
Dissecting the Supramolecular Dispersion of Fullerenes by Proteins/Peptides: Amino Acid Ranking and Driving Forces for Binding to C60

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
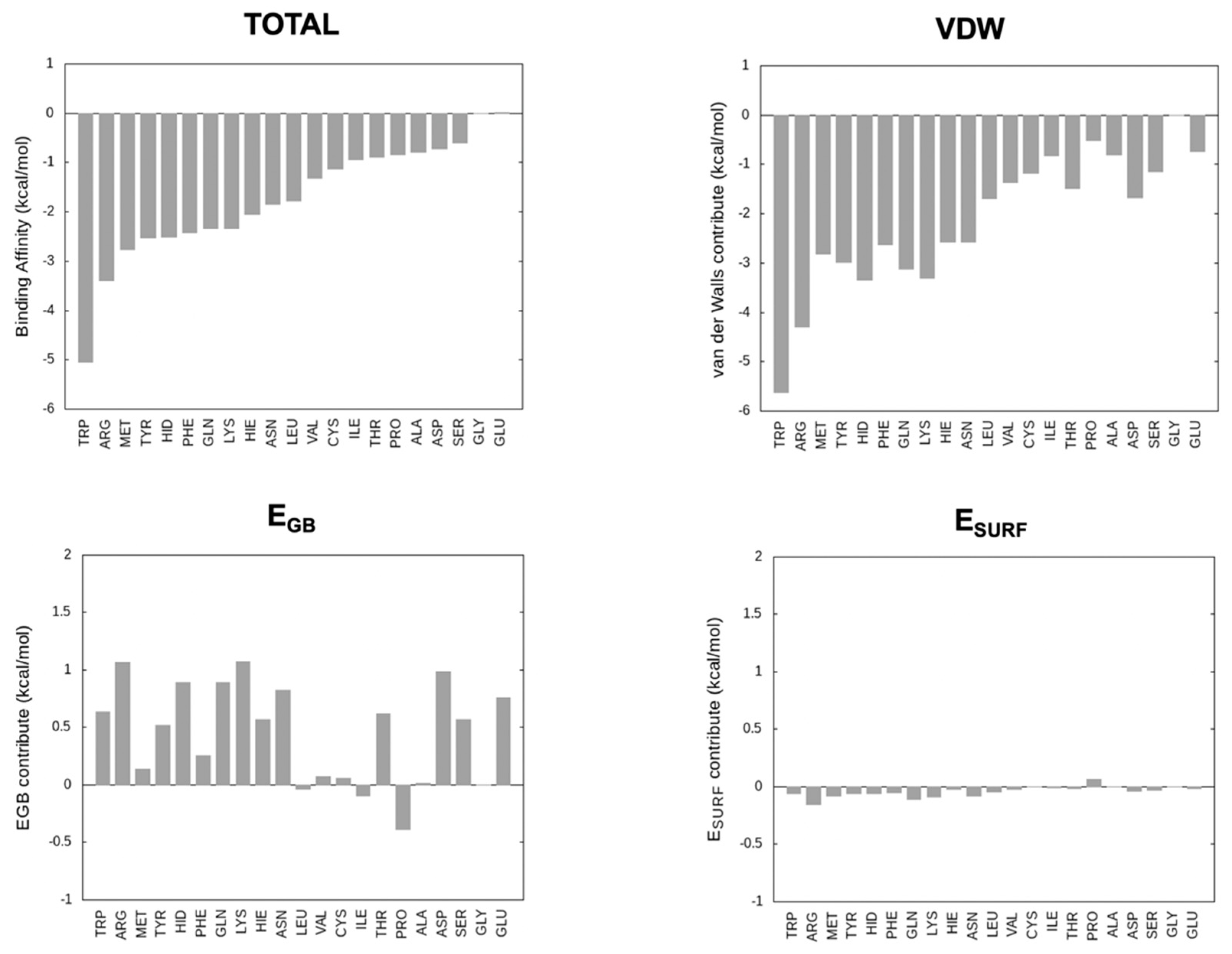
2.1. Ranking the Binding Energies
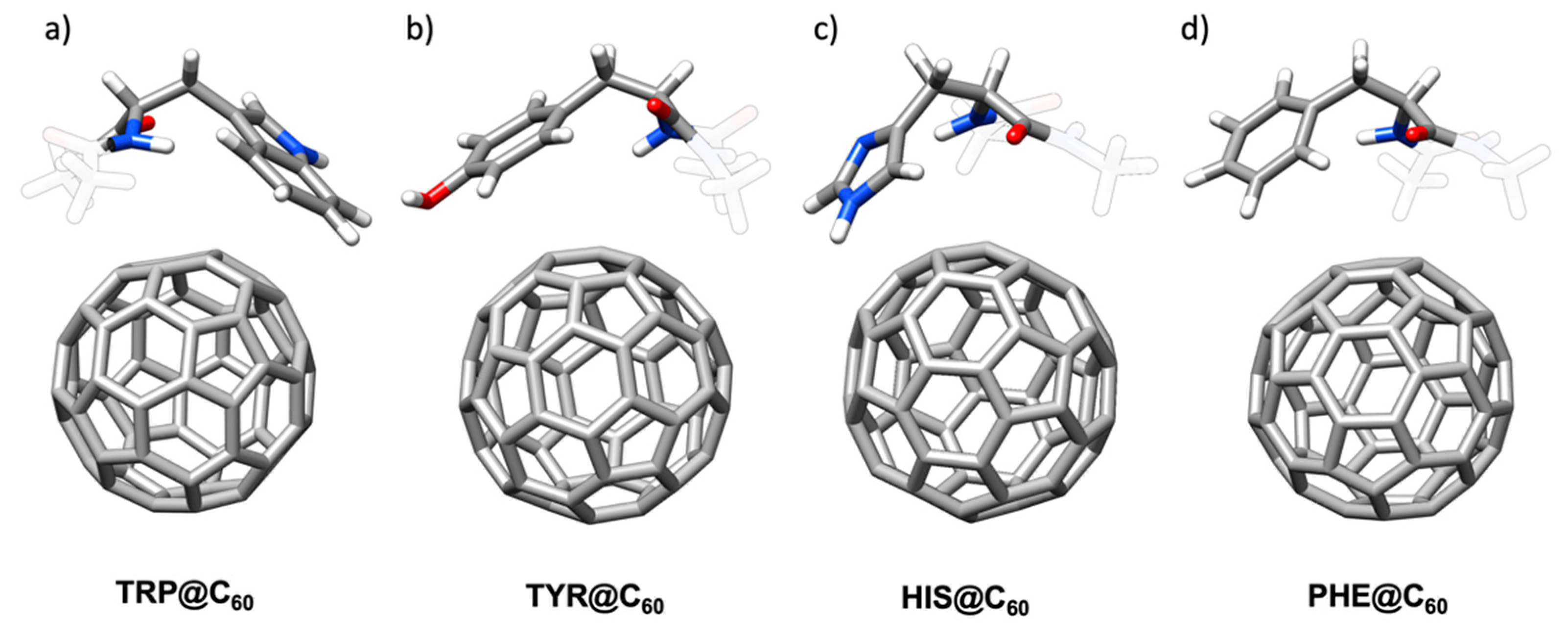
2.1.1. Aromatic Amino Acids
2.1.2. Charged Amino Acids
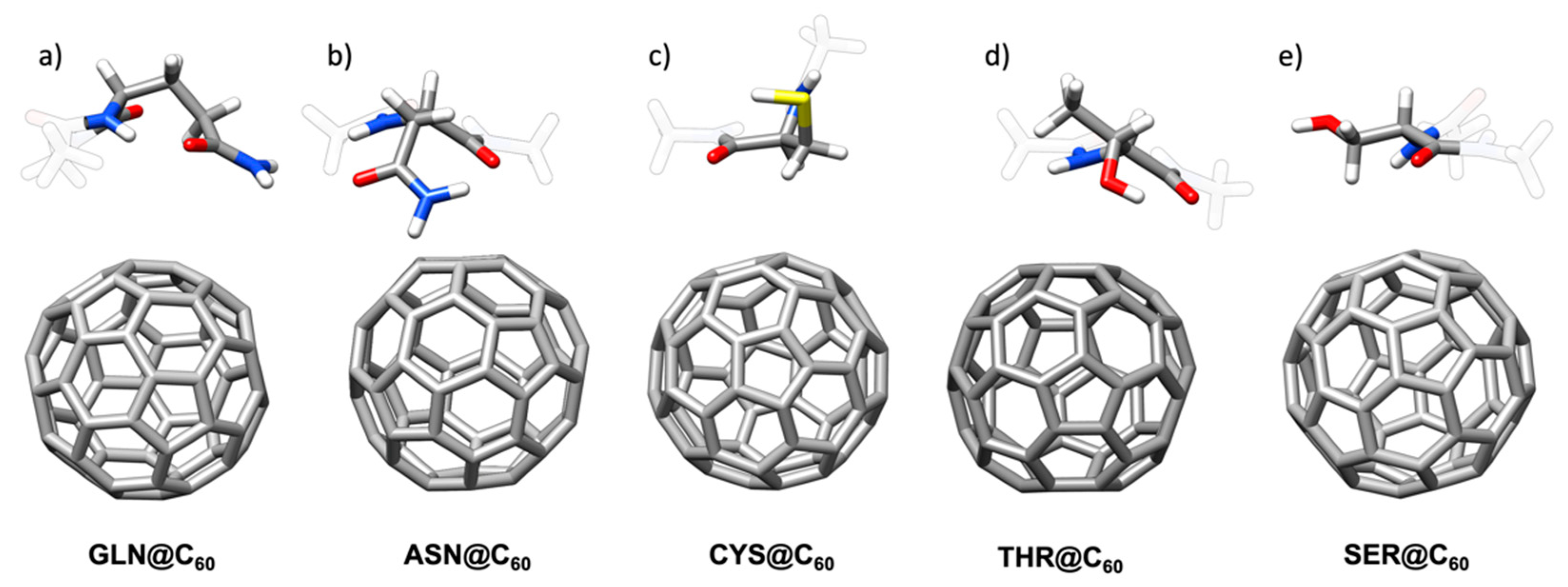
2.1.3. Polar Amino Acids
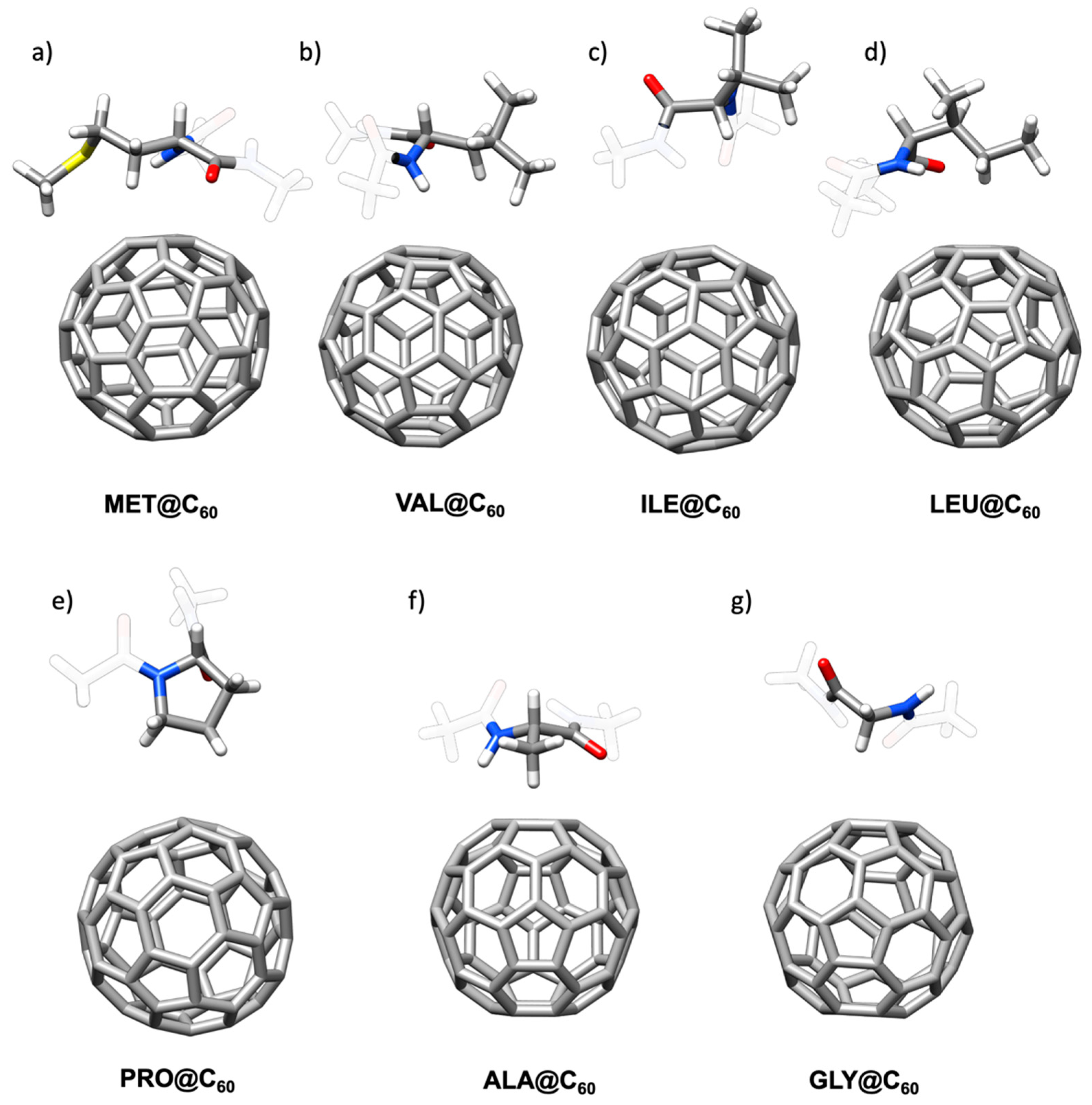
2.1.4. Hydrophobic Amino Acids
2.2. Conformations of Amino Acids in Water and Upon Interaction with C60
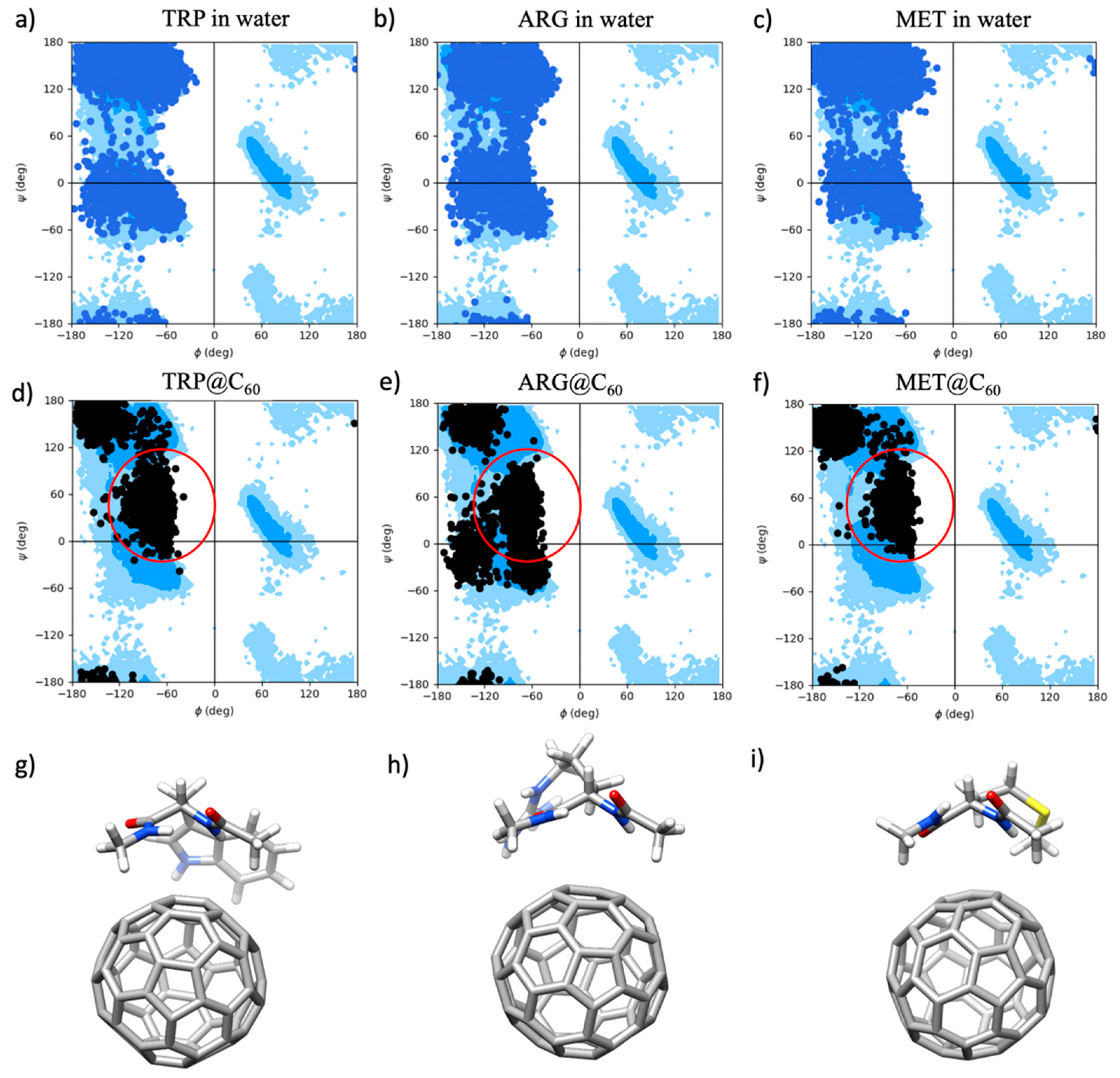
2.2.1. Analysis of the Torsional Angles of the Amino Acid Backbone
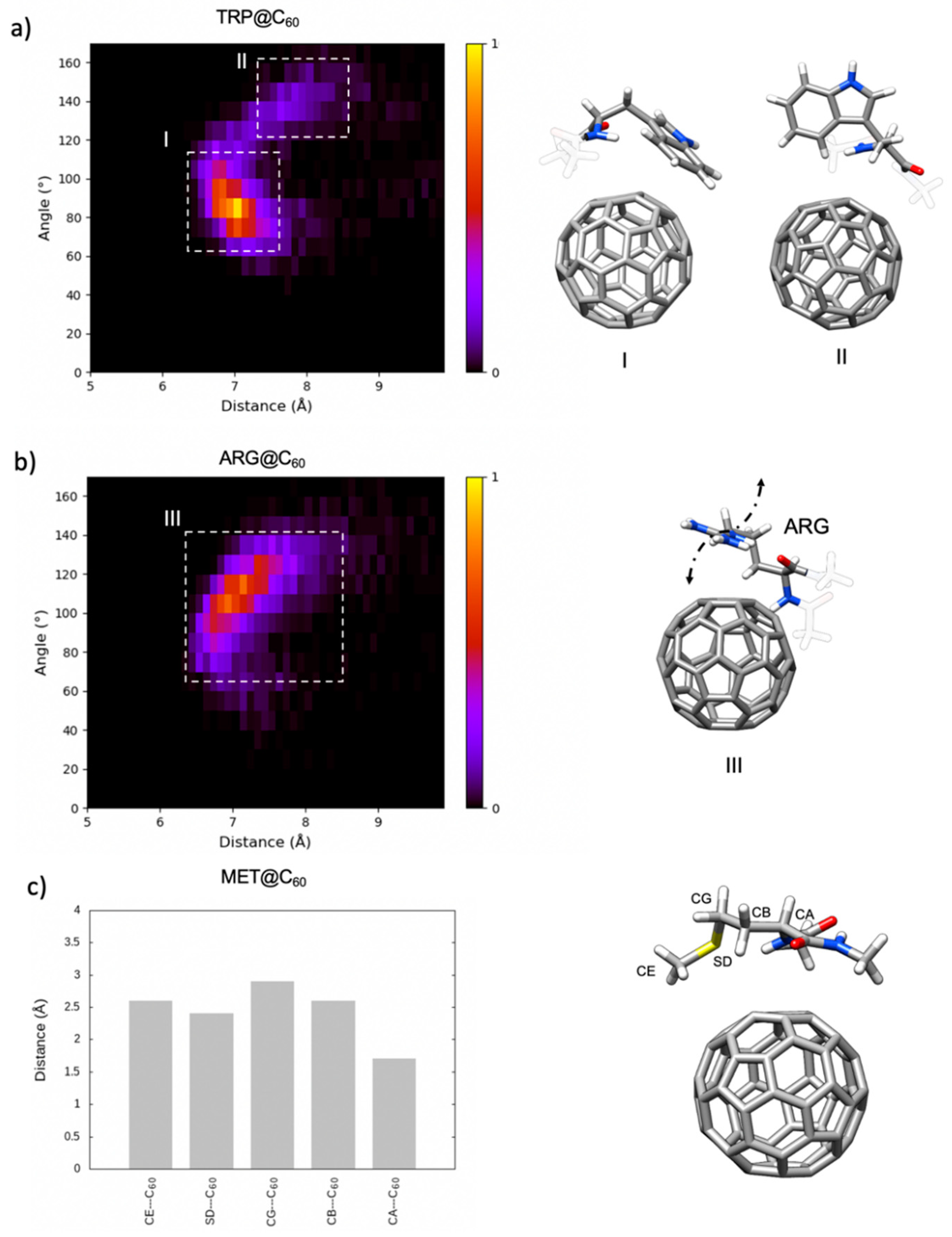
2.2.2. Analysis of the Side Chain Conformation of the Amino Acids upon C60 Binding
3. Materials and Methods
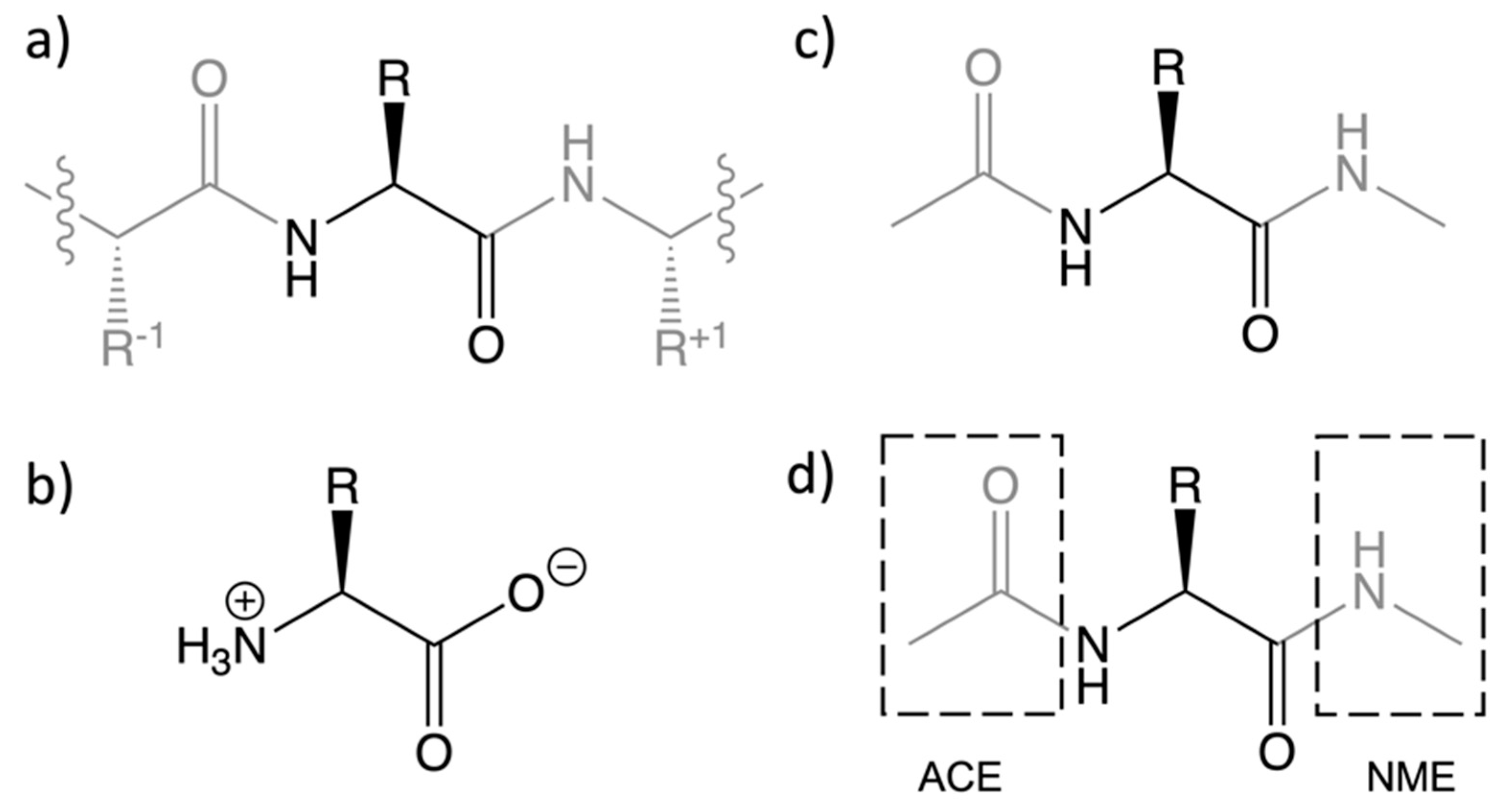
3.1. System Setup
3.2. MD Simulations
3.3. Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) and Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) Analysis
3.4. Conformational Analysis of the Amino Acids
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Goodarzi, S.; Da Ros, T.; Conde, J.; Sefat, F.; Mozafari, M. Fullerene: Biomedical engineers get to revisit an old friend. Mater. Today 2017, 20, 460–480. [Google Scholar] [CrossRef] [Green Version]
- Castro, E.; Garcia, A.H.; Zavala, G.; Echegoyen, L. Fullerenes in biology and medicine. J. Mater. Chem. B 2017, 5, 6523–6535. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, A.; Zhou, Z.; Connor, J.; Madhankumar, A.; Pamujula, S.; Sayes, C.M.; Kepley, C.L. Application of fullerenes in nanomedicine: An update. Nanomedicine 2013, 8, 1191–1208. [Google Scholar] [CrossRef] [Green Version]
- Mchedlov-Petrossyan, N.O. Fullerenes in Liquid Media: An Unsettling Intrusion into the Solution Chemistry. Chem. Rev. 2013, 113, 5149–5193. [Google Scholar] [CrossRef]
- Nakamura, E.; Isobe, H. Functionalized Fullerenes in Water. The First 10 Years of Their Chemistry, Biology, and Nanoscience. Acc. Chem. Res. 2003, 36, 807–815. [Google Scholar] [CrossRef]
- Bosi, S.; Da Ros, T.; Spalluto, G.; Prato, M. Fullerene derivatives: An attractive tool for biological applications. Eur. J. Med. Chem. 2003, 38, 913–923. [Google Scholar] [CrossRef]
- Dugan, L.L.; Turetsky, D.M.; Du, C.; Lobner, D.; Wheeler, M.; Almli, C.R.; Shen, C.K.F.; Luh, T.Y.; Choi, D.W.; Lin, T.S. Carboxyfullerenes as neuroprotective agents. Proc. Natl. Acad. Sci. USA 1997, 94, 9434–9439. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.M.Y.; Chyi, B.Y.; Wang, S.D.; Yu, H.H.; Kanakamma, P.P.; Luh, T.Y.; Chou, C.K.; Ho, L.T. Carboxyfullerene prevents iron-induced oxidative stress in rat brain. J. Neurochem. 1999, 72, 1634–1640. [Google Scholar] [CrossRef] [PubMed]
- Straface, E.; Natalini, B.; Monti, D.; Franceschi, C.; Schettini, G.; Bisaglia, M.; Fumelli, C.; Pincelli, C.; Pellicciari, R.; Malorni, W. C3-Fullero-tris-methanodicarboxylic acid protects epithelial cells from radiation-induced anoikia by influencing cell adhesion ability. FEBS Lett. 1999, 454, 335–340. [Google Scholar]
- Bozdaganyan, M.E.; Orekhov, P.S.; Shaytan, A.K.; Shaitan, K.V. Comparative computational study of interaction of C60-fullerene and tris-malonyl-C60-fullerene isomers with lipid bilayer: Relation to their antioxidant effect. PLoS ONE 2014, 9, e102487. [Google Scholar] [CrossRef]
- Yang, X.; Ebrahimi, A.; Li, J.; Cui, Q. Fullerene-biomolecule conjugates and their biomedicinal applications. Int. J. Nanomed. 2013, 9, 77–92. [Google Scholar]
- Bianco, A.; Da, T.; Prato, M.; Toniolo, C.; Ros, T. Da Fullerene-based Amino Acids and Peptides. J. Pept. Sci. 2001, 7, 208–219. [Google Scholar] [CrossRef]
- Barron, A.R. [60] Fullerene-peptides: Bio-nano conjugates with structural and chemical diversity. J. Enzyme Inhib. Med. Chem. 2016, 31, 164–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennepalli, S.; Pyne, S.G.; Keller, P.A. [60] Fullerenyl amino acids and peptides: A review of their synthesis and applications. RSC Adv. 2014, 4, 46383–46398. [Google Scholar] [CrossRef] [Green Version]
- Pochkaeva, E.I.; Podolsky, N.E.; Zakusilo, D.N.; Petrov, A.V.; Charykov, N.A.; Vlasov, T.D.; Penkova, A.V.; Vasina, L.V.; Murin, I.V.; Sharoyko, V.V.; et al. Fullerene derivatives with amino acids, peptides and proteins: From synthesis to biomedical application. Prog. Solid State Chem. 2020, 57, 100255. [Google Scholar] [CrossRef]
- Kurz, A.; Halliwell, C.M.; Davis, J.J.; Allen, H.; Hill, O.; Canters, G.W. A fullerene-modified protein. Chem. Commun. 1998, 433–434. [Google Scholar] [CrossRef]
- Chen, B.X.; Wilson, S.R.; Das, M.; Coughlin, D.J.; Erlanger, B.F. Antigenicity of fullerenes: Antibodies specific for fullerenes and their characteristics. Proc. Natl. Acad. Sci. USA 1998, 95, 10809–10813. [Google Scholar] [CrossRef] [Green Version]
- Guldi, D.M.; Prato, M. Excited-state properties of C60 fullerene derivatives. Acc. Chem. Res. 2000, 33, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Dallavalle, M.; Leonzio, M.; Calvaresi, M.; Zerbetto, F. Explaining Fullerene Dispersion by using Micellar Solutions. ChemPhysChem 2014, 15, 2998–3005. [Google Scholar] [CrossRef]
- Di Giosia, M.; Nicolini, F.; Ferrazzano, L.; Soldà, A.; Valle, F.; Cantelli, A.; Marforio, T.D.; Bottoni, A.; Zerbetto, F.; Montalti, M.; et al. Stable and Biocompatible Monodispersion of C 60 in Water by Peptides. Bioconjug. Chem. 2019, 30, 808–814. [Google Scholar] [CrossRef]
- Di Giosia, M.; Zerbetto, F.; Calvaresi, M. Incorporation of Molecular Nanoparticles Inside Proteins: The Trojan Horse Approach in Theranostics. Acc. Mater. Res. 2021, 2, 594–605. [Google Scholar] [CrossRef]
- Belgorodsky, B.; Fadeev, L.; Kolsenik, J.; Gozin, M. Formation of a soluble stable complex between pristine C60-fullerene and a native blood protein. ChemBioChem 2006, 7, 1783–1789. [Google Scholar] [CrossRef]
- Di Giosia, M.; Soldà, A.; Seeger, M.; Cantelli, A.; Arnesano, F.; Nardella, M.I.; Mangini, V.; Valle, F.; Montalti, M.; Zerbetto, F.; et al. A Bio-Conjugated Fullerene as a Subcellular-Targeted and Multifaceted Phototheranostic Agent. Adv. Funct. Mater. 2021, 31, 1–8. [Google Scholar] [CrossRef]
- Calvaresi, M.; Arnesano, F.; Bonacchi, S.; Bottoni, A.; Calò, V.; Conte, S.; Falini, G.; Fermani, S.; Losacco, M.; Montalti, M.; et al. C60@Lysozyme: Direct observation by nuclear magnetic resonance of a 1:1 fullerene protein adduct. ACS Nano 2014, 8, 1871–1877. [Google Scholar] [CrossRef] [PubMed]
- Soldà, A.; Cantelli, A.; Di Giosia, M.; Montalti, M.; Zerbetto, F.; Rapino, S.; Calvaresi, M. C60@lysozyme: A new photosensitizing agent for photodynamic therapy. J. Mater. Chem. B 2017, 5, 6608–6615. [Google Scholar] [CrossRef] [PubMed]
- Di Giosia, M.; Bomans, P.H.H.; Bottoni, A.; Cantelli, A.; Falini, G.; Franchi, P.; Guarracino, G.; Friedrich, H.; Lucarini, M.; Paolucci, F.; et al. Proteins as supramolecular hosts for C60: A true solution of C60 in water. Nanoscale 2018, 10, 9908–9916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- di Giosia, M.; Valle, F.; Cantelli, A.; Bottoni, A.; Zerbetto, F.; Calvaresi, M. C60 bioconjugation with proteins: Towards a palette of carriers for all pH ranges. Materials (Basel) 2018, 11, 691. [Google Scholar] [CrossRef] [Green Version]
- Vance, S.J.; Desai, V.; Smith, B.O.; Kennedy, M.W.; Cooper, A. Aqueous solubilization of C60 fullerene by natural protein surfactants, latherin and ranaspumin-2. Biophys. Chem. 2016, 214–215, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Di Costanzo, L.; Geremia, S. Atomic details of carbon-based nanomolecules interacting with proteins. Molecules 2020, 25, 3555. [Google Scholar] [CrossRef]
- Calvaresi, M.; Zerbetto, F. Baiting proteins with C60. ACS Nano 2010, 4, 2283–2299. [Google Scholar] [CrossRef]
- Calvaresi, M.; Zerbetto, F. Fullerene sorting proteins. Nanoscale 2011, 3, 2873–2881. [Google Scholar] [CrossRef]
- Ahmed, L.; Rasulev, B.; Kar, S.; Krupa, P.; Mozolewska, M.A.; Leszczynski, J. Inhibitors or toxins? Large library target-specific screening of fullerene-based nanoparticles for drug design purpose. Nanoscale 2017, 9, 10263–10276. [Google Scholar] [CrossRef]
- Kim, K.-H.; Ko, D.-K.; Kim, Y.-T.; Kim, N.H.; Paul, J.; Zhang, S.-Q.; Murray, C.B.; Acharya, R.; DeGrado, W.F.; Kim, Y.H.; et al. Protein-directed self-assembly of a fullerene crystal. Nat. Commun. 2016, 7, 11429. [Google Scholar] [CrossRef] [Green Version]
- Liutkus, M.; López-Andarias, A.; Mejías, S.H.; López-Andarias, J.; Gil-Carton, D.; Feixas, F.; Osuna, S.; Matsuda, W.; Sakurai, T.; Seki, S.; et al. Protein-directed crystalline 2D fullerene assemblies. Nanoscale 2020, 12, 3614–3622. [Google Scholar] [CrossRef] [Green Version]
- Calvaresi, M.; Bottoni, A.; Zerbetto, F. Thermodynamics of Binding between Proteins and Carbon Nanoparticles: The Case of C60@Lysozyme. J. Phys. Chem. C 2015, 119, 28077–28082. [Google Scholar] [CrossRef] [Green Version]
- Trozzi, F.; Marforio, T.D.; Bottoni, A.; Zerbetto, F.; Calvaresi, M. Engineering the Fullerene-protein Interface by Computational Design: The Sum is More than its Parts. Isr. J. Chem. 2017, 57, 547–552. [Google Scholar] [CrossRef]
- Maoyong, S.; Guibin, J.; Junfa, Y.; Hailin, W. Inhibition of polymerase activity by pristine fullerene nanoparticles can be mitigated by abundant proteins. Chem. Commun. 2010, 46, 1404–1406. [Google Scholar] [CrossRef]
- Fjodorova, N.; Novič, M.; Venko, K.; Rasulev, B. A comprehensive cheminformatics analysis of structural features affecting the binding activity of fullerene derivatives. Nanomaterials 2020, 10, 90. [Google Scholar] [CrossRef] [Green Version]
- Roy, P.; Bag, S.; Chakraborty, D.; Dasgupta, S. Exploring the Inhibitory and Antioxidant Effects of Fullerene and Fullerenol on Ribonuclease, A. ACS Omega 2018, 3, 12270–12283. [Google Scholar] [CrossRef] [Green Version]
- Serda, M.; Szewczyk, G.; Krzysztyńska-Kuleta, O.; Korzuch, J.; Dulski, M.; Musioł, R.; Sarna, T. Developing [60] Fullerene Nanomaterials for Better Photodynamic Treatment of Non-Melanoma Skin Cancers. ACS Biomater. Sci. Eng. 2020, 6, 5930–5940. [Google Scholar] [CrossRef] [PubMed]
- De Leon, A.; Jalbout, A.F.; Basiuk, V.A. Fullerene-amino acid interactions. A theoretical study. Chem. Phys. Lett. 2008, 452, 306–314. [Google Scholar] [CrossRef]
- Basiuk, V.A.; González-Luciano, E. Noncovalent interactions of amino acids with fullerene C60: A dispersion-corrected DFT study. Fullerenes Nanotub. Carbon Nanostructures 2016, 24, 371–379. [Google Scholar] [CrossRef]
- Ganazzoli, F.; Raffaini, G. Classical atomistic simulations of protein adsorption on carbon nanomaterials. Curr. Opin. Colloid Interface Sci. 2019, 41, 11–26. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Calvaresi, M.; Furini, S.; Domene, C.; Bottoni, A.; Zerbetto, F. Blocking the passage: C60 geometrically clogs K+ channels. ACS Nano 2015, 9, 4827–4834. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zou, Y.; Zhang, Q.; Chen, P.; Liu, Y.; Qian, Z. Distinct binding dynamics, sites and interactions of fullerene and fullerenols with amyloid-β peptides revealed by molecular dynamics simulations. Int. J. Mol. Sci. 2019, 20, 2048. [Google Scholar] [CrossRef] [Green Version]
- Braden, B.C.; Goldbaum, F.A.; Chen, B.X.; Kirschner, A.N.; Wilson, S.R.; Erlanger, B.F. X-ray crystal structure of an anti-Buckminsterfullerene antibody Fab fragment: Biomolecular recognition of C60. Proc. Natl. Acad. Sci. USA 2000, 97, 12193–12197. [Google Scholar] [CrossRef] [Green Version]
- Osipov, E.M.; Hendrickson, O.D.; Tikhonova, T.V.; Zherdev, A.V.; Solopova, O.N.; Sveshnikov, P.G.; Dzantiev, B.B.; Popov, V.O. Structure of the anti-C60 fullerene antibody fab fragment: Structural determinants of fullerene binding. Acta Nat. 2019, 11, 58–65. [Google Scholar] [CrossRef]
- Bai, C.; Lao, Z.; Chen, Y.; Tang, Y.; Wei, G. Pristine and Hydroxylated Fullerenes Prevent the Aggregation of Human Islet Amyloid Polypeptide and Display Different Inhibitory Mechanisms. Front. Chem. 2020, 8, 1–11. [Google Scholar] [CrossRef]
- Zuo, G.; Kang, S.G.; Xiu, P.; Zhao, Y.; Zhou, R. Interactions between proteins and carbon-based nanoparticles: Exploring the origin of nanotoxicity at the molecular level. Small 2013, 9, 1546–1556. [Google Scholar] [CrossRef]
- Bai, Y.; Wu, X.; Ouyang, P.; Shi, M.; Li, Q.; Maimaiti, T.; Lan, S.; Yang, S.T.; Chang, X.L. Surface modification mediates the interaction between fullerene and lysozyme: Protein structure and antibacterial activity. Environ. Sci. Nano 2021, 8, 76–85. [Google Scholar] [CrossRef]
- Wu, X.; Yang, S.T.; Wang, H.; Wang, L.; Hu, W.; Cao, A.; Liu, Y. Influences of the size and hydroxyl number of fullerenes/fullerenols on their interactions with proteins. J. Nanosci. Nanotechnol. 2010, 10, 6298–6304. [Google Scholar] [CrossRef]
- Yang, S.T.; Wang, H.; Guo, L.; Gao, Y.; Liu, Y.; Cao, A. Interaction of fullerenol with lysozyme investigated by experimental and computational approaches. Nanotechnology 2008, 19, 395101. [Google Scholar] [CrossRef]
- Giełdoń, A.; Witt, M.M.; Gajewicz, A.; Puzyn, T. Rapid insight into C60 influence on biological functions of proteins. Struct. Chem. 2017, 28, 1775–1788. [Google Scholar] [CrossRef]
- Hirano, A.; Kameda, T. Aromaphilicity Index of Amino Acids: Molecular Dynamics Simulations of the Protein Binding Affinity for Carbon Nanomaterials. ACS Appl. Nano Mater. 2021, 4, 2486–2495. [Google Scholar] [CrossRef]
- Dasetty, S.; Barrows, J.K.; Sarupria, S. Adsorption of amino acids on graphene: Assessment of current force fields. Soft Matter 2019, 15, 2359–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, A.; Tanaka, T.; Kataura, H.; Kameda, T. Arginine side chains as a dispersant for individual single-wall carbon nanotubes. Chem. A Eur. J. 2014, 20, 4922–4930. [Google Scholar] [CrossRef] [PubMed]
- Iwashita, K.; Shiraki, K.; Ishii, R.; Tanaka, T.; Hirano, A. Arginine suppresses the adsorption of lysozyme onto single-wall carbon nanotubes. Chem. Lett. 2016, 45, 952–954. [Google Scholar] [CrossRef]
- Calvaresi, M.; Hoefinger, S.; Zerbetto, F. Probing the structure of lysozyme-carbon-nanotube hybrids with molecular dynamics. Chem. A Eur. J. 2012, 18, 4308–4813. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.; Coppens, M.-O.; Garde, S. Role of Arginine in Mediating Protein–Carbon Nanotube Interactions. Langmuir 2015, 31, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Garg, M.; Shah, D.; Rajagopalan, R. Solubilization of aromatic and hydrophobic moieties by arginine in aqueous solutions. J. Chem. Phys. 2010, 133, 054902. [Google Scholar] [CrossRef]
- Di Giosia, M.; Valle, F.; Cantelli, A.; Bottoni, A.; Zerbetto, F.; Fasoli, E.; Calvaresi, M. High-throughput virtual screening to rationally design protein—Carbon nanotube interactions. Identification and preparation of stable water dispersions of protein—Carbon nanotube hybrids and efficient design of new functional materials. Carbon 2019, 147, 70–82. [Google Scholar] [CrossRef]
- Di Giosia, M.; Marforio, T.D.; Cantelli, A.; Valle, F.; Zerbetto, F.; Su, Q.; Wang, H.; Calvaresi, M. Inhibition of α-chymotrypsin by pristine single-wall carbon nanotubes: Clogging up the active site. J. Colloid Interface Sci. 2020, 571, 174–184. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER16; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Zhang, L.; Lemonnier, J.-F.; Acocella, A.; Calvaresi, M.; Zerbetto, F.; Leigh, D.A. Effects of knot tightness at the molecular level. Proc. Natl. Acad. Sci. USA 2019, 116, 2452–2457. [Google Scholar] [CrossRef] [Green Version]
- Di Silvio, S.; Bologna, F.; Milli, L.; Giuri, D.; Zanna, N.; Castellucci, N.; Monari, M.; Calvaresi, M.; Górecki, M.; Angelici, G.; et al. Elusive π-helical peptide foldamers spotted by chiroptical studies. Org. Biomol. Chem. 2020, 18, 865–877. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marforio, T.D.; Calza, A.; Mattioli, E.J.; Zerbetto, F.; Calvaresi, M. Dissecting the Supramolecular Dispersion of Fullerenes by Proteins/Peptides: Amino Acid Ranking and Driving Forces for Binding to C60. Int. J. Mol. Sci. 2021, 22, 11567. https://doi.org/10.3390/ijms222111567
Marforio TD, Calza A, Mattioli EJ, Zerbetto F, Calvaresi M. Dissecting the Supramolecular Dispersion of Fullerenes by Proteins/Peptides: Amino Acid Ranking and Driving Forces for Binding to C60. International Journal of Molecular Sciences. 2021; 22(21):11567. https://doi.org/10.3390/ijms222111567
Chicago/Turabian StyleMarforio, Tainah Dorina, Alessandro Calza, Edoardo Jun Mattioli, Francesco Zerbetto, and Matteo Calvaresi. 2021. "Dissecting the Supramolecular Dispersion of Fullerenes by Proteins/Peptides: Amino Acid Ranking and Driving Forces for Binding to C60" International Journal of Molecular Sciences 22, no. 21: 11567. https://doi.org/10.3390/ijms222111567
APA StyleMarforio, T. D., Calza, A., Mattioli, E. J., Zerbetto, F., & Calvaresi, M. (2021). Dissecting the Supramolecular Dispersion of Fullerenes by Proteins/Peptides: Amino Acid Ranking and Driving Forces for Binding to C60. International Journal of Molecular Sciences, 22(21), 11567. https://doi.org/10.3390/ijms222111567