
Chlorogenic Acid Decreases Glutamate Release from Rat Cortical Nerve Terminals by P/Q-Type Ca2+ Channel Suppression: A Possible Neuroprotective Mechanism

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
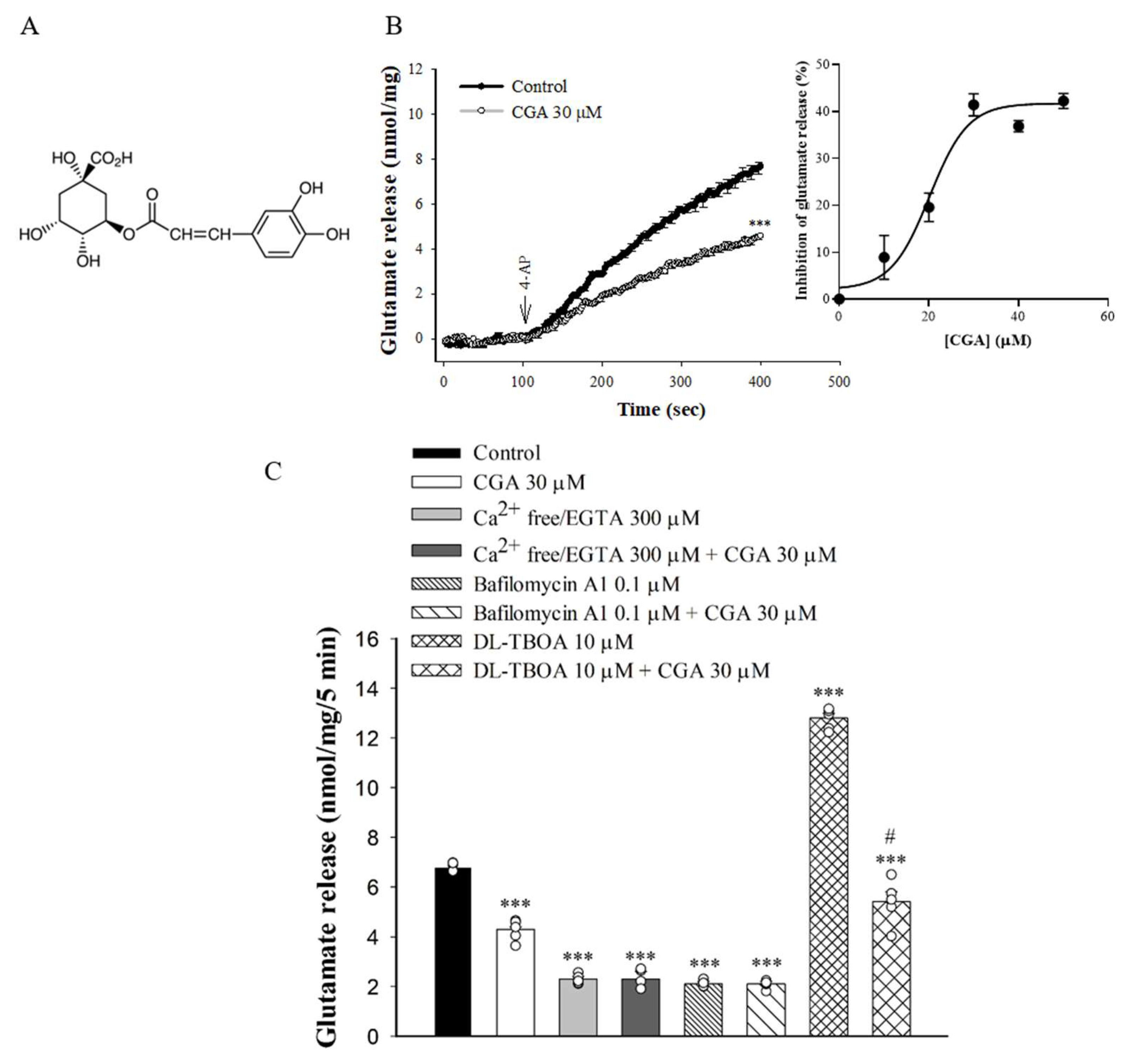
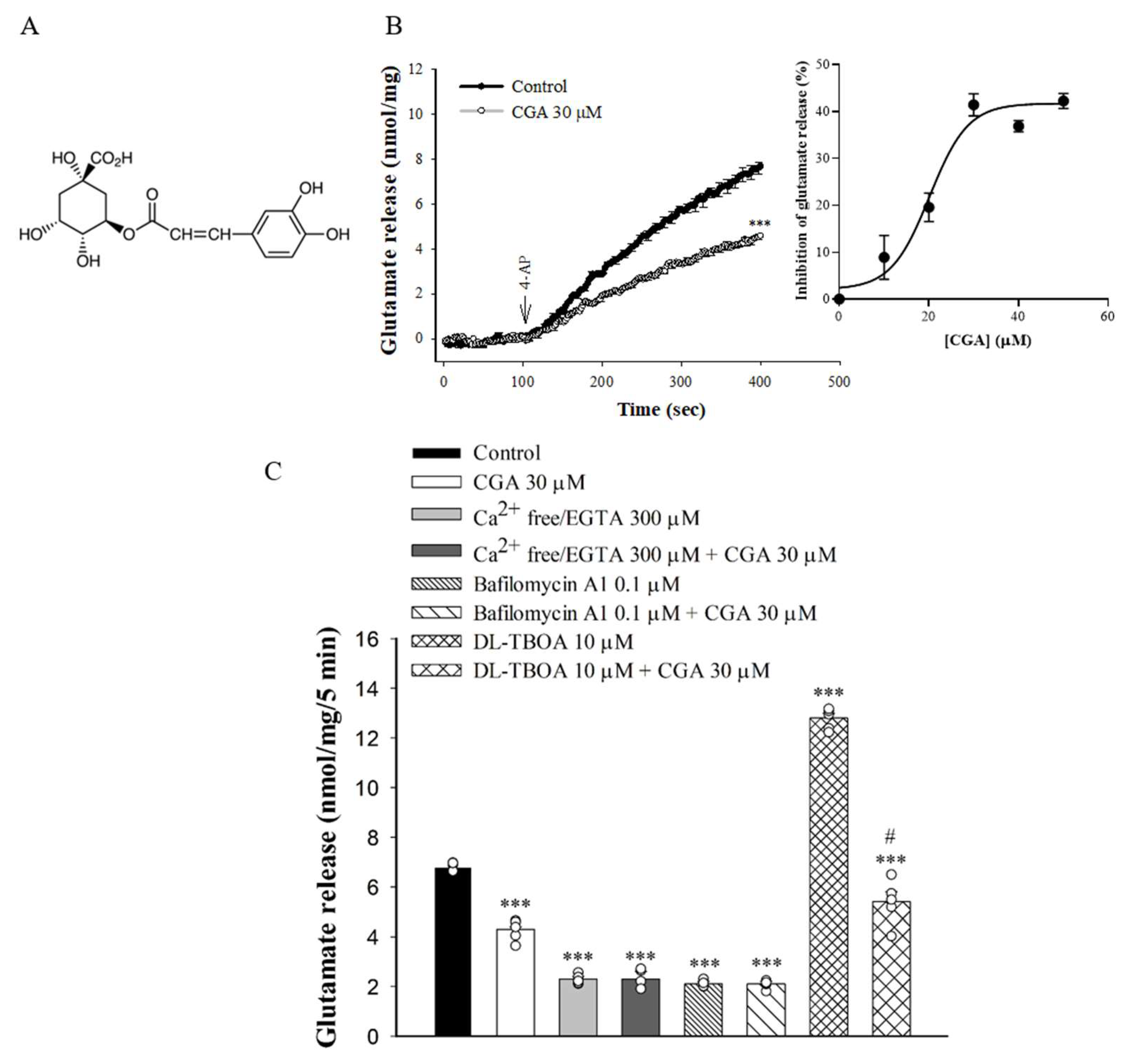
2.1. Effect of CGA on 4-Aminopyridine (4-AP)-Evoked Glutamate Release from Rat Cerebrocorticalsynaptosomes
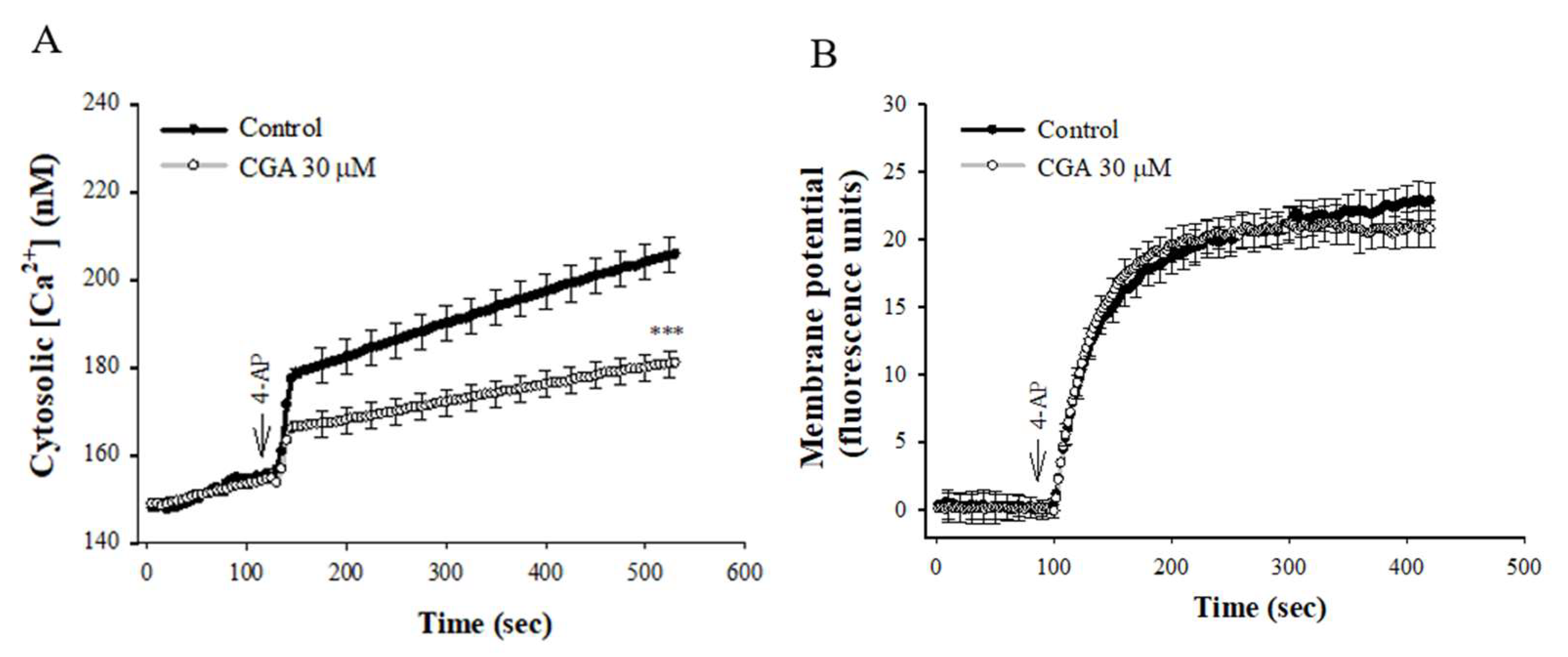
2.2. Effect of CGA on 4-AP-Induced Increase in [Ca2+]C and Membrane Potential Depolarization in the Synaptosomes
2.3. Effect of CGA on Glutamate Release in the Presence of Voltage-Dependent Ca2+ Channel Blockers or Intracellular Ca2+ Release Inhibitors
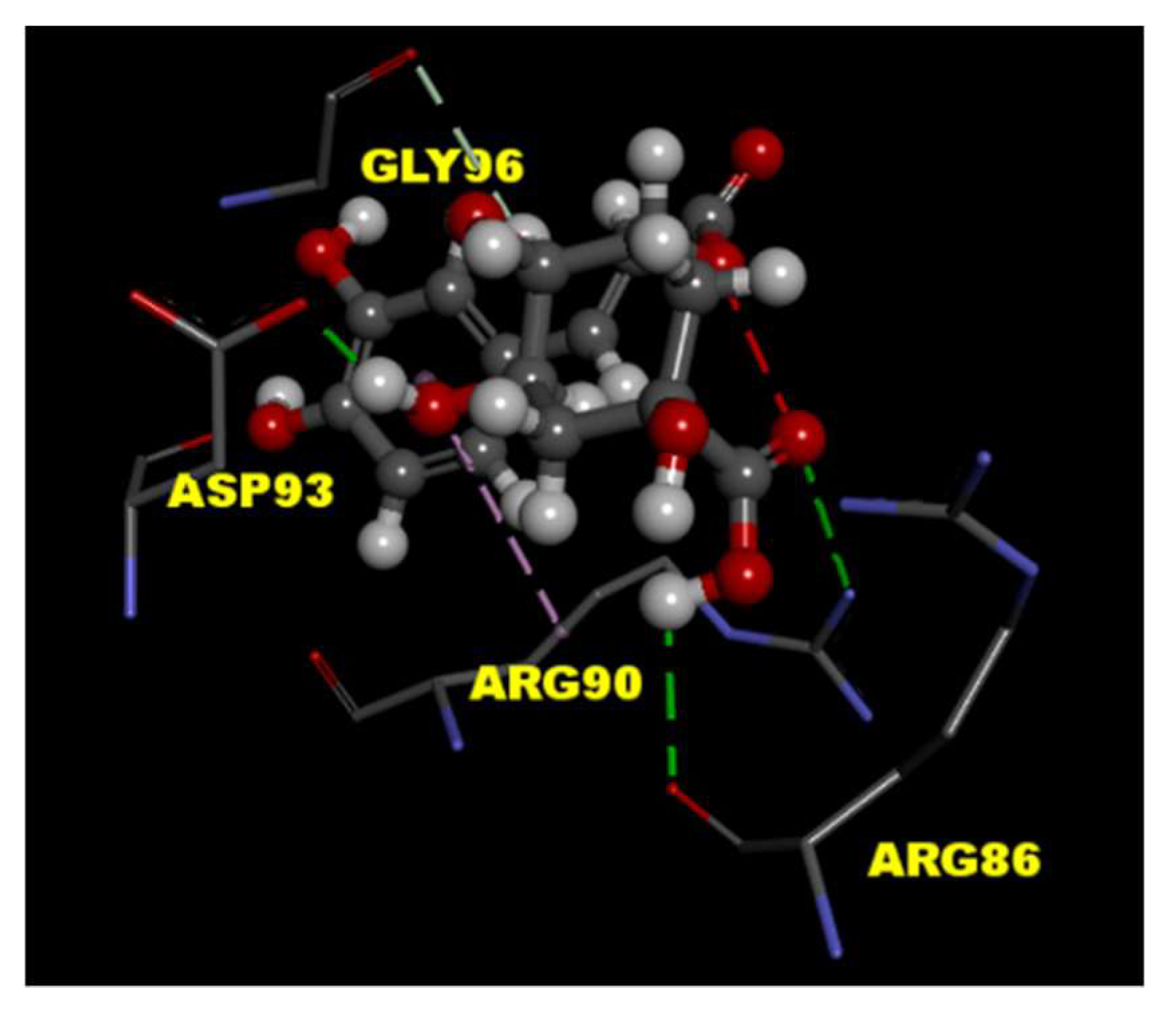
2.4. CGA Interacts with the P/Q-Type Ca2+ Channel
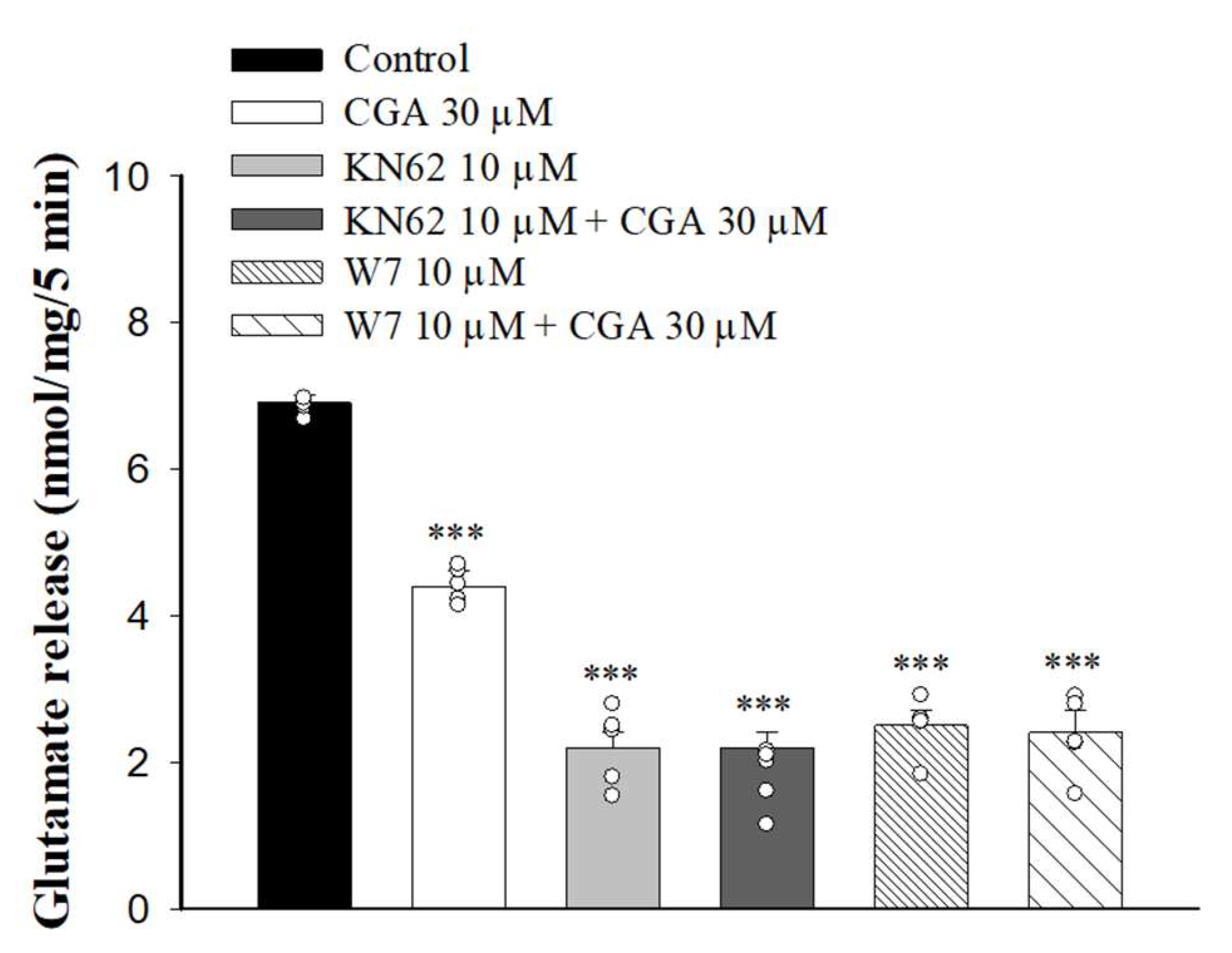
2.5. Effect of CGA on Glutamate Release in the Presence of Calmodulin and Ca2+/Calmodulin-Dependent Kinase II (CaMKII) Inhibitors
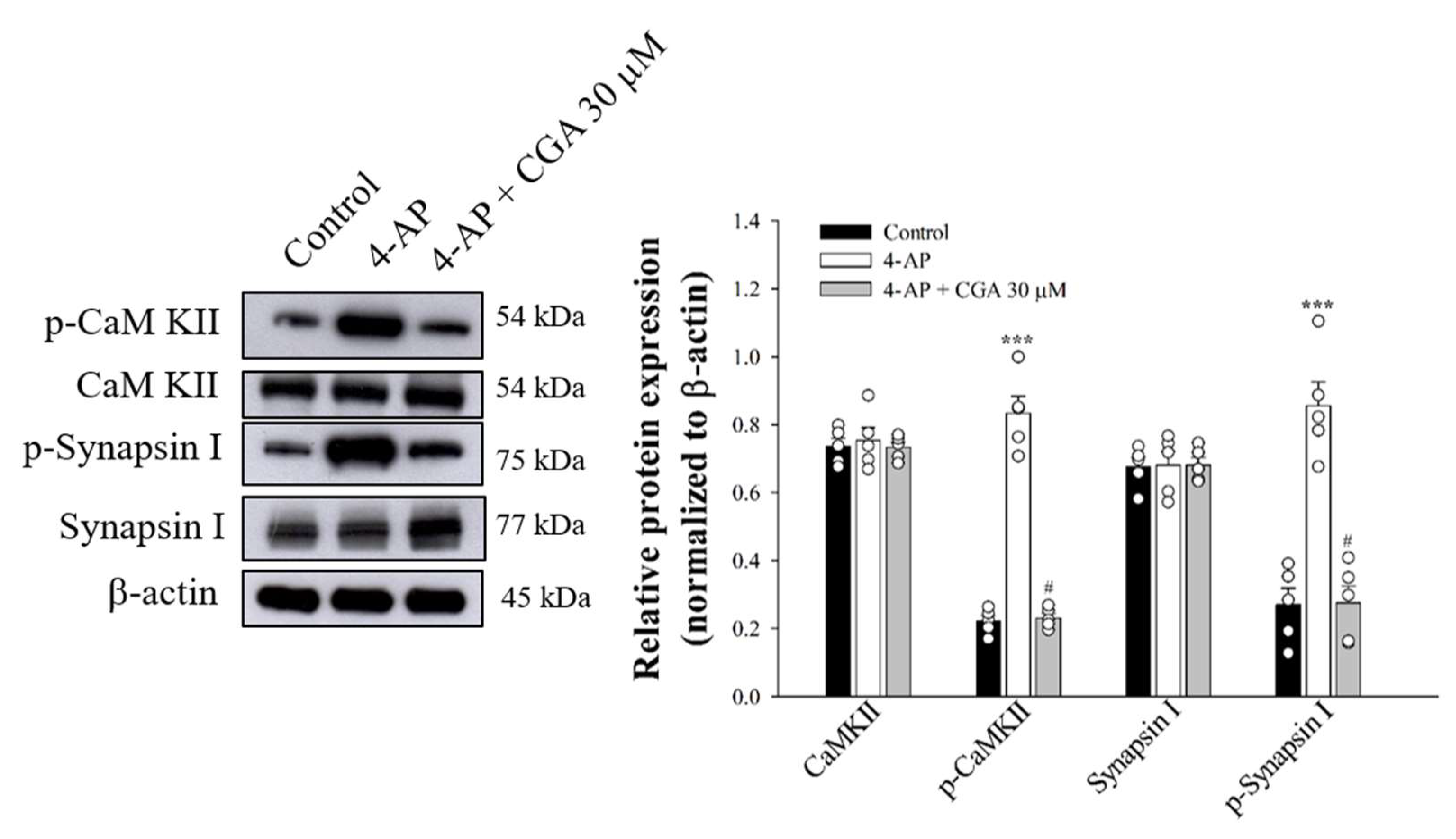
2.6. Effect of CGA on the Phosphorylation of CaMKII and Its Substrate, Synapsin I, in Cerebrocorticalsynaptosomes
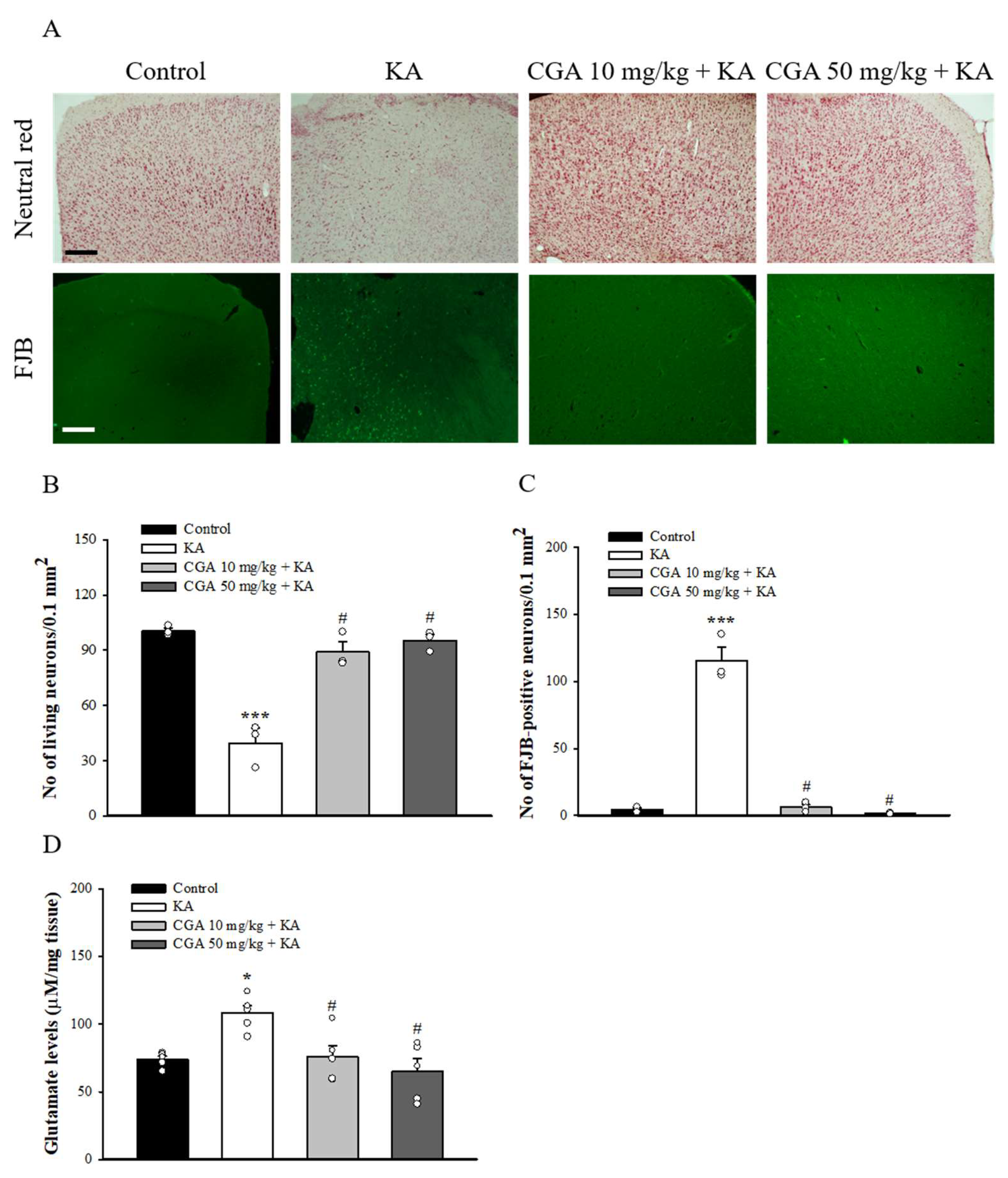
2.7. Effect of CGA on the Neuronal Damage and Glutamate Elevation in the Cortex of Rats with KA
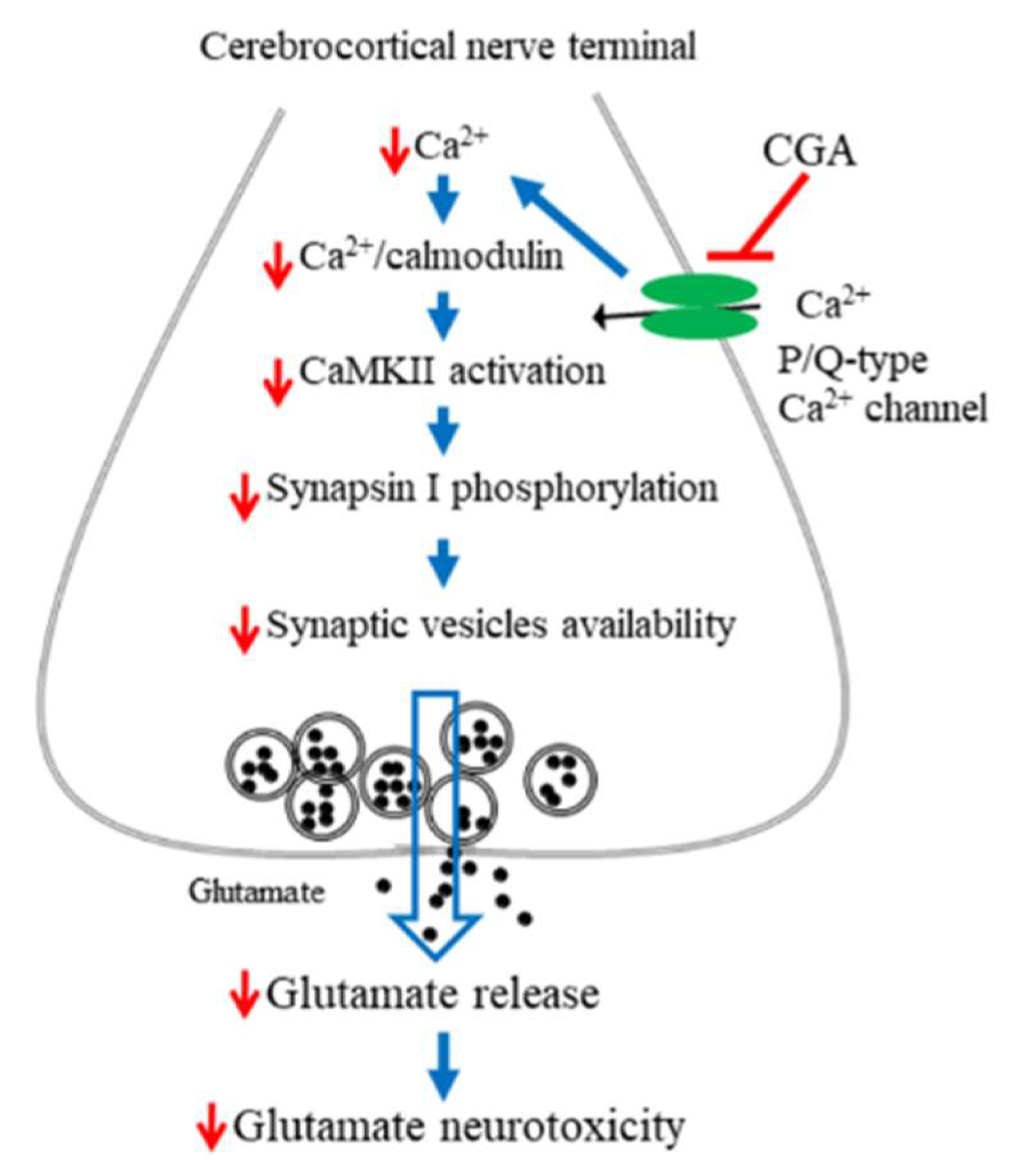
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Materials
4.3. Preparation of Synaptosomes
4.4. Glutamate Release Determination
4.5. Determination of Cytosolic Free Ca2+ Concentration ([Ca2+]C)
4.6. Determination of Synaptosomal Membrane Potential
4.7. Molecular Docking Study
4.8. Western Blot
4.9. Histological Staining
4.10. Determination of Glutamate in Brain Tissue
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Headley, P.M.; Grillner, S. Excitatory amino acids and synaptic transmission: The evidence for a physiological function. Trends Pharmacol. Sci. 1990, 11, 205–211. [Google Scholar] [CrossRef]
- Javitt, D.C. Glutamate as a therapeutic target in psychiatric disorders. Mol. Psychiatry 2004, 9, 984–997. [Google Scholar] [CrossRef] [Green Version]
- Meldrum, B.; Garthwaite, J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol. Sci. 1990, 11, 379–387. [Google Scholar] [CrossRef]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [Green Version]
- González, J.C.; Egea, J.; Del Carmen Godino, M.; Fernandez-Gomez, F.J.; Sánchez-Prieto, J.; Gandía, L.; García, A.G.; Jordán, J.; Hernández-Guijo, J.M. Neuroprotectant minocycline depresses glutamatergic neurotransmission and Ca2+signalling in hippocampal neurons. Eur. J. Neurosci. 2007, 26, 2481–2495. [Google Scholar] [CrossRef]
- Lazarevic, V.; Yang, Y.; Ivanova, D.; Fejtova, A.; Svenningsson, P. Riluzole attenuates the efficacy of glutamatergic transmission by interfering with the size of the readily releasable neurotransmitter pool. Neuropharmacology 2018, 143, 38–48. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, B.; She, Y.; Song, X. Dexmedetomidine ameliorates lidocaine-induced spinal neurotoxicity via inhibiting glutamate release and the PKC pathway. Neurotoxicology 2018, 69, 77–83. [Google Scholar] [CrossRef]
- Vauzour, D.; Rodriguez-Mateos, A.; Corona, G.; Oruna-Concha, M.J.; Spencer, J.P. Polyphenols and human health: Prevention of disease and mechanisms of action. Nutrients 2010, 2, 1106–1131. [Google Scholar] [CrossRef] [Green Version]
- Gorzynik-Debicka, M.; Przychodzen, P.; Cappello, F.; Kuban-Jankowska, A.; Marino Gammazza, A.; Knap, N.; Wozniak, M.; Gorska-Ponikowska, M. Potential health benefits of olive oil and plant polyphenols. Int. J. Mol. Sci. 2018, 19, 686. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.Y.; Lu, C.W.; Huang, S.K.; Wang, S.J. Ferulic acid suppresses glutamate release through inhibition of voltage-dependent calcium entry in rat cerebrocortical nerve terminals. J. Med. Food 2013, 16, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, S.; Kan, J.; Zhang, J.; Zhou, L.; Huang, Y.; Zhang, Y. Chinese herbal medicine interventions in neurological disorder therapeutics by regulating glutamate signaling. Curr. Neuropharmacol. 2020, 18, 260–276. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Hsieh, P.W.; Kuo, J.R.; Wang, S.J. Rosmarinic acid, a bioactive phenolic compound, inhibits glutamate release from rat cerebrocortical synaptosomes through GABA(A) receptor activation. Biomolecules 2021, 11, 1029. [Google Scholar] [CrossRef] [PubMed]
- Naveed, M.; Hejazi, V.; Abbas, M.; Kamboh, A.A.; Khan, G.J.; Shumzaid, M.; Ahmad, F.; Babazadeh, D.; FangFang, X.; Modarresi-Ghazani, F.; et al. Chlorogenic acid (CGA): A pharmacological review and call for further research. Biomed. Pharmacother. 2018, 97, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Tian, Z.; Cui, Y.; Liu, Z.; Ma, X. Chlorogenic acid: A comprehensive review of the dietary sources, processing effects, bioavailability, beneficial properties, mechanisms of action, and future directions. Compr. Rev. Food Sci. Food Saf. 2020, 19, 3130–3158. [Google Scholar] [CrossRef]
- Song, J.; Zhou, N.; Ma, W.; Gu, X.; Chen, B.; Zeng, Y.; Yang, L.; Zhou, M. Modulation of gut microbiota by chlorogenic acid pretreatment on rats with adrenocorticotropic hormone induced depression-like behavior. Food Funct. 2019, 10, 2947–2957. [Google Scholar] [CrossRef]
- Gao, L.; Li, X.; Meng, S.; Ma, T.; Wan, L.; Xu, S. Chlorogenic acid alleviates Aβ25-35-induced autophagy and cognitive impairment via the mTOR/TFEB signaling pathway. Drug Des. Dev. Ther. 2020, 14, 1705–1716. [Google Scholar] [CrossRef]
- Hermawati, E.; Arfian, N.; Mustofa, M.; Partadiredja, G. Chlorogenic acid ameliorates memory loss and hippocampal cell death after transient global ischemia. Eur. J. Neurosci. 2020, 51, 651–669. [Google Scholar] [CrossRef]
- Lee, T.K.; Kang, I.J.; Kim, B.; Sim, H.J.; Kim, D.W.; Ahn, J.H.; Lee, J.C.; Ryoo, S.; Shin, M.C.; Cho, J.H.; et al. Experimental pretreatment with chlorogenic acid prevents transient ischemia-induced cognitive decline and neuronal damage in the hippocampus through anti-oxidative and anti-inflammatory effects. Molecules 2020, 25, 3578. [Google Scholar] [CrossRef]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Dilnashin, H.; Singh, R.; Singh, S.P. Neuroprotective effect of chlorogenic acid on mitochondrial dysfunction-mediated apoptotic death of DA neurons in a Parkinsonian mouse model. Oxid. Med. Cell. Longev. 2020, 2020, 6571484. [Google Scholar] [CrossRef]
- Wang, X.; Fan, X.; Yuan, S.; Jiao, W.; Liu, B.; Cao, J.; Jiang, W. Chlorogenic acid protects against aluminium-induced cytotoxicity through chelation and antioxidant actions in primary hippocampal neuronal cells. Food Funct. 2017, 8, 2924–2934. [Google Scholar] [CrossRef]
- Rebai, O.; Amri, M. Chlorogenic acid prevents AMPA-mediated excitotoxicity in optic nerve oligodendrocytes through a PKC and caspase-dependent pathways. Neurotox. Res. 2018, 34, 559–573. [Google Scholar] [CrossRef]
- Youn, Y.; Jeon, S.H.; Jin, H.Y.; Che, D.N.; Jang, S.I.; Kim, Y.S. Chlorogenic acid-rich Solanum melongena extract has protective potential against rotenone-induced neurotoxicity in PC-12 cells. J. Food Biochem. 2019, 43, e12999. [Google Scholar] [CrossRef]
- Meldrum, B.S. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J. Nutr. 2000, 130, 1007s–1015s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikami, Y.; Yamazawa, T. Chlorogenic acid, a polyphenol in coffee, protects neurons against glutamate neurotoxicity. Life Sci. 2015, 139, 69–74. [Google Scholar] [CrossRef]
- Rebai, O.; Belkhir, M.; Sanchez-Gomez, M.V.; Matute, C.; Fattouch, S.; Amri, M. differential molecular targets for neuroprotective effect of chlorogenic acid and its related compounds against glutamate induced excitotoxicity and oxidative stress in rat cortical neurons. Neurochem. Res. 2017, 42, 3559–3572. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, S.; Simonyi, A.; Sun, G.Y.; Sun, A.Y. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol. Neurobiol. 2005, 31, 3–16. [Google Scholar] [CrossRef]
- Nicholls, D.G. Presynaptic modulation of glutamate release. Prog. Brain Res. 1998, 116, 15–22. [Google Scholar] [CrossRef]
- Zhou, Q.; Petersen, C.C.; Nicoll, R.A. Effects of reduced vesicular filling on synaptic transmission in rat hippocampal neurones. J. Physiol. 2000, 525, 195–206. [Google Scholar] [CrossRef]
- Nicholls, D.G. Release of glutamate, aspartate, and gamma-aminobutyric acid from isolated nerve terminals. J. Neurochem. 1989, 52, 331–341. [Google Scholar] [CrossRef]
- Vázquez, E.; Sánchez-Prieto, J. Presynaptic modulation of glutamate release targets different calcium channels in rat cerebrocortical nerve terminals. Eur. J. Neurosci. 1997, 9, 2009–2018. [Google Scholar] [CrossRef]
- Berridge, M.J. Neuronal calcium signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Barrie, A.P.; Nicholls, D.G.; Sanchez-Prieto, J.; Sihra, T.S. An ion channel locus for the protein kinase C potentiation of transmitter glutamate release from guinea pig cerebrocortical synaptosomes. J. Neurochem. 1991, 57, 1398–1404. [Google Scholar] [CrossRef]
- Coffey, E.T.; Sihra, T.S.; Nicholls, D.G.; Pocock, J.M. Phosphorylation of synapsin I and MARCKS in nerve terminals is mediated by Ca2+ entry via an Aga-GI sensitive Ca2+ channel which is coupled to glutamate exocytosis. FEBS Lett. 1994, 353, 264–268. [Google Scholar] [CrossRef] [Green Version]
- Heitman, E.; Ingram, D.K. Cognitive and neuroprotective effects of chlorogenic acid. Nutr. Neurosci. 2017, 20, 32–39. [Google Scholar] [CrossRef]
- Fernandes, M.Y.D.; Dobrachinski, F.; Silva, H.B.; Lopes, J.P.; Gonçalves, F.Q.; Soares, F.A.A.; Porciúncula, L.O.; Andrade, G.M.; Cunha, R.A.; Tomé, A.R. Neuromodulation and neuroprotective effects of chlorogenic acids in excitatory synapses of mouse hippocampal slices. Sci. Rep. 2021, 11, 10488. [Google Scholar] [CrossRef]
- Wu, L.G.; Saggau, P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997, 20, 204–212. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Sihra, T.S.; Sanchez-Prieto, J. Calcium-dependent and -independent release of glutamate from synaptosomes monitored by continuous fluorometry. J. Neurochem. 1987, 49, 50–57. [Google Scholar] [CrossRef]
- Nichols, R.A.; Sihra, T.S.; Czernik, A.J.; Nairn, A.C.; Greengard, P. Calcium/calmodulin-dependent protein kinase II increases glutamate and noradrenaline release from synaptosomes. Nature 1990, 343, 647–651. [Google Scholar] [CrossRef]
- Llinás, R.; Gruner, J.A.; Sugimori, M.; McGuinness, T.L.; Greengard, P. Regulation by synapsin I and Ca(2+)-calmodulin-dependent protein kinase II of the transmitter release in squid giant synapse. J. Physiol. 1991, 436, 257–282. [Google Scholar] [CrossRef]
- Hinds, H.L.; Goussakov, I.; Nakazawa, K.; Tonegawa, S.; Bolshakov, V.Y. Essential function of alpha-calcium/calmodulin-dependent protein kinase II in neurotransmitter release at a glutamatergic central synapse. Proc. Natl. Acad. Sci. USA 2003, 100, 4275–4280. [Google Scholar] [CrossRef] [Green Version]
- Ferkany, J.W.; Zaczek, R.; Coyle, J.T. Kainic acid stimulates excitatory amino acid neurotransmitter release at presynaptic receptors. Nature 1982, 298, 757–759. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. Limbic seizure and brain damage produced by kainic acid: Mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 1985, 14, 375–403. [Google Scholar] [CrossRef]
- Smani, D.; Sarkar, S.; Raymick, J.; Kanungo, J.; Paule, M.G.; Gu, Q. Downregulation of 14-3-3 proteins in a kainic acid-induced neurotoxicity model. Mol. Neurobiol. 2018, 55, 122–129. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef]
- Penkowa, M.; Florit, S.; Giralt, M.; Quintana, A.; Molinero, A.; Carrasco, J.; Hidalgo, J. Metallothionein reduces central nervous system inflammation, neurodegeneration, and cell death following kainic acid-induced epileptic seizures. J. Neurosci. Res. 2005, 79, 522–534. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, T.Y.; Chiu, K.M.; Lee, M.Y.; Huang, J.H.; Wang, S.J. Silymarin inhibits glutamate release and prevents against kainic acid-induced excitotoxic injury in rats. Biomedicines 2020, 8, 486. [Google Scholar] [CrossRef]
- Aseervatham, G.S.B.; Suryakala, U.; Sundaram, S.; Bose, P.C.; Sivasudha, T. Expression pattern of NMDA receptors reveals antiepileptic potential of apigenin 8-C-glucoside and chlorogenic acid in pilocarpine induced epileptic mice. Biomed. Pharmacother. 2016, 82, 54–64. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Sihra, T.S. Synaptosomes possess an exocytotic pool of glutamate. Nature 1986, 321, 772–773. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, T.Y.; Huang, S.K.; Wang, S.J. Echinacoside inhibits glutamate release by suppressing voltage-dependent Ca2+ entry and protein kinase C in rat cerebrocortical nerve terminals. Int. J. Mol. Sci. 2016, 17, 1006. [Google Scholar] [CrossRef] [Green Version]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Akerman, K.E.; Scott, I.G.; Heikkilä, J.E.; Heinonen, E. Ionic dependence of membrane potential and glutamate receptor-linked responses in synaptoneurosomes as measured with a cyanine dye, DiS-C2-(5). J. Neurochem. 1987, 48, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.W.; Lin, T.Y.; Pan, T.L.; Wang, P.W.; Chiu, K.M.; Lee, M.Y.; Wang, S.J. Asiatic acid prevents cognitive deficits by inhibiting calpain activation and preserving synaptic and mitochondrial function in rats with kainic acid-induced seizure. Biomedicines 2021, 9, 284. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, Y.-C.; Kuo, Y.-H.; Hsieh, P.-W.; Hsieh, T.-Y.; Kuo, J.-R.; Wang, S.-J. Chlorogenic Acid Decreases Glutamate Release from Rat Cortical Nerve Terminals by P/Q-Type Ca2+ Channel Suppression: A Possible Neuroprotective Mechanism. Int. J. Mol. Sci. 2021, 22, 11447. https://doi.org/10.3390/ijms222111447
Hung Y-C, Kuo Y-H, Hsieh P-W, Hsieh T-Y, Kuo J-R, Wang S-J. Chlorogenic Acid Decreases Glutamate Release from Rat Cortical Nerve Terminals by P/Q-Type Ca2+ Channel Suppression: A Possible Neuroprotective Mechanism. International Journal of Molecular Sciences. 2021; 22(21):11447. https://doi.org/10.3390/ijms222111447
Chicago/Turabian StyleHung, Yi-Chieh, Yi-Hsiu Kuo, Pei-Wen Hsieh, Ting-Yang Hsieh, Jinn-Rung Kuo, and Su-Jane Wang. 2021. "Chlorogenic Acid Decreases Glutamate Release from Rat Cortical Nerve Terminals by P/Q-Type Ca2+ Channel Suppression: A Possible Neuroprotective Mechanism" International Journal of Molecular Sciences 22, no. 21: 11447. https://doi.org/10.3390/ijms222111447
APA StyleHung, Y. -C., Kuo, Y. -H., Hsieh, P. -W., Hsieh, T. -Y., Kuo, J. -R., & Wang, S. -J. (2021). Chlorogenic Acid Decreases Glutamate Release from Rat Cortical Nerve Terminals by P/Q-Type Ca2+ Channel Suppression: A Possible Neuroprotective Mechanism. International Journal of Molecular Sciences, 22(21), 11447. https://doi.org/10.3390/ijms222111447