH-associated protein 1 |
| KO | Knockout |
| LDH | Lactate dehydrogenase |
| LSD | Lysosomal storage disorder |
| MA | Malonic acid |
| MCA | 2-methylcitrate |
| MCT | Medium chain triglyceride |
| MEF2C | Myocyte enhancer factor 2 |
| MMA | Methylmalonic acid |
| MMAemia | Methylmalonic acidaemia |
| MMF | Monomethylfumarate |
| MMUT | L-methylmalonyl-CoA mutase |
| MnSOD | Manganese superoxide dismutase |
| MPS | Mucopolysaccharidosis |
| MPTP | 1-methyl-4-phenyl-1,2,3,6,-tetrahydropyridine |
| mPTP | Mitochondrial permeability transition pore |
| MRC | Mitochondrial respiratory chain |
| mRNA | Messenger RNA |
| MS | Multiple sclerosis |
| mtDNA | Mitochondrial DNA |
| NAD+ | Nicotinamide adenine dinucleotide |
| NCD | Non-communicable disease |
| NPC | Niemann Pick Type C |
| Nrf1 | Nuclear respiratory factor 1 |
| Nrf2 | Nuclear factor erythroid 2-related factor |
| NQO1 | NADH quinone oxidoreductase |
| OMM | Outer mitochondrial membrane |
| OS | Oxidative stress |
| OXPHOS | Oxidative phosphorylation |
| PCA | Protocatechuic acid |
| PD | Parkinson’s disease |
| PGC1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PINK1 | PTEN-induced kinase 1 |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PPQ | Pyrroloquinoline quinone |
| PTEN | Phosphatase and tensin homolog |
| ROS | Reactive oxygen species |
| RNS | Reactive nitrogen species |
| SAM | Senescence-accelerated mouse |
| Ser/Thr | Serine/threonine |
| SIRT | Silent mating type information regulation 2 homolog |
| SNpc | Substantia nigra pars compacta |
| SOD | Superoxide dismutase |
| TCA cycle | Tricarboxylic acid cycle |
| TFEB | Transcription factor EB |
| TZD | Thiazolidinedione |
| UPS | Ubiquitin-proteasome system |
| USP | Ubiquitin-specific peptidase |
| VDAC1 | Voltage-dependent anion-selective channel 1 |
| ΔΨm | Mitochondrial membrane potential |
| βHB | β-hydroxybutyrate |
References
- Ramanan, V.K.; Saykin, A.J. Pathways to neurodegeneration: Mechanistic insights from GWAS in Alzheimer’s disease, Parkinson’s disease and related disorders. Am. J. Neurodegener. Dis. 2013, 2, 145–175. [Google Scholar] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Rockenstein, E.; Crews, L.; Masliah, E. Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromol. Med. 2003, 4, 21–36. [Google Scholar] [CrossRef]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Murphy, M.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, M.; Kim, S.J. Reactive oxygen/nitrogen species and their functional correlations in neuro-degenerative diseases. J. Neural Transm. 2012, 119, 891–910. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef]
- Deas, E.; Plun-Favreau, H.; Wood, N.W. PINK1 function in health and disease. EMBO Mol. Med. 2009, 1, 152–165. [Google Scholar] [CrossRef]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox. Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Moreira, P.I.; Zhu, X.; Wang, X.; Lee, H.-G.; Nunomura, A.; Petersen, R.B.; Perry, G.; Smith, M.A. Mitochondria: A therapeutic target in neurodegeneration. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1802, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxidative Med. Cell. Longev. 2019, 2019, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolson, G.L. Mitochondrial Dysfunction and Chronic Disease: Treatment with Natural Supplements. Integr. Med. 2014, 13, 35–43. [Google Scholar]
- Gao, F.; Zhang, J. Mitochondrial quality control and neurodegenerative diseases. Neuronal. Signal. 2018, 2, NS20180062. [Google Scholar] [CrossRef] [Green Version]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial dysfunction in neurodegenerative diseases and the po-tential countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824. [Google Scholar] [CrossRef] [Green Version]
- Luciani, A.; Schumann, A.; Berquez, M.; Chen, Z.; Nieri, D.; Failli, M.; Debaix, H.; Fest, B.P.; Tokoyami, N.; Raimondi, A.; et al. Impaired mitophagy links mitochondrial disease to epithelial stress in Methylmalonyl-CoA mutase deficiency. Nat. Commun. 2020, 11, 970. [Google Scholar] [CrossRef] [Green Version]
- Sayre, L.M.; Perry, G.; Smith, M.A. Oxidative stress and neurotoxicity. Chem Res. Toxicol. 2008, 21, 172–188. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.V. Mitochondrial diseases. Lancet 2012, 379, 1825–1834. [Google Scholar] [CrossRef]
- Lezi, E.; Swerdlow, R.H. Mitochondria in Neurodegeneration; Springer: Berlin/Heidelberg, Germany, 2011; Volume 942, pp. 269–286. [Google Scholar] [CrossRef] [Green Version]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxidative Med. Cell. Longev. 2020, 2020, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, G.; Johnson, J.A. The Nrf2-ARE pathway: A valuable therapeutic target for the treatment of neuro-degenerative diseases. Recent Pat. CNS Drug Discov. 2012, 7, 218–229. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, J.; Von Stockum, S.; Marchesan, E.; Caicci, F.; Ferrari, V.; Rakovic, A.; Klein, C.; Antonini, A.; Bubacco, L.; Ziviani, E. USP 14 inhibition corrects an in vivo model of impaired mitophagy. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Leo, E.E.M.; Campos, M.R.S. Systemic Oxidative Stress: A Key Point in Neurodegeneration—A Review. J. Nutr. Heal. Aging 2019, 23, 694–699. [Google Scholar] [CrossRef]
- Wertheim-Tysarowska, K.; Gos, M.; Sykut-Cegielska, J.; Bal, J. Genetic analysis in inherited metabolic disorders—From diagnosis to treatment. Own experience, current state of knowledge and perspectives. Dev. Period. Med. 2015, 19, 413–431. [Google Scholar]
- García-Cazorla, A.; Saudubray, J.-M. Cellular neurometabolism: A tentative to connect cell biology and metabolism in neurology. J. Inherit. Metab. Dis. 2018, 41, 1043–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepien, K.M.; Heaton, R.; Rankin, S.; Murphy, A.; Bentley, J.; Sexton, D.; Hargreaves, I.P. Evidence of Oxidative Stress and Secondary Mitochondrial Dysfunction in Metabolic and Non-Metabolic Disorders. J. Clin. Med. 2017, 6, 71. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.; Fiskum, G.; Chinopoulos, C.; Lorenzo, B.J.; Browne, S.E.; Patel, M.S.; Beal, M.F. Mitochondrial -Ketoglutarate Dehydrogenase Complex Generates Reactive Oxygen Species. J. Neurosci. 2004, 24, 7779–7788. [Google Scholar] [CrossRef] [Green Version]
- Adam-Vizi, V. Production of Reactive Oxygen Species in Brain Mitochondria: Contribution by Electron Transport Chain and Non–Electron Transport Chain Sources. Antioxid. Redox. Signal. 2005, 7, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2016, 360, 201–205. [Google Scholar] [CrossRef]
- Park, J.-S.; Davis, R.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, Parkin and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Konovalova, J.; Gerasymchuk, D.; Parkkinen, I.; Chmielarz, P.; Domanskyi, A. Interplay between MicroRNAs and Oxidative Stress in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6055. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.L.; Chou, A.H.; Wu, A.S.; Chen, S.Y.; Weng, Y.H.; Kao, Y.C.; Yeah, T.H.; Chu, P.J.; Lu, C.S. PARK6 PINK1 mutants are defective in maintaining mitochondrial membrane potential and inhibiting ROS formation of substantia nigra dopaminergic neurons. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 674–684. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Andersson, U.; Scarpulla, R.C. PGC-1-Related Coactivator, a Novel, Serum-Inducible Coactivator of Nuclear Respiratory Factor 1-Dependent Transcription in Mammalian Cells. Mol. Cell. Biol. 2001, 21, 3738–3749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2016, 20, 5574–5593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.; Silvaggi, J.M.; Kiselak, T.; Zhneg, K.; Clore, E.L.; Dai, Y.; Bass, C.E.; Simon, D.K. Pgc-1α overexpression downregulates Pitx3 and increases susceptibility to MPTP toxicity associated with decreased Bdnf. PLoS ONE 2012, 7, e48925. [Google Scholar] [CrossRef] [Green Version]
- Small, D.M.; Morais, C.; Coombes, J.; Bennett, N.C.; Johnson, D.W.; Gobe, G.C. Oxidative stress-induced alterations in PPAR-γ and associated mitochondrial destabilization contribute to kidney cell apoptosis. Am. J. Physiol. Physiol. 2014, 307, F814–F822. [Google Scholar] [CrossRef]
- Tyagi, S.; Sharma, S.; Gupta, P.; Saini, A.S.; Kaushal, C. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Corona, J.C.; Duchen, M.R. PPARγ as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic. Biol. Med. 2016, 100, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miglio, G.; Rosa, A.C.; Rattazzi, L.; Collino, M.; Lombardi, G.; Fantozzi, R. PPARγ stimulation promotes mitochondrial biogenesis and prevents glucose deprivation-induced neuronal cell loss. Neurochem. Int. 2009, 55, 496–504. [Google Scholar] [CrossRef]
- Hybertson, B.; Gao, B.; Doan, A. The clinical potential of influencing Nrf2 signaling in degenerative and immunological disorders. Clin. Pharmacol. Adv. Appl. 2014, 6, 19–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tufekci, K.U.; Bayin, E.C.; Genc, S.; Genc, K. The Nrf2/ARE pathway: A promising target to counteract mi-tochondrial dysfunction in Parkinson’s disease. Parkinson’s Dis. 2011, 2011, 314082. [Google Scholar] [CrossRef] [Green Version]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Camps, J.; García-Heredia, A.; Hernández-Aguilera, A.; Joven, J. Paraoxonases, mitochondrial dysfunction and non-communicable diseases. Chem. Interact. 2016, 259, 382–387. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunctional and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, C.; Lim, K.-L. Genetic Insights into Sporadic Parkinson’s Disease Pathogenesis. Curr. Genom. 2013, 14, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Barodia, S.K.; Creed, R.B.; Goldberg, M.S. Parkin and PINK1 functions in oxidative stress and neurodegener-ation. Brain Res. Bull. 2018, 133, 51–59. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.Y.; Kishinevsky, S.; Mazzulli, J.R.; Graziotto, J.; Mrejeru, A.; Mosharov, E.V.; Puspita, L.; Valiulahi, P.; Sulzer, D.; Milner, T.A.; et al. Parkin and PINK1 patient iPSC-derived mid-brain dopamine neurons exhibit mitochondrial dysfunction and α-synuclein accumulation. Stem Cell Rep. 2016, 7, 664–677. [Google Scholar] [CrossRef] [Green Version]
- Koyano, F.; Matsuda, N. Molecular mechanisms underlying PINK1 and Parkin catalysed ubiquitylation of substrates on damaged mitochondria. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 2791–2796. [Google Scholar] [CrossRef] [Green Version]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.; Whitworth, A.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2010, 20, 867–879. [Google Scholar] [CrossRef]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.-H.; Becker, D.; Voos, W.; Leuner, K.; Müller, W.E.; Kudin, A.P.; et al. Parkinson Phenotype in Aged PINK1-Deficient Mice Is Accompanied by Progressive Mitochondrial Dysfunction in Absence of Neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Cepeda, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; et al. Parkin-deficient Mice Exhibit Nigrostriatal Deficits but Not Loss of Dopaminergic Neurons. J. Biol. Chem. 2003, 278, 43628–43635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, D.N.; Primiani, C.T.; Cookson, M.R. The effects of variants in the PARK2 (parkin), PINK1 and PARK7 (DJ-1) genes along with evidence for their pathogenicity. Curr. Protein Pept. Sci. 2018, 18, 702–714. [Google Scholar] [CrossRef]
- Bertolin, G.; Jacoupy, M.; Traver, S.; Ferrando-Miguel, R.; Georges, T.S.; Grenier, K.; Ardila-Osorio, H.; Muriel, M.-P.; Takahashi, H.; Lees, A.J.; et al. Parkin maintains mitochondrial levels of the protective Parkinson’s disease-related enzyme 17-β hydroxysteroid dehydrogenase type. Cell Death Differ. 2015, 22, 1563–1576. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Vicente, M. Neuronal Mitophagy in Neurodegenerative Diseases. Front. Mol. Neurosci. 2017, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Gluck, M.R.; Zeevalk, G.D. Inhibition of brain mitochondrial respiration by dopamine and its metabolites: Implications for Parkinson’s disease and catecholamine-associated diseases. J. Neurochem. 2004, 91, 788–795. [Google Scholar] [CrossRef]
- Stichel, C.C.; Zhu, X.-R.; Bader, V.; Linnartz, B.; Schmidt, S.; Lübbert, H. Mono- and double-mutant mouse models of Parkinson’s disease display severe mitochondrial damage. Hum. Mol. Genet. 2007, 16, 2377–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meiser, J.; Weindl, D.; Hiller, K. Complexity of dopamine metabolism. Cell Commun. Signal. 2013, 11, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Izaizumi, Y.; Okada, Y.; Okano, H. Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain. 2012, 5, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-H.; Burgess, J.D.; Faroqi, A.H.; DeMeo, N.N.; Fiesel, F.C.; Springer, W.; Delenclos, M.; McLean, P.J. Alpha-synuclein-induced mitochondrial dysfunction is mediated via a sirtuin 3-dependent pathway. Mol. Neurodegener. 2020, 15, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Choubey, V.; Safiulina, D.; Vaarmann, A.; Cagalinec, M.; Wareski, P.; Kuum, M.; Zharkovsky, A.; Kaasik, A. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 2011, 286, 10814–10824. [Google Scholar] [CrossRef] [Green Version]
- Ryan, B.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Ta-lantova, M.; et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 2013, 155, 1351–1364. [Google Scholar] [CrossRef] [Green Version]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1α. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Reeve, A.K.; Ludtmann, M.H.R.; Angelova, P.R.; Simcox, E.M.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.; Turn-bull, D.M.; Abramov, A.Y. Aggregated α-synuclein and complex I deficiency: Exploration of their relation-ship in differentiated neurons. Cell Death Dis. 2015, 6, 1820. [Google Scholar] [CrossRef]
- Faustini, G.; Bono, F.; Valerio, A.; Pizzi, M.; Spano, P.F.; Bellucci, A. Mitochondria and α-synuclein: Friends or foes in the pathogenesis of Parkinson’s Disease? Genes 2017, 8, 377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi-Fakhari, D.; Saffari, A.; Wahlster, L.; Lu, J.; Byrne, S.; Hoffmann, G.F.; Jungbluth, H.; Sahin, M. Congenital disorders of autophagy: An emerging novel class of inborn errors of neuro-metabolism. Brain 2016, 139, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Saudubray, J.-M.; Garcia-Cazorla, A. An overview of inborn errors of metabolism affecting the brain: From neurodevelopment to neurodegenerative disorders. Dialog-Clin. Neurosci. 2018, 20, 301–326. [Google Scholar] [CrossRef] [Green Version]
- Duarte, J.M.N.; Schuck, P.F.; Wenk, G.L.; Ferreira, G.C. Metabolic disturbances in diseases with neurological involvement. Aging Dis. 2014, 5, 238–255. [Google Scholar]
- Zhou, X.; Cui, Y.; Han, J. Methylmalonic acidaemia: Current status and research priorities. Intractable Rare Dis Res. 2018, 7, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Keyfi, F.; Talebi, S.; Varasteh, A.-R. Methylmalonic Acidemia Diagnosis by Laboratory Methods. Rep. Biochem. Mol. Biol. 2016, 5, 1–14. [Google Scholar]
- Morath, M.A.; Okun, J.G.; Muller, I.B.; Sauer, S.W.; Horster, F.; Hoffmann, G.F.; Kolker, S. Neurodegeneration and chronic renal failure in methylmalonic aciduria—A pathophysiological approach. J. Inherit. Metab. Dis. 2008, 31, 35–43. [Google Scholar] [CrossRef]
- Richard, E.; Gallego-Villar, L.; Rivera-Barahona, A.; Oyarzábal, A.; Pérez, B.; Rodríguez-Pombo, P.; Desviat, L.R. Altered Redox. Homeostasis in Branched-Chain Amino Acid Disorders, Organic Acidurias, and Homocystinuria. Oxid. Med. Cell. Longev. 2018, 2018, 1–17. [Google Scholar] [CrossRef]
- Brusque, A.M.; Borba, R.; Schuck, P.F.; Dalcin, K.B.; Ribeiro, C.A.J.; Silva, C.G.; Wannmacher, C.M.D.; Du-tra-Filho, C.S.; Wyse, A.T.S.; Briones, P.; et al. Inhibition of the mitochondrial respiratory chain com-plex activities in rat cerebral cortex by methylmalonic acid. Neurochem. Int. 2002, 40, 593–601. [Google Scholar] [CrossRef]
- Kolker, S.; Okun, J.G. Methylmalonic acid—An endogenous toxin? Cell Mol. Life Sci. 2005, 62, 621–624. [Google Scholar] [CrossRef]
- Richard, E.; Alvarez-Barrientos, A.; Perez, B.; Desviat, L.; Ugarte, M. Methylmalonic acidaemia leads to increased production of reactive oxygen species and induction of apoptosis through the mitochondrial/caspase pathway. J. Pathol. 2007, 213, 453–461. [Google Scholar] [CrossRef]
- Atkuri, K.R.; Cowan, T.M.; Kwan, T.; Ng, A.; Herzenberg, L.A.; Enns, G.M. Inherited disorders affecting mito-chondrial function are associated with glutathione deficiency and hypocitrullinemia. Proc. Natl. Acad. Sci. USA 2009, 106, 3941–3945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, D.R.; Kowaltowski, A.J.; Wajner, M.; Castilho, R.F. Mitochondrial energy metabolism in neurodegen-eration associated with methylmalonic acidaemia. J. Bioenerg. Biomembr. 2011, 43, 39–46. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, B.; Nelson, D.; Silver, I.; Erecinska, M.; Chesselet, M.-F. Methylmalonate toxicity in primary neuronal cultures. Neuroscience 1998, 86, 279–290. [Google Scholar] [CrossRef]
- Okun, J.G.; Hörster, F.; Farkas, L.M.; Feyh, P.; Hinz, A.; Sauer, S.; Hoffmann, G.F.; Unsicker, K.; Mayatepek, E.; Kölker, S. Neurodegeneration in Methylmalonic Aciduria Involves Inhibition of Complex II and the Tricarboxylic Acid Cycle, and Synergistically Acting Excitotoxicity. J. Biol. Chem. 2002, 277, 14674–14680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Berquez, M.; Luciani, A. Mitochondria, mitophagy and metabolic disease: Towards assembling the puzzle. Cell Stress 2020, 4, 147–150. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas. MUT. Available online: https://www.proteinatlas.org/ENSG00000146085-MUT/tissue (accessed on 28 May 2021).
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic investigations of the mitochondrial complex I inhibitor rotenone in the context of pharmacological and safety evaluation. Sci. Rep. 2017, 7, 45465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashtan, C.; Abousedira, M.; Rozen, S.; Manivel, J.C.; McCann, M.; Tuchman, M. Chronic Administration of Methylmalonic Acid (MMA) to Rats Causes Proteinuria and Renal Tubular Injury. Pediatr. Res. 1998, 43, 309. [Google Scholar] [CrossRef] [Green Version]
- Platt, F.M.; Boland, B.; Van Der Spoel, A.C. Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Kloska, A.; Tylki-Szymanska, A.; Wegrzyn, G. Lysosomal storage diseases—An overview. Postepy Biochem. 2011, 57, 128–132. [Google Scholar] [PubMed]
- Marques, A.R.A.; Saftig, P. Lysosomal storage disorders—Challenges, concepts and avenues for therapy: Beyond rare diseases. J. Cell Sci. 2019, 132, 221739. [Google Scholar] [CrossRef]
- Plotegher, N.; Duchen, M.R. Mitochondrial Dysfunction and Neurodegeneration in Lysosomal Storage Disorders. Trends Mol. Med. 2017, 23, 116–134. [Google Scholar] [CrossRef]
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef]
- Stepien, K.M.; Roncaroli, F.; Turton, N.; Hendriksz, C.J.; Roberts, M.; Heaton, R.A.; Hargreaves, I. Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review. J. Clin. Med. 2020, 9, 2596. [Google Scholar] [CrossRef]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.; Richard-Londt, A.; Brandner, S.; Waddington, S.; Schapira, A.; Duchen, M.R. Mitochondria and Quality Control Defects in a Mouse Model of Gaucher Disease—Links to Parkinson’s Disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Cleeter, M.W.; Chau, K.; Gluck, C.; Mehta, A.; Hughes, D.A.; Duchen, M.; Wood, N.; Hardy, J.; Cooper, J.M.; Schapira, A.H. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 2013, 62, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deganuto, M.; Pittis, M.G.; Pines, A.; Dominissini, S.; Kelley, M.R.; Garcia, R.; Quadrifoglio, F.; Bembi, B.; Tell, G. Altered intracellular redox status in Gaucher disease fibroblasts and impairment of adaptive response against oxidative stress. J. Cell. Physiol. 2007, 212, 223–235. [Google Scholar] [CrossRef]
- Wos, M.; Szczepanowska, J.; Pikula, S.; Tylki-Szymanska, A.; Zablocki, K.; Bandorowicz-Pikula, J. Mitochondrial dysfunction in fibroblasts derived from patients with Niemann-Pick type C disease. Arch. Biochem. Biophys. 2016, 593, 50–59. [Google Scholar] [CrossRef]
- Linsinger, G.; Wilhelm, S.; Wagner, H.; Hacker, G. Uncouplers of Oxidative Phosphorylation Can Enhance a Fas Death Signal. Mol. Cell. Biol. 1999, 19, 3299–3311. [Google Scholar] [CrossRef] [Green Version]
- Moren, C.; Juarez-Flores, D.L.; Chain, K.Y.; Gegg, M.; Garrabou, G.; Gonzalez-Casacuberta, I.; Guitart-Mampel, M.; Tolosa, E.; Marti, M.J.; Cardellach, F.; et al. GBA mutation promotes early mitochondrial dysfunction in 3D neurosphere models. Aging 2019, 11, 10338–10355. [Google Scholar] [CrossRef]
- De la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; Garrido-Maraver, J.; Cordero, M.D.; Paz, M.V.; Pavón, A.D.; Alcocer-Gómez, E.; De Lavera, I.; Ybot-Gonzalez, P.; et al. Pharmacological Chaperones and Coenzyme Q10 Treatment Improves Mutant β-Glucocerebrosidase Activity and Mitochondrial Function in Neuronopathic Forms of Gaucher Disease. Sci. Rep. 2015, 5, 10903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Stubblefield, B.; Cookson, M.R.; Goldin, E.; Velayati, A.; Tayebi, N.; Sidransky, E. Aggregation of α-synuclein in brain samples from subjects with glucocerebrosidase mutations. Mol. Genet. Metab. 2011, 104, 185–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar α-synuclein oli-gomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Tolnay, M.; Love, S.; Goedert, M. Microtubule-associated protein tau, heparan sulphate and α-synuclein in several neurodegenerative diseases with dementia. Acta Neuropathol. 1999, 97, 585–594. [Google Scholar] [CrossRef]
- Hamano, K.; Hayashi, M.; Shioda, K.; Fukatsu, R.; Mizutani, S. Mechanisms of neurodegeneration in mu-copolysaccharidoses II and IIIB: Analysis of human brain tissue. Acta Neuropathol. 2008, 115, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Heo, T.H.; Kim, S.J. Altered levels of α-synuclein and sphingolipids in Batten disease lympho-blast cells. Gene 2014, 539, 181–185. [Google Scholar] [CrossRef]
- Pchelina, S.; Nuzhnyi, E.; Emelyanov, A.; Boukina, T.; Usenko, T.; Nikolaev, M.; Salogub, G.; Yakimovskii, A.; Zakharova, E.Y. Increased plasma oligomeric alpha-synuclein in patients with lysosomal storage diseases. Neurosci. Lett. 2014, 583, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, R.M.; Corum, D.; Beeson, C.C.; Schnellmann, R.G. Mitochondrial biogenesis as a pharmaco-logical target: A new approach to acute and chronic diseases. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 1–15. [Google Scholar]
- Cameron, R.B.; Beeson, C.C.; Schnellmann, R.G. Development of therapeutics that induce mitochondrial biogenesis for the treatment of acute and chronic degenerative diseases. J. Med. Chem. 2016, 59, 10411–10434. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884–890. [Google Scholar] [CrossRef] [Green Version]
- Lindholm, D.; Mäkelä, J.; Di Liberto, V.; Mudò, G.; Belluardo, N.; Eriksson, O.; Saarma, M. Current disease modifying approaches to treat Parkinson’s disease. Cell. Mol. Life Sci. 2016, 73, 1365–1379. [Google Scholar] [CrossRef]
- Pannu, N.; Bhatnagar, A. Resveratrol: From enhanced biosynthesis and bioavailability to multitargeting chronic diseases. Biomed. Pharmacother. 2018, 109, 2237–2251. [Google Scholar] [CrossRef]
- Naoi, M.; Wu, Y.; Shamoto-Nagai, M.; Maruyama, W. Mitochondria in neuroprotection by phytochemicals: Bioactive polyphenols modulate mitochondrial apoptosis system, function and structure. Int. J. Mol. Sci. 2019, 20, 2451. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G. Mitochondrial implication in apoptosis. Towards an endosymbiont hypothesis of apoptosis evolution. Cell Death Differ. 1997, 4, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, J.C.S.; Deus, C.M.; Borges, F.; Oliveira, P. Mitochondria: Targeting mitochondrial reactive oxygen species with mitochondriotropic polyphenolic-based antioxidants. Int. J. Biochem. Cell Biol. 2018, 97, 98–103. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Schluesener, H. Natural polyphenols against neurodegenerative disorders: Potentials and pitfalls. Ageing Res. Rev. 2012, 11, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Gueguen, N.; Desquiret-Dumas, V.; Leman, G.; Chupin, S.; Baron, S.; Nivet-Antoine, V.; Vessières, E.; Ayer, A.; Henrion, D.; Lenaers, G.; et al. Resveratrol Directly Binds to Mitochondrial Complex I and Increases Oxidative Stress in Brain Mitochondria of Aged Mice. PLoS ONE 2015, 10, e0144290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robb, E.L.; Winkelmolen, L.; Visanji, N.; Brotchie, J.; Stuart, J.A. Dietary resveratrol administration in-creases MnSOD expression and activity in mouse brain. Biochem. Biophys. Res. Commun. 2008, 372, 254–259. [Google Scholar] [CrossRef]
- Xia, N.; Daiber, A.; Förstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1633–1646. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Wang, Y.-Y.; Liu, H.; Lu, Y.-F.; Wu, Q.; Liu, J.; Shi, J.-S. Resveratrol Produces Neurotrophic Effects on Cultured Dopaminergic Neurons through Prompting Astroglial BDNF and GDNF Release. Evid. Based Complement. Altern. Med. 2012, 2012, 937605. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-Activated AMPK/SIRT1/Autophagy in Cellular Models of Parkinson’s Disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’Aquila, C.; De Mari, M.; et al. Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 902–915. [Google Scholar] [CrossRef] [Green Version]
- Tellone, E.; Galtieri, A.; Russo, A.; Giardina, B.; Ficarra, S. Resveratrol: A focus on several neurodegenerative diseases. Oxid. Med. Cell Longev. 2015, 2015, 392169. [Google Scholar] [CrossRef] [Green Version]
- Perrone, L.; Squillaro, T.; Napolitano, F.; Terracciano, C.; Sampaolo, S.; Melone, M.A.B. The autophagy signalling pathway: A potential multifunctional therapeutic target of curcumin in neurological and neuromuscular diseases. Nutrients 2019, 11, 1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäger, R.; Lowery, R.P.; Calvanese, A.V.; Joy, J.M.; Purpura, M.; Wilson, J.M. Comparative absorption of curcumin formulations. Nutr. J. 2014, 13, 11. [Google Scholar] [CrossRef] [Green Version]
- Negrette-Guzman, M.; Garcia-Nino, W.R.; Tapia, E.; Zazueta, C.; Huerta-Yepez, S.; Leon-Contreras, J.C.; Hernandez-Pando, R.; Aparicio-Trejo, O.E.; Madero, M.; Pedraza-Chaverri, J. Curcumin attenuates gentami-cin-induced kidney mitochondrial alterations: Possible role of a mitochondrial biogenesis mechanism. Evid. Based Complement. Altern. Med. 2015, 2015, 917435. [Google Scholar] [CrossRef]
- Van der Merwe, C.; van Dyk, H.C.; Engelbrecht, L.; van der Westhuizen, F.H.; Kinnear, C.; Loos, B.; Bardien, S. Curcumin rescues a PINK1 knock down SH-SY5Y cellular model of Parkinson’s Disease from mitochondrial dysfunction and cell death. Mol. Neurobiol. 2017, 54, 2752–2762. [Google Scholar] [CrossRef]
- Zbarsky, V.; Datla, K.P.; Parkar, S.; Rai, D.K.; Aruoma, O.I.; Dexter, D.T. Neuroprotective properties of the natural phenolic antioxidants curcumin and naringenin but not quercetin and fisetin in a 6-OHDA model of Parkinson’s disease. Free Radic. Res. 2005, 39, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Du, X.X.; Jiang, H.; Xie, J.X. Curcumin attenuates 6-hydroxydopamine-induced cytotoxicity by anti-oxidation and nuclear factor-kappa B modulation in MES23.5 cells. Biochem. Pharmacol. 2009, 78, 178–183. [Google Scholar] [CrossRef]
- Raza, H.; John, A.; Brown, E.M.; Benedict, S.; Kambal, A. Alterations in mitochondrial respiratory functions, redox metabolism and apoptosis by oxidant 4-hydroxynonenal and antioxidants curcumin and melatonin in PC12 cells. Toxicol. Appl. Pharmacol. 2008, 226, 161–168. [Google Scholar] [CrossRef]
- Wang, M.S.; Boddapati, S.; Emadi, S.; Sierks, M.R. Curcumin reduces α-synuclein induced cytotoxicity in Parkinson’s disease cell model. BMC Neurosci. 2010, 11, 57. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Yu, Y.; Li, X.; Ross, C.A.; Smith, W.W. Curcumin protects against A53T alpha-synuclein-induced toxicity in a PC12 inducible cell model for Parkinsonism. Pharmacol. Res. 2011, 63, 439–444. [Google Scholar] [CrossRef]
- Williams, I.M.; Wallom, K.L.; Smith, D.A.; Al Eisa, N.; Smith, C.; Platt, F.M. Improved neuroprotection using miglustat, curcumin and ibuprofen as a triple combination therapy in Niemann-Pick disease type C1 mice. Neurobiol. Dis. 2014, 67, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Noh, Y.H.; Kim, K.Y.; Shim, M.S.; Choi, S.H.; Ellisman, M.H.; Weinreb, R.N.; Perkins, G.A.; Ju, W.K. Inhibi-tion of oxidative stress by coenzyme Q10 increases mitochondrial mass and improves bioenergetic function in optic nerve head astrocytes. Cell Death Dis. 2013, 4, 820. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Sawashita, J.; Nishio, S.Y.; Hashimoto, S.; Suzuki, N.; Yoshimura, H.; Tsuruoka, M.; Wang, Y.; Liu, Y.; Luo, H.; et al. Ubiquinol-10 supplementation activates mitochondria functions to decelerate senescence in senescence-accelerated mice. Antioxid. Redox. Signal. 2014, 20, 2606–2620. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A. A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front. Pharmacol. 2015, 6, 206. [Google Scholar] [CrossRef] [Green Version]
- Somayajulu, M.; McCarthy, S.; Hung, M.; Sikorska, M.; Borowy-Borowski, H.; Pandey, S. Role of mitochondria in neuronal cell death induced by oxidative stress; neuroprotection by coenzyme Q10. Neurobiol. Dis. 2005, 18, 618–627. [Google Scholar] [CrossRef]
- Stone, L.H.; Chappuis, E.; Wright, C.; Kelly, R.F.; McFalls, E.O. CoQ10 enhances PGC1α and increases ex-pression of mitochondrial antioxidant proteins in chronically ischemic swine myocardium. Nutr. Metab. 2019, 16, 92. [Google Scholar] [CrossRef] [Green Version]
- Papucci, L.; Schiavone, N.; Witort, E.; Donnini, M.; Lapucci, A.; Tempestini, A.; Formigli, L.; Zecchi-Orlandini, S.; Orlandini, G.; Carella, G.; et al. Coenzyme Q10 prevents apoptosis by inhibiting mito-chondrial depolarization independently of its free radical scavenging property. J. Biol. Chem. 2003, 278, 28220–28228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta (BBA)-Biomembr. 2004, 1660, 171–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Littarru, G.P.; Tiano, L. Bioenergetic and antioxidant properties of Coenzyme Q10: Recent developments. Mol. Biotechnol. 2007, 37, 31–37. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Nukada, H.; Urakami, T.; Murphy, M.P. Antioxidant and pro-oxidant properties of pyrroloquinoline quinone (PQQ): Implications for its function in biological systems. Biochem. Pharmacol. 2003, 65, 67–74. [Google Scholar] [CrossRef]
- Chowanadisai, W.; Bauerly, K.A.; Tchaparian, E.; Wong, A.; Cortopassi, G.A.; Rucker, R.B. Pyrroloquinoline quinone stimulates mitochondrial biogenesis through cAMP response element-binding protein phosphorylation and increased PGc-1alpha expression. J. Biol. Chem. 2010, 285, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.B.; Chowanadisai, W.; Mishchuk, D.O.; Satre, M.A.; Slupsky, C.M.; Rucker, R.B. Dietary pyrrol-oquinoline quinone (PQQ) alters indicators of inflammation and mitochondrial-related metabolism in human subjects. J. Nutr. Biochem. 2013, 24, 2076–2084. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Chen, J.; Guo, H.; Lu, J.; Zhou, J.; Guo, X.; Shi, Y.; Zhang, Y.; Yu, S.; Zhang, Q.; et al. Pyrroloquinoline quinone promotes mitochondrial biogenesis in rotenone-induced Parkinson’s disease model via AMPK activation. Acta Pharmacol. Sin. 2020, 42, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Muscoli, C.; Fresta, M.; Cardile, V.; Palumbo, M.; Renis, M.; Puglisi, G.; Paolino, D.; Nistico, S.; Rotiroti, D.; Mollace, V. Ethanol-induced injury in rat primary cortical astrocytes involves oxidative stress: Effect of idebenone. Neurosci. Lett. 2002, 329, 21–24. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Khan, A.; Zheng, H.; Yuan, C.; Jiang, H. Advances in drug therapy for mitochondrial diseases. Ann. Transl. Med. 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Yan, A.; Liu, Z.; Song, L.; Wang, X.; Zhang, Y.; Wu, N.; Lin, J.; Liu, Y.; Liu, Z. Idebenone alleviates neuroinflammation and modulates microglial polarization in LPS-stimulated BV2 cells and MPTP-induced Parkinson’s Disease mice. Front. Cell Neurosci. 2019, 12, 529. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, L.; Turnaturi, R.; Parenti, C.; Pasquinucci, L. Idebenone: Novel Strategies to improve its systemic and local efficacy. Nanomaterials 2018, 8, 87. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Wu, J.S.; Tsai, H.D.; Huang, C.Y.; Chen, J.J.; Sun, G.Y.; Lin, T.N. Peroxisome prolifera-tor-activated receptor gamma (PPAR-γ) and neurodegenerative disorders. Mol. Neurobiol. 2012, 46, 114–124. [Google Scholar] [CrossRef]
- Hwang, H.-H.; Moon, P.-G.; Lee, J.-E.; Kim, J.-G.; Lee, W.; Ryu, S.H.; Baek, M.-C. Identification of the target proteins of rosiglitazone in 3T3-L1 adipocytes through proteomic analysis of cytosolic and secreted proteins. Mol. Cells 2011, 31, 239–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, T.W.; Lee, J.Y.; Shim, W.S.; Kang, E.S.; Kim, S.K.; Ahn, C.W.; Lee, H.C.; Cha, B.-S. Rosiglitazone protects human neuroblastoma SH-SY5Y cells against acetaldehyde-induced cytotoxicity. Biochem. Biophys. Res. Commun. 2006, 340, 221–227. [Google Scholar] [CrossRef]
- Smith, U. Pioglitazone: Mechanism of action. Int. J. Clin. Pract. Suppl. 2001, 121, 13–18. [Google Scholar]
- García-Bueno, B.; Caso, J.R.; Pérez-Nievas, B.G.; Lorenzo, P.; Leza, J.C. Effects of peroxisome prolifera-tor-activated receptor gamma agonists on brain glucose and glutamate transporters after stress in rats. Neuropsychopharmacology 2007, 32, 1251–1260. [Google Scholar] [CrossRef]
- Khabbush, A.; Orford, M.; Tsai, Y.C.; Rutherford, T.; O’Donnell, M.; Eaton, S.; Heales, S.J.R. Neuronal decanoic acid oxidation is markedly lower than that of octanoic acid: A mechanistic insight into the medium-chain triglyceride ketogenic diet. Epilepsia 2017, 58, 1423–1429. [Google Scholar] [CrossRef]
- Dabke, P.; Das, A.M. Mechanism of Action of Ketogenic Diet Treatment: Impact of Decanoic Acid and Beta—Hydroxybutyrate on Sirtuins and Energy Metabolism in Hippocampal Murine Neurons. Nutrients 2020, 12, 2379. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.D.; Kanabus, M.; Erson, G.; Hargreaves, I.P.; Rutherford, T.; O’Donnell, M.; Cross, H.; Rahman, S.; Eaton, S.; Heales, S.J.R. The ketogenic diet component decanoic acid increases mitochondrial cristae synthase and complex I activity in neuronal cells. J. Neurochem. 2014, 129, 426–433. [Google Scholar] [CrossRef]
- Damiano, F.; De Benedetto, G.E.; Longo, S.; Giannotti, L.; Fico, D.; Siculella, L.; Giudetti, A.M. Decanoic Acid and Not Octanoic Acid Stimulates Fatty Acid Synthesis in U87MG Glioblastoma Cells: A Metabolomics Study. Front. Neurosci. 2020, 14, 783. [Google Scholar] [CrossRef]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell. Longev. 2019, 2019, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.R.; Shaikh, M.A.; Haq, S.H.I.U.; Nazir, S. Neuroprotective role of chrysin in attenuating loss of dopaminergic neurons and improving motor, learning and memory functions in rats. Int. J. Health Sci. 2018, 12, 35–43. [Google Scholar]
- Angelopoulou, E.; Pyrgelis, E.-S.; Piperi, C. Neuroprotective potential of chrysin in Parkinson’s disease: Molecular mechanisms and clinical implications. Neurochem. Int. 2019, 132, 104612. [Google Scholar] [CrossRef]
- Burton, N.C.; Kensler, T.W.; Guilarte, T.R. In vivo modulation of the Parkinsonian phenotype by Nrf. NeuroToxicology 2006, 27, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Thangarajan, S.; Ramachandran, S.; Krishnamurthy, P. Chrysin exerts neuroprotective effects against 3-Nitropropionic acid induced behavioral despair-mitochondrial dysfunction and striatal apoptosis via upregulating Bcl-2 gene and downregulating Bax-Bad genes in male wistar rats. Biomed. Pharmacother. 2016, 84, 514–525. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, G.; Szeto, S.S.; Chong, C.M.; Quan, Q.; Huang, C.; Cui, W.; Guo, B.; Wang, Y.; Han, Y.; et al. Examining the neuroprotective effects of protocatechuic acid and chrysin on in vitro and in vivo models of Parkinson disease. Free Radic. Biol. Med. 2015, 84, 331–343. [Google Scholar] [CrossRef]
- Yadav, S.K.; Soin, D.; Ito, K.; Dhib-Jalbut, S. Insight into the mechanism of action of dimethyl fumarate in multiple sclerosis. J. Mol. Med. 2019, 97, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, M.; Kaidery, N.A.; Yang, L.; Calingasan, N.; Smirnova, N.; Gaisin, A.; Gaisina, I.; Gazaryan, I.; Hushpulian, D.M.; Kaddour-Djebbar, I.; et al. Distinct Nrf2 Signaling Mechanisms of Fumaric Acid Esters and Their Role in Neuroprotection against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Experimental Parkinson’s-Like Disease. J. Neurosci. 2016, 36, 6332–6351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lastres-Becker, I.; Garcia-Yague, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kugler, S.; Rabano, A.; Cuadrado, A. Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson’s Disease. Antioxid. Redox. Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scannevin, R.G.; Chollate, S.; Jung, M.Y.; Shackett, M.; Patel, H.; Bista, P.; Zeng, W.; Ryan, S.; Yamamoto, M.; Lukashev, M.; et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther. 2012, 341, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Carlström, K.E.; Chinthakindi, P.K.; Espinosa, B.; Al Nimer, F.; Arnér, E.S.J.; Arvidsson, P.I.; Piehl, F.; Johansson, K. Characterization of More Selective Central Nervous System Nrf2-Activating Novel Vinyl Sulfoximine Compounds Compared to Dimethyl Fumarate. Neurotherapeutics 2020, 17, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Kyles, P.; Tyagi, S.C.; Tyagi, N. Autophagy of mitochondria: A promising therapeu-tic target for neurodegenerative disease. Cell Biochem. Biophys. 2015, 70, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Pfoh, R.; Lacdao, I.K.; Saridakis, V. Deubiquitinases and the new therapeutic opportunities offered to cancer. Endocr. Relat. Cancer 2015, 22, T35–T54. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, C.; Roy, M.; Mondal, R.; Chakraborty, J. USP14 as a therapeutic target against neurodegenera-tion: A rat brain perspective. Front. Cell Dev. Biol. 2020, 8, 727. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, Y.; Ding, S.; Li, J.; Song, N.; Ren, Y.; Hong, D.; Wu, C.; Li, B.; Wang, F.; et al. Small molecule inhibitors reveal allosteric regulation of USP14 via steric blockade. Cell Res. 2018, 28, 1186–1194. [Google Scholar] [CrossRef]
- Xu, D.; Shan, B.; Sun, H.; Xiao, J.; Zhu, K.; Xie, X.; Li, X.; Liang, W.; Lu, X.; Qian, L.; et al. USP14 regulates autophagy by suppressing K63 ubiquitination of Beclin. Genes Dev. 2016, 30, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Sadoshima, J. Molecular Mechanisms of Mitochondrial Autophagy/Mitophagy in the Heart. Circ. Res. 2015, 116, 1477–1490. [Google Scholar] [CrossRef]
- Bingol, B.; Tea, J.; Phu, L.; Reichelt, M.; Bakalarski, C.; Song, Q.; Foreman, O.; Kirkpatrick, D.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Goker-Alpan, O.; Stubblefield, B.K.; Giasson, B.I.; Sidransky, E. Glucocerebrosidase is present in α-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 2010, 120, 641–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wipke, B.T.; Hoepner, R.; Strassburger-Krogias, K.; Thomas, A.M.; Gianni, D.; Szak, S.; Brennan, M.S.; Pistor, M.; Gold, R.; Chan, A.; et al. Different Fumaric Acid Esters Elicit Distinct Pharmacologic Responses. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e950. [Google Scholar] [CrossRef]
- Liu, K.; Jing, M.J.; Liu, C.; Yan, D.Y.; Ma, Z.; Wang, C.; Deng, Y.; Liu, W.; Xu, B. Effect of trehalose on man-ganese-induced mitochondrial dysfunction and neuronal cell damage in mice. Basic Clin. Pharmacol. Toxicol. 2019, 125, 536–547. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta (BBA)-Bioenerg. 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhi, X.; Pan, L.; Zhou, P. Trehalose inhibits A53T mutant α-synuclein overexpression and neuro-toxicity in transduced PC12 cells. Molecules 2017, 22, 1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotfi, P.; Tse, D.Y.-Y.; Di Ronza, A.; Seymour, M.L.; Martano, G.; Cooper, J.; Pereira, F.A.; Passafaro, M.; Wu, S.M.; Sardiello, M. Trehalose reduces retinal degeneration, neuroinflammation and storage burden caused by a lysosomal hydrolase deficiency. Autophagy 2018, 14, 1419–1434. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millichap, L.E.; Damiani, E.; Tiano, L.; Hargreaves, I.P. Targetable Pathways for Alleviating Mitochondrial Dysfunction in Neurodegeneration of Metabolic and Non-Metabolic Diseases. Int. J. Mol. Sci. 2021, 22, 11444. https://doi.org/10.3390/ijms222111444
Millichap LE, Damiani E, Tiano L, Hargreaves IP. Targetable Pathways for Alleviating Mitochondrial Dysfunction in Neurodegeneration of Metabolic and Non-Metabolic Diseases. International Journal of Molecular Sciences. 2021; 22(21):11444. https://doi.org/10.3390/ijms222111444
Chicago/Turabian StyleMillichap, Lauren Elizabeth, Elisabetta Damiani, Luca Tiano, and Iain P. Hargreaves. 2021. "Targetable Pathways for Alleviating Mitochondrial Dysfunction in Neurodegeneration of Metabolic and Non-Metabolic Diseases" International Journal of Molecular Sciences 22, no. 21: 11444. https://doi.org/10.3390/ijms222111444
APA StyleMillichap, L. E., Damiani, E., Tiano, L., & Hargreaves, I. P. (2021). Targetable Pathways for Alleviating Mitochondrial Dysfunction in Neurodegeneration of Metabolic and Non-Metabolic Diseases. International Journal of Molecular Sciences, 22(21), 11444. https://doi.org/10.3390/ijms222111444