
Rejuvenating the Aging Heart by Enhancing the Expression of the Cisd2 Prolongevity Gene


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Enhancing Cisd2 in Aging Hearts via Inducible Cardiac-Specific Cisd2 Overexpression (Cisd2 icOE)
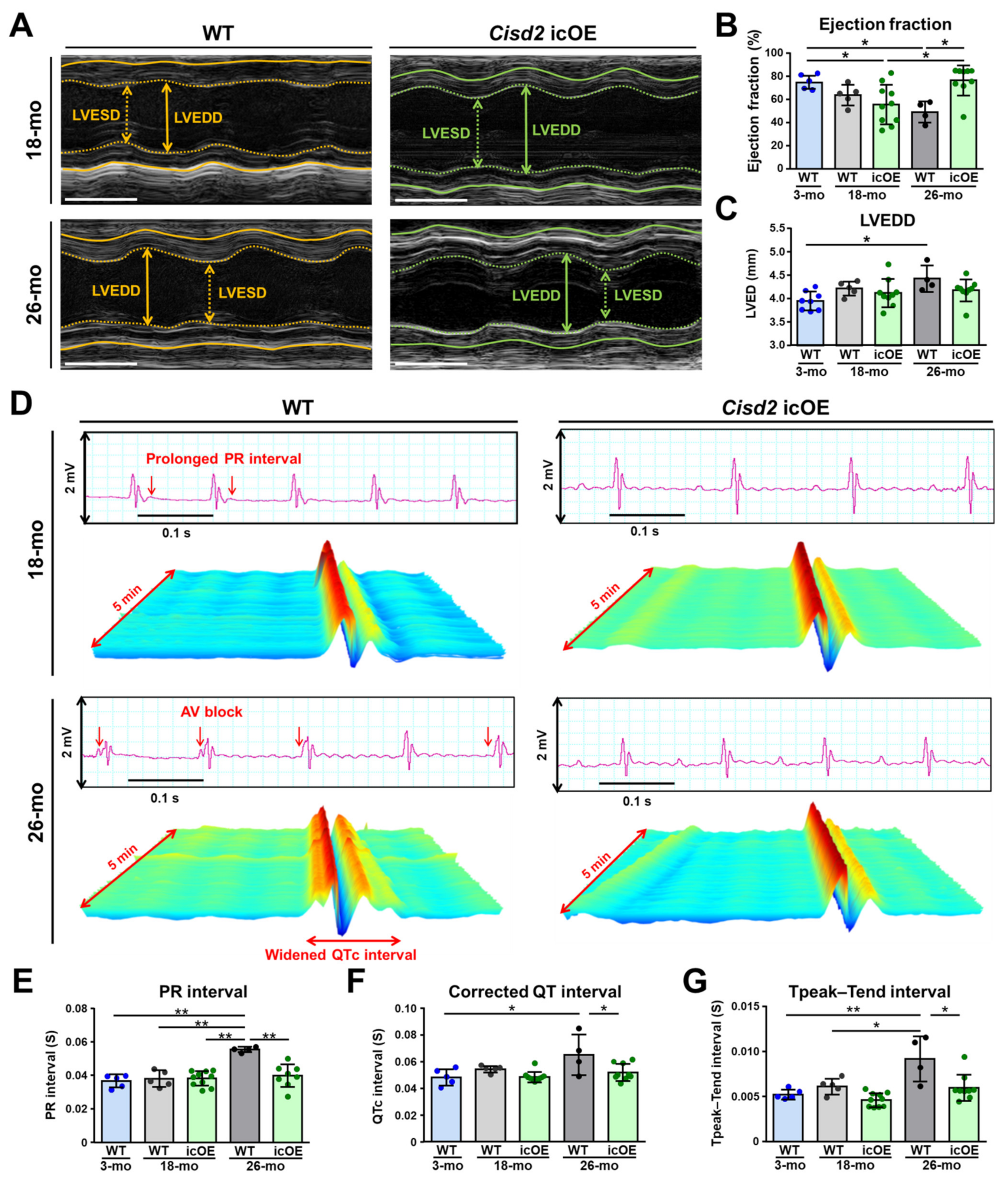
2.2. Cisd2 icOE Prevents an Exacerbation of the Age-Associated Electromechanical Dysfunction
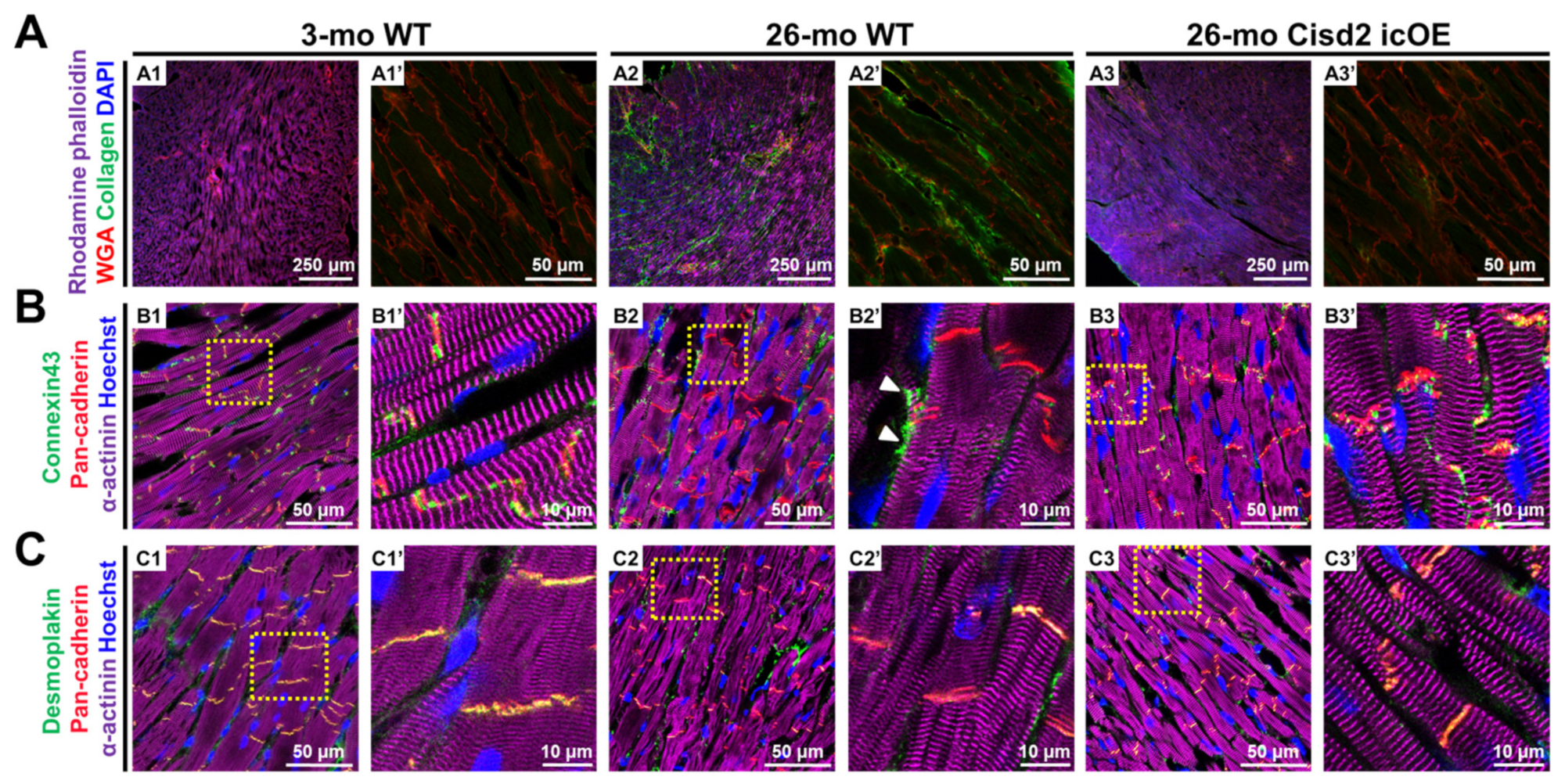
2.3. Cisd2 icOE Reverses Age-Associated Cardiac Fibrosis and Intercalated Disc Defects
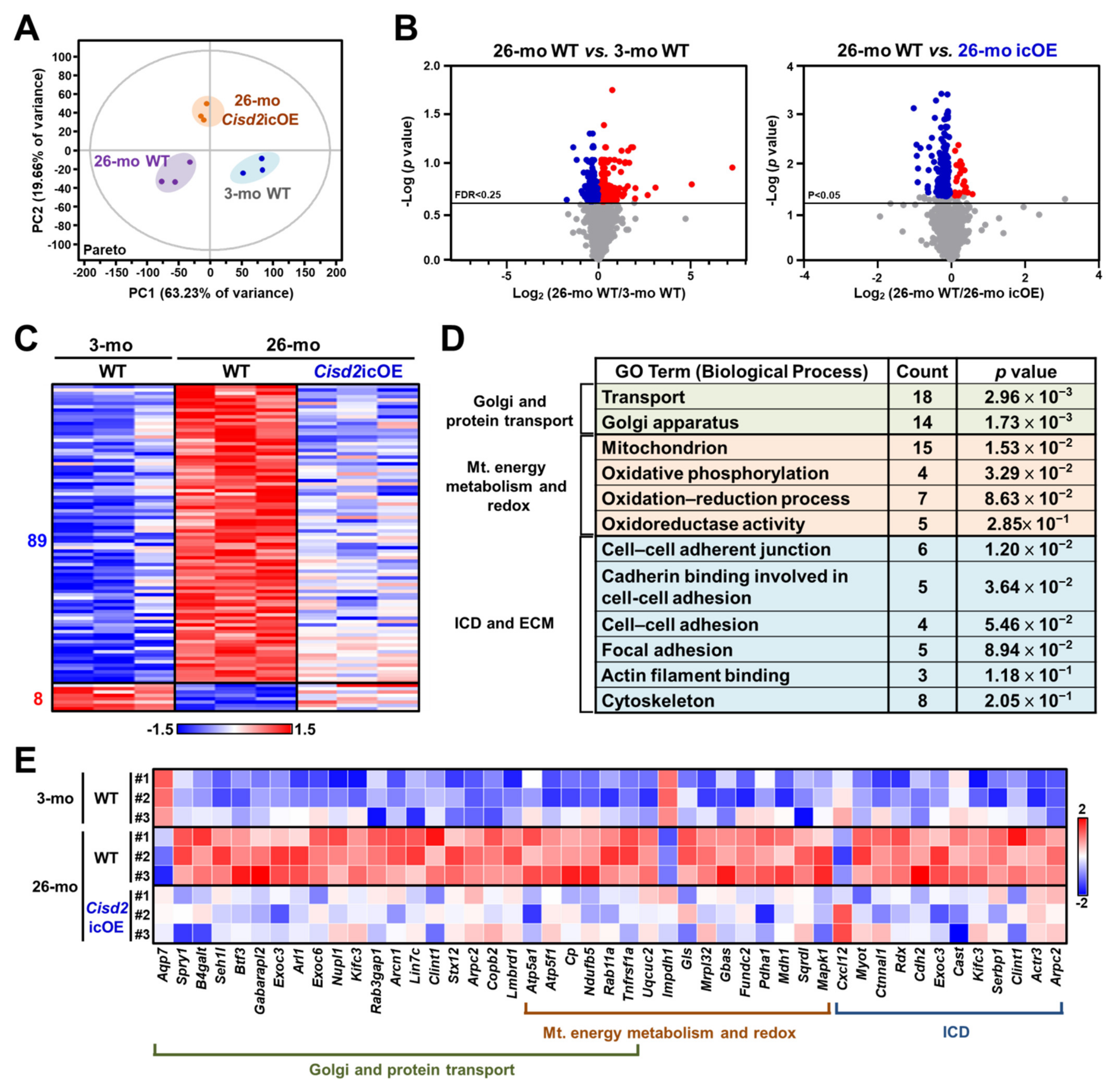
2.4. Cisd2 icOE Changes the Cardiac Transcriptomic Profile from an Old-Age Pattern to a Younger Pattern
2.5. Analyses of Biological Functions and Pathways Associated with the DEGs Reverted by Cisd2 icOE
3. Discussion
3.1. Current Strategies for Heart Rejuvenation and Their Limitations
3.1.1. Exercising at an Old Age
3.1.2. Cell Therapy
3.1.3. Calorie Restriction
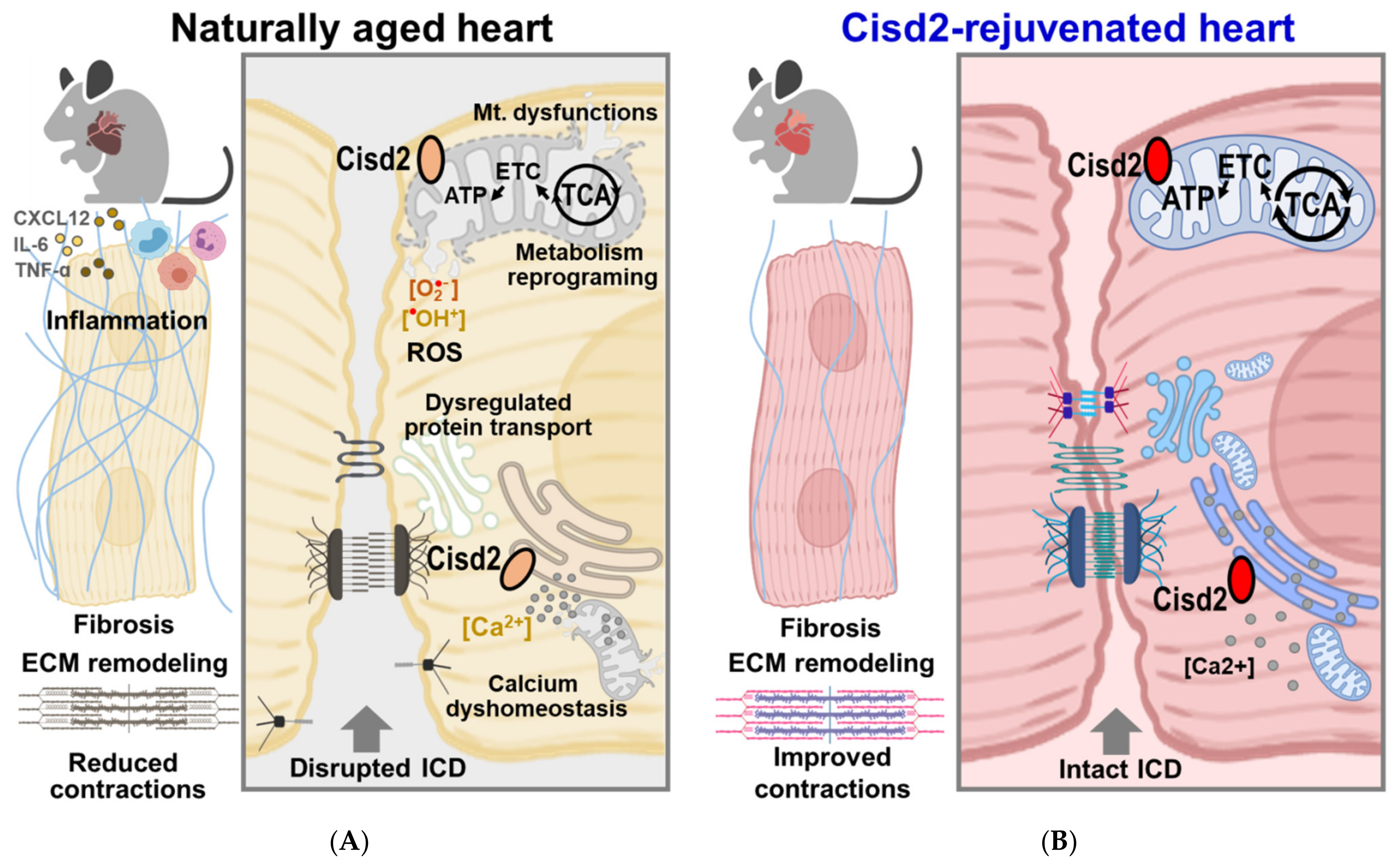
3.2. Comparing the Aging Heart and the Cisd2-Mediated Rejuvenated Heart in Terms of Energy Supply, Protein Transport, and Heart Contraction
3.3. Re-Expression of Cisd2 at a Late Age Stage Activates the Sirtuin Pathways, which then Rejuvenate the Aged Myocardium
3.4. A Potential Role for Cardiac-Resident Macrophages (CRMs) in the Heart of Cisd2 icOE Mice
3.5. Conclusion and Perspectives: Cisd2 as a Potential Molecular Target for Cardiac Rejuvenation
4. Materials and Methods
4.1. Mice
4.2. Cisd2 Induction
4.3. Western Blotting
4.4. Transthoracic Echocardiography
4.5. Electrocardiography (ECG)
4.6. Immunofluorescence (IF), Confocal Microscopy, and Cx43/Pan-Cadherin Co-Localization Coefficiency
4.7. RNA Isolation from Tissue, RNA Sequencing, and Pathway Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.-S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Danaei, G.; Ding, E.L.; Mozaffarian, D.; Taylor, B.; Rehm, J.; Murray, C.J.L.; Ezzati, M. The preventable causes of death in the United States: Comparative risk assessment of dietary, lifestyle, and metabolic risk factors. PLoS Med. 2009, 6, e1000058. [Google Scholar] [CrossRef]
- Kubli, D.A.; Sussman, M.A. Editorial commentary: Mitochondrial autophagy in cardiac aging is all fluxed up. Trends Cardiovasc. Med. 2017, 28, 261–262. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.; Verzosa, G.C.; Pezeshki, A.-M.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nat. Cell Biol. 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Korski, K.I.; Kubli, D.A.; Wang, B.J.; Khalafalla, F.G.; Monsanto, M.M.; Firouzi, F.; Echeagaray, O.H.; Kim, T.; Adamson, R.M.; Dembitsky, W.P.; et al. Hypoxia Prevents Mitochondrial Dysfunction and Senescence in Human c-Kit+ Cardiac Progenitor Cells. Stem Cells 2019, 37, 555–567. [Google Scholar] [CrossRef]
- Rodríguez, M.I.; Carretero, M.; Escames, G.; Lopez, L.C.; Maldonado, M.D.; Tan, D.-X.; Reiter, R.J.; Acuña-Castroviejo, D. Chronic melatonin treatment prevents age-dependent cardiac mitochondrial dysfunction in senescence-accelerated mice. Free. Radic. Res. 2007, 41, 15–24. [Google Scholar] [CrossRef]
- Häseli, S.; Deubel, S.; Jung, T.; Grune, T.; Ott, C. Cardiomyocyte Contractility and Autophagy in a Premature Senescence Model of Cardiac Aging. Oxidative Med. Cell. Longev. 2020, 2020, 8141307. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Feng, D.; Xu, J. Cardioprotective effects of microRNA-18a on acute myocardial infarction by promoting cardiomy-ocyte autophagy and suppressing cellular senescence via brain derived neurotrophic factor. Cell Biosci. 2019, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.-H.; Chou, Y.-J.; Kao, C.-H.; Tsai, T.-F. Mitochondria and Calcium Homeostasis: Cisd2 as a Big Player in Cardiac Ageing. Int. J. Mol. Sci. 2020, 21, 9238. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Guberman, M.; Kirshenbaum, L.A. Mitochondrial quality control: The role of mitophagy in aging. Trends Cardiovasc. Med. 2018, 28, 246–260. [Google Scholar] [CrossRef]
- Dutta, D.; Calvani, R.; Bernabei, R.; Leeuwenburgh, C.; Marzetti, E. Contribution of impaired mitochondrial autophagy to cardiac aging: Mechanisms and therapeutic opportunities. Circ. Res. 2012, 110, 1125–1138. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.-Q.; Huang, Y.-L.; Teng, Y.-C.; Wang, T.-W.; Kao, C.-H.; Yeh, C.-H.; Tsai, T.-F. CISD2 maintains cellular homeostasis. Biochim. et Biophys. Acta Bioenerg. 2021, 1868, 118954. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Kao, C.-H.; Wang, C.-H.; Wu, C.-Y.; Tsai, C.-Y.; Liu, F.-C.; Yang, C.-W.; Wei, Y.-H.; Hsu, M.-T.; Tsai, S.-F.; et al. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 2009, 23, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.-Q.; Chen, Y.-F.; Chen, J.-R.; Jou, Y.-S.; Wu, P.-C.; Kao, C.-H.; Wang, C.-H.; Huang, Y.-L.; Chen, C.-F.; Huang, T.-S.; et al. CISD2 Haploinsufficiency Disrupts Calcium Homeostasis, Causes Nonalcoholic Fatty Liver Disease, and Promotes Hepatocellular Carcinoma. Cell Rep. 2017, 21, 2198–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, C.-H.; Shen, Z.-Q.; Hsiung, S.-Y.; Wu, P.-C.; Teng, Y.-C.; Chou, Y.-J.; Fang, S.-W.; Chen, C.-F.; Yan, Y.-T.; Kao, L.-S.; et al. Cisd2 is essential to delaying cardiac aging and to maintaining heart functions. PLoS Biol. 2019, 17, e3000508. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-F.; Kao, C.-H.; Kirby, R.; Tsai, T.-F. Cisd2 mediates mitochondrial integrity and life span in mammals. Autophagy 2009, 5, 1043–1045. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-H.; Chen, Y.-F.; Wu, C.-Y.; Wu, P.-C.; Huang, Y.-L.; Kao, C.-H.; Lin, C.-H.; Kao, L.-S.; Tsai, T.-F.; Wei, Y.-H. Cisd2 modulates the differentiation and functioning of adipocytes by regulating intracellular Ca2+ homeostasis. Hum. Mol. Genet. 2014, 23, 4770–4785. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.L.; Shen, Z.Q.; Wu, C.Y.; Teng, Y.C.; Liao, C.C.; Kao, C.H.; Chen, L.K.; Lin, C.H.; Tsai, T.F. Comparative pro-teomic profiling reveals a role for Cisd2 in skeletal muscle aging. Aging Cell 2018, 17, e12705. [Google Scholar] [CrossRef]
- Chen, Y.F.; Chou, T.Y.; Lin, I.H.; Chen, C.G.; Kao, C.H.; Huang, G.J.; Chen, L.K.; Wang, P.N.; Lin, C.P.; Tsai, T.F. Upreg-ulation of Cisd2 attenuates Alzheimer’s-related neuronal loss in mice. J. Pathol. 2020, 250, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-Y.; Chen, Y.-F.; Wang, C.-H.; Kao, C.-H.; Zhuang, H.-W.; Chen, C.-C.; Chen, L.-K.; Kirby, R.; Wei, Y.-H.; Tsai, S.-F.; et al. A persistent level of Cisd2 extends healthy lifespan and delays aging in mice. Hum. Mol. Genet. 2012, 21, 3956–3968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohal, D.S.; Nghiem, M.; Crackower, M.A.; Witt, S.A.; Kimball, T.R.; Tymitz, K.M.; Penninger, J.M.; Molkentin, J.D. Tem-porally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res. 2001, 89, 20–25. [Google Scholar] [CrossRef]
- Peng, F.; Xie, F.; Muzik, O. Alteration of Copper Fluxes in Brain Aging: A Longitudinal Study in Rodent Using 64CuCl2-PET/CT. Aging Dis. 2018, 9, 109–118. [Google Scholar] [CrossRef]
- Strait, J.B.; Lakatta, E.G. Aging-Associated Cardiovascular Changes and Their Relationship to Heart Failure. Heart Fail. Clin. 2012, 8, 143–164. [Google Scholar] [CrossRef] [Green Version]
- Nagibin, V.; Egan Benova, T.; Viczenczova, C.; Szeiffova Bacova, B.; Dovinova, I.; Barancik, M.; Tribulova, N. Ageing related down-regulation of myocardial connexin-43 and up-regulation of MMP-2 may predict propensity to atrial fibrillation in ex-perimental animals. Physiol. Res. 2016, 65, S91–S100. [Google Scholar] [CrossRef]
- Wright, K.J.; Thomas, M.M.; Betik, A.C.; Belke, D.; Hepple, R.T. Exercise training initiated in late middle age attenuates cardiac fibrosis and advanced glycation end-product accumulation in senescent rats. Exp. Gerontol. 2014, 50, 9–18. [Google Scholar] [CrossRef]
- Teng, Y.-C.; Wang, J.-Y.; Chi, Y.-H.; Tsai, T.-F. Exercise and the Cisd2 Prolongevity Gene: Two Promising Strategies to Delay the Aging of Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 9059. [Google Scholar] [CrossRef] [PubMed]
- Banks, L.; Buchan, T.A.; Dizonno, V. Aerobic exercise attenuates ageing of the athletic heart. J. Physiol. 2016, 594, 3183–3184. [Google Scholar] [CrossRef] [Green Version]
- Vagnozzi, R.J.; Kasam, R.K.; Sargent, M.A.; Molkentin, J.D. Cardiac Cell Therapy Fails to Rejuvenate the Chronically Scarred Rodent Heart. Circulation 2021, 144, 328–331. [Google Scholar] [CrossRef]
- Bolli, R. Cell therapy for acute myocardial infarction: Requiescat in Pace. Eur. Heart J. 2020, 41, 3711–3714. [Google Scholar] [CrossRef] [PubMed]
- Rafatian, G.; Kamkar, M.; Parent, S.; Michie, C.; Risha, Y.; Molgat, A.S.D.; Seymour, R.; Suuronen, E.J.; Davis, D.R. Mybl2 rejuvenates heart explant-derived cells from aged donors after myocardial infarction. Aging Cell 2020, 19, e13174. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The fibroblast awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [Green Version]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Su, Y.-J.; Wang, P.-W.; Weng, S.-W. The Role of Mitochondria in Immune-Cell-Mediated Tissue Regeneration and Ageing. Int. J. Mol. Sci. 2021, 22, 2668. [Google Scholar] [CrossRef] [PubMed]
- Weixler, V.; Lapusca, R.; Grangl, G.; Guariento, A.; Saeed, M.Y.; Cowan, D.B.; Del Nido, P.J.; McCully, J.D.; Friehs, I. Au-togenous mitochondria transplantation for treatment of right heart failure. J. Thorac. Cardiovasc. Surg. 2021, 162, e111–e121. [Google Scholar] [CrossRef]
- Mietsch, M.; Hinkel, R. "Empowering" Cardiac Cells via Stem Cell Derived Mitochondrial Transplantation- Does Age Matter? Int. J. Mol. Sci. 2021, 22, 1824. [Google Scholar] [CrossRef]
- Liang, W.J.; Gustafsson, B. The Aging Heart: Mitophagy at the Center of Rejuvenation. Front. Cardiovasc. Med. 2020, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-D.; Rebrin, I.; Forster, M.J.; Sohal, R.S. Effects of age and caloric restriction on mitochondrial protein oxidative damage in mice. Mech. Ageing Dev. 2012, 133, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Y.; Lv, S.; Huang, M.; Lv, Y.; Yu, J.; Liu, J.; Tang, T.; Qi, H.; Di, W.; Ding, G. Opposing effects on cardiac function by calorie restriction in different-aged mice. Aging Cell 2017, 16, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.-F.; Karunadharma, P.P.; Chiao, Y.A.; Basisty, N.; Crispin, D.; Hsieh, E.J.; Chen, T.; Gu, H.; Djukovic, D.; Raftery, D.; et al. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell 2014, 13, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Kao, C.H.; Chen, Y.F.; Wei, Y.H.; Tsai, T.F. Cisd2 mediates lifespan: Is there an interconnection among Ca(2)(+) homeostasis, autophagy, and lifespan? Free Radic Res. 2014, 48, 1109–1114. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial Metabolism in Aging Heart. Circ. Res. 2016, 118, 1593–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuka, S.; Tatarkova, Z.; Račay, P.; Lehotský, J.; Dobrota, D.; Kaplan, P. Effect of aging on formation of reactive oxygen species by mitochondria of rat heart. Gen. Physiol. Biophys. 2014, 32, 415–420. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Miguel, V.E.; Lujan, C.; Espie-Caullet, T.; Martinez-Martinez, D.; Moore, S.; Backes, C.; Gonzalez, S.; Galimov, E.R.; Brown, A.E.X.; Halic, M.; et al. Increased fidelity of protein syn-thesis extends lifespan. Cell Metab. 2021, S1550–S4131. [Google Scholar] [CrossRef]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res. Rev. 2011, 10, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisimova, A.; Alexandrov, A.I.; Makarova, N.E.; Gladyshev, V.N.; Dmitriev, S.E. Protein synthesis and quality control in aging. Aging 2018, 10, 4269–4288. [Google Scholar] [CrossRef] [PubMed]
- Henning, R.; Brundel, B.J.J.M. Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 2017, 14, 637–653. [Google Scholar] [CrossRef]
- Alavez, S.; Vantipalli, M.C.; Zucker, D.J.; Klang, I.M.; Lithgow, G.J. Amyloid-binding compounds maintain protein home-ostasis during ageing and extend lifespan. Nature 2011, 472, 226–229. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Hu, X.; Henning, R.H.; Brundel, B.J. Keeping up the balance: Role of HDACs in cardiac proteostasis and ther-apeutic implications for atrial fibrillation. Cardiovasc. Res. 2016, 109, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H. Mechanisms and disease implications of sirtuin-mediated autophagic regulation. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regu-lates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef]
- Lavu, S.; Boss, O.; Elliott, P.J.; Lambert, P.D. Sirtuins—novel therapeutic targets to treat age-associated diseases. Nat. Rev. Drug Discov. 2008, 7, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Vakhrusheva, O.; Smolka, C.; Gajawada, P.; Kostin, S.; Boettger, T.; Kubin, T.; Braun, T.; Bober, E. Sirt7 Increases Stress Resistance of Cardiomyocytes and Prevents Apoptosis and Inflammatory Cardiomyopathy in Mice. Circ. Res. 2008, 102, 703–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.; Ivanov, S.; Satpathy, A.; et al. Embryonic and Adult-Derived Resident Cardiac Macrophages Are Maintained through Distinct Mechanisms at Steady State and during Inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Chen, R.; Chakrabarti, S.; Su, Z. Resident macrophages as potential therapeutic targets for cardiac ageing and injury. Clin. Transl. Immunol. 2020, 9, e1167. [Google Scholar] [CrossRef]
- Wagner, J.U.; Dimmeler, S. Cellular cross-talks in the diseased and aging heart. J. Mol. Cell. Cardiol. 2019, 138, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Meschiari, C.A.; Ero, O.K.; Pan, H.; Finkel, T.; Lindsey, M.L. The impact of aging on cardiac extracellular matrix. GeroScience 2017, 39, 7–18. [Google Scholar] [CrossRef]
- Hayden, E.C. Anti-ageing pill pushed as bona fide drug. Nat. Cell Biol. 2015, 522, 265–266. [Google Scholar] [CrossRef]
- Nadon, N.L.; Strong, R.; Miller, R.A.; Harrison, D.E. NIA Interventions Testing Program: Investigating Putative Aging Intervention Agents in a Genetically Heterogeneous Mouse Model. EBioMedicine 2016, 21, 3–4. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Strong, R.; Miller, R.A.; Antebi, A.; Astle, C.M.; Bogue, M.; Denzel, M.; Fernandez, E.; Flurkey, K.; Hamilton, K.L.; Lamming, D.W.; et al. Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer. Aging Cell 2016, 15, 872–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, C.; Holden, S.E.; Jenkins-Jones, S.; Morgan, C.L.; Halcox, J.; Schernthaner, G.; Mukherjee, J.; Currie, C.J. Can people with type 2 diabetes live longer than those without? A comparison of mortality in people initiated with metformin or sulphonylurea monotherapy and matched, non-diabetic controls. Diabetes, Obes. Metab. 2014, 16, 1165–1173. [Google Scholar] [CrossRef] [Green Version]
- Minor, R.K.; Baur, J.; Gomes, A.P.; Ward, T.M.; Csiszar, A.; Mercken, E.M.; Abdelmohsen, K.; Shin, Y.-K.; Canto, C.; Scheibye-Knudsen, M.; et al. SRT1720 improves survival and healthspan of obese mice. Sci. Rep. 2011, 1, 70. [Google Scholar] [CrossRef] [Green Version]
- Mercken, E.M.; Mitchell, S.J.; Martin-Montalvo, A.; Minor, R.K.; Almeida, M.; Gomes, A.P.; Scheibye-Knudsen, M.; Palacios, H.H.; Licata, J.J.; Zhang, Y.; et al. SRT 2104 extends survival of male mice on a standard diet and preserves bone and muscle mass. Aging Cell 2014, 13, 787–796. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Venkataraman, K.; Hollingsworth, A.; Piche, M.; Tai, T.C. Polyphenols: Benefits to the Cardiovascular System in Health and in Aging. Nutrients 2013, 5, 3779–3827. [Google Scholar] [CrossRef] [PubMed]
- Soriano, P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999, 21, 70–71. [Google Scholar] [CrossRef]
- Nakamura, E.; Nguyen, M.T.; Mackem, S. Kinetics of tamoxifen-regulated Cre activity in mice using a cartilage-specific CreER(T) to assay temporal activity windows along the proximodistal limb skeleton. Dev. Dyn. 2006, 235, 2603–2612. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, C.-H.; Chou, Y.-J.; Chu, T.-K.; Tsai, T.-F. Rejuvenating the Aging Heart by Enhancing the Expression of the Cisd2 Prolongevity Gene. Int. J. Mol. Sci. 2021, 22, 11487. https://doi.org/10.3390/ijms222111487
Yeh C-H, Chou Y-J, Chu T-K, Tsai T-F. Rejuvenating the Aging Heart by Enhancing the Expression of the Cisd2 Prolongevity Gene. International Journal of Molecular Sciences. 2021; 22(21):11487. https://doi.org/10.3390/ijms222111487
Chicago/Turabian StyleYeh, Chi-Hsiao, Yi-Ju Chou, Ting-Kuan Chu, and Ting-Fen Tsai. 2021. "Rejuvenating the Aging Heart by Enhancing the Expression of the Cisd2 Prolongevity Gene" International Journal of Molecular Sciences 22, no. 21: 11487. https://doi.org/10.3390/ijms222111487
APA StyleYeh, C.-H., Chou, Y.-J., Chu, T.-K., & Tsai, T.-F. (2021). Rejuvenating the Aging Heart by Enhancing the Expression of the Cisd2 Prolongevity Gene. International Journal of Molecular Sciences, 22(21), 11487. https://doi.org/10.3390/ijms222111487