
Breast Cancer CAFs: Spectrum of Phenotypes and Promising Targeting Avenues



{kind=link}
Abstract
:1. Introduction
2. CAF Sources
3. CAFs Heterogeneity
3.1. Phenotypical Heterogeneity (CAF Subtypes)
3.2. Functional Heterogeneity
3.2.1. ECM Remodeling
3.2.2. Immune Regulation
3.2.3. Vascular Regulation
3.2.4. Metabolic Adaptations
3.2.5. Tumor Stemness and Chemoresistance
3.2.6. Motility and Invasiveness
3.2.7. Genetic and Epigenetic Alterations
4. CAFs and Prognosis
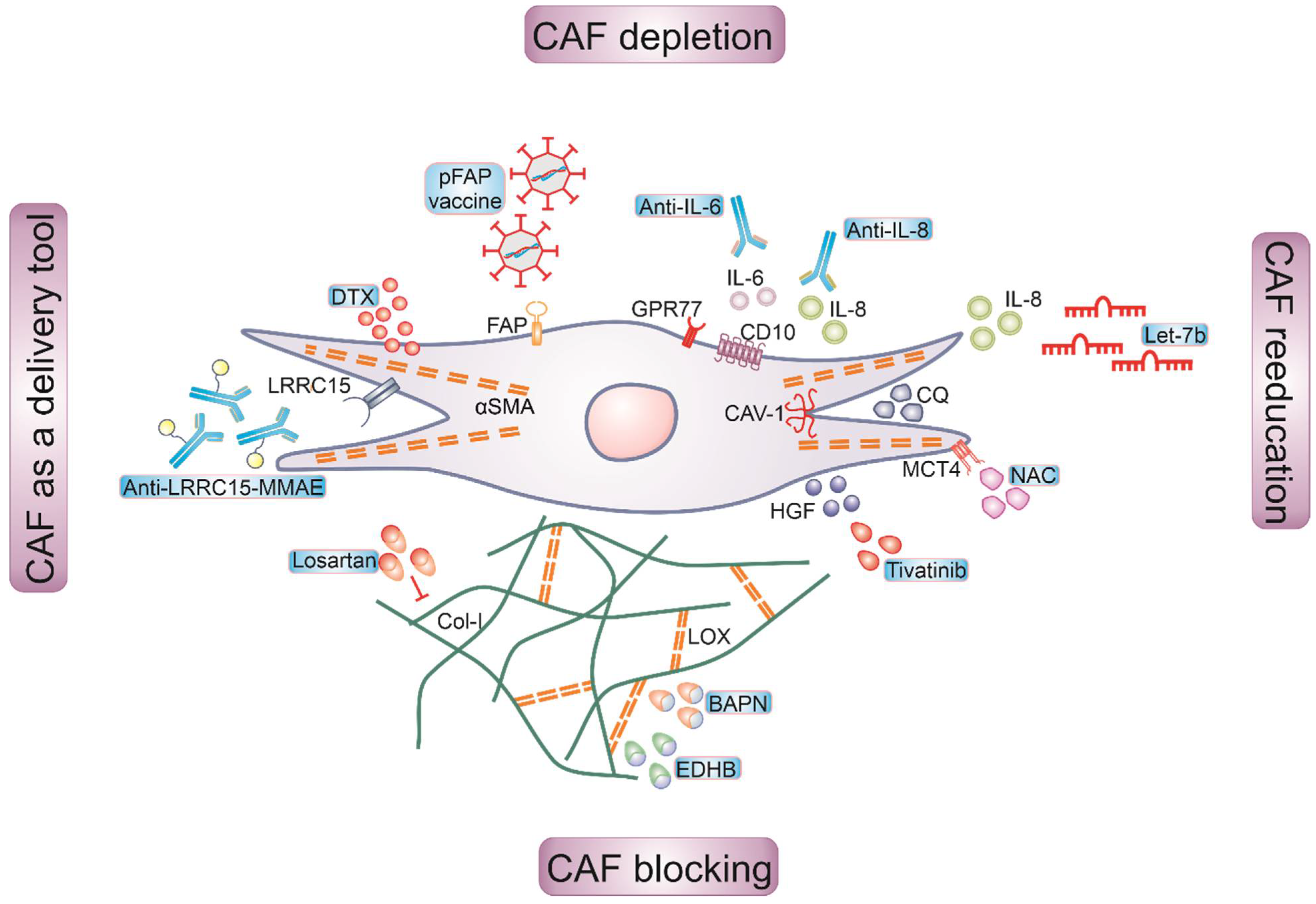
5. Targeting of CAFs for Cancer Therapy
5.1. Depleting CAFs
5.2. Reeducating CAFs
5.3. Blocking CAF Functions
5.4. CAFs as a Drug Delivery Tool
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Xu, J.; Lan, H. Tumor-Associated Macrophages in Tumor Metastasis: Biological Roles and Clinical Therapeutic Applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.-J. Panel members Personalizing the Treatment of Women with Early Breast Cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, Breast Cancer Subtypes, and Survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbeck, N.; Gnant, M. Breast Cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Basal-like Breast Cancer: A Critical Review. J. Clin. Oncol. 2008, 26, 2568–2581. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Morales-Vasquez, F.; Hortobagyi, G.N. Overview of Resistance to Systemic Therapy in Patients with Breast Cancer. Adv. Exp. Med. Biol. 2007, 608, 1–22. [Google Scholar] [CrossRef]
- Cardoso, F.; Costa, A.; Senkus, E.; Aapro, M.; André, F.; Barrios, C.H.; Bergh, J.; Bhattacharyya, G.; Biganzoli, L.; Cardoso, M.J.; et al. 3rd ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 3). Ann. Oncol. 2017, 28, 16–33. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting Neoantigens to Augment Antitumour Immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell 2020, 37, 443–455. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Ruocco, M.R.; Avagliano, A.; Granato, G.; Imparato, V.; Masone, S.; Masullo, M.; Nasso, R.; Montagnani, S.; Arcucci, A. Involvement of Breast Cancer-Associated Fibroblasts in Tumor Development, Therapy Resistance and Evaluation of Potential Therapeutic Strategies. Curr. Med. Chem. 2018, 25, 3414–3434. [Google Scholar] [CrossRef]
- Bu, L.; Baba, H.; Yasuda, T.; Uchihara, T.; Ishimoto, T. Functional Diversity of Cancer-associated Fibroblasts in Modulating Drug Resistance. Cancer Sci. 2020, 111, 3468–3477. [Google Scholar] [CrossRef]
- Sorrell, J.M.; Caplan, A.I. Fibroblasts—A Diverse Population at the Center of It All. Int. Rev. Cell Mol. Biol. 2009, 276, 161–214. [Google Scholar] [PubMed]
- Desmoulière, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming Growth Factor-Beta 1 Induces Alpha-Smooth Muscle Actin Expression in Granulation Tissue Myofibroblasts and in Quiescent and Growing Cultured Fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carthy, J.M. TGFβ Signaling and the Control of Myofibroblast Differentiation: Implications for Chronic Inflammatory Disorders. J. Cell. Physiol. 2018, 233, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.-I.; Lau, L.F. Resolution of Organ Fibrosis. J. Clin. Investing. 2018, 128, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J.; Boyd, N.F. Mammographic Density. Potential Mechanisms of Breast Cancer Risk Associated with Mammographic Density: Hypotheses Based on Epidemiological Evidence. Breast Cancer Res. 2008, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Brücher, B.L.; Lyman, G.; van Hillegersberg, R.; Pollock, R.E.; Lordick, F.; Yang, H.-K.; Ushijima, T.; Yeoh, K.-G.; Skricka, T.; Polkowski, W.; et al. Imagine a World without Cancer. BMC Cancer 2014, 14, 186. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, T.W.; Byrne, C.; Colditz, G.; Connolly, J.L.; Schnitt, S.J. Radial Scars in Benign Breast-Biopsy Specimens and the Risk of Breast Cancer. N. Eng. J. Med. 1999, 340, 430–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorak, H.F. Tumors: Wounds That Do Not Heal. N. Eng. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, G.S.; Poutahidis, T.; Erdman, S.E.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Cancer-Associated Fibroblasts Drive the Progression of Metastasis through Both Paracrine and Mechanical Pressure on Cancer Tissue. Mol. Cancer Res. 2012, 10, 1403–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, C.J.; Huang, Y.-H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massagué, J. TGF-β Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated Regulation of Myeloid Cells by Tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Malanchi, I. Neutrophils Support Lung Colonization of Metastasis-Initiating Breast Cancer Cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Pietras, K.; Ostman, A. Hallmarks of Cancer: Interactions with the Tumor Stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G. Spatially and Functionally Distinct Subclasses of Breast Cancer-Associated Fibroblasts Revealed by Single Cell RNA Sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct Populations of Inflammatory Fibroblasts and Myofibroblasts in Pancreatic Cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Madar, S.; Goldstein, I.; Rotter, V. ’Cancer Associated Fibroblasts’—More than Meets the Eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef]
- Elwakeel, E.; Brüne, B.; Weigert, A. PGE2 in Fibrosis and Cancer: Insights into Fibroblast Activation. Prostagl. Other Lipid Med. 2019, 143, 106339. [Google Scholar] [CrossRef] [PubMed]
- Elwakeel, E.; Brüggemann, M.; Fink, A.F.; Schulz, M.H.; Schmid, T.; Savai, R.; Brüne, B.; Zarnack, K.; Weigert, A. Weigert Phenotypic Plasticity of Fibroblasts during Mammary Carcinoma Development. Int. J. Mol. Sci. 2019, 20, 4438. [Google Scholar] [CrossRef] [Green Version]
- Cirri, P.; Chiarugi, P. Cancer-Associated-Fibroblasts and Tumour Cells: A Diabolic Liaison Driving Cancer Progression. Cancer Metast. Rev. 2012, 31, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Strell, C.; Paulsson, J.; Jin, S.-B.; Tobin, N.P.; Mezheyeuski, A.; Roswall, P.; Mutgan, C.; Mitsios, N.; Johansson, H.; Wickberg, S.M. Impact of Epithelial–Stromal Interactions on Peritumoral Fibroblasts in Ductal Carcinoma in Situ. J. Natl. Cancer Inst. 2019, 111, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-β and Stromal Cell-Derived Factor-1 (SDF-1) Signaling Drives the Evolution of Tumor-Promoting Mammary Stromal Myofibroblasts. PNAS 2010, 107, 20009–20014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharon, Y.; Raz, Y.; Cohen, N.; Ben-Shmuel, A.; Schwartz, H.; Geiger, T.; Erez, N. Tumor-Derived Osteopontin Reprograms Normal Mammary Fibroblasts to Promote Inflammation and Tumor Growth in Breast Cancer. Cancer Res. 2015, 75, 963–973. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.-S.; Shaked, Y.; Tsai, K.K. Targeting the Interplay Between Cancer Fibroblasts, Mesenchymal Stem Cells, and Cancer Stem Cells in Desmoplastic Cancers. Front. Oncol. 2019, 9, 688. [Google Scholar] [CrossRef] [Green Version]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone Marrow-Derived Fibroblasts Are a Functionally Distinct Stromal Cell Population in Breast Cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, P.J.; Ebrahimsade, S.; Ramaswamy, A.; Moll, R. CD34+ Fibrocytes in Invasive Ductal Carcinoma, Ductal Carcinoma in Situ, and Benign Breast Lesions. Virchows Arch. 2002, 440, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Anderberg, C.; Pietras, K. On the Origin of Cancer-Associated Fibroblasts. Cell Cycle 2009, 8, 1461–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- William Petersen, O.; Lind Nielsen, H.; Gudjonsson, T.; Villadsen, R.; Rønnov-Jessen, L.; Bissell, M.J. The Plasticity of Human Breast Carcinoma Cells Is More than Epithelial to Mesenchymal Conversion. Breast Cancer Res. 2001, 3, 213–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochet, L.; Lehuédé, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S.; et al. Adipocyte-Derived Fibroblasts Promote Tumor Progression and Contribute to the Desmoplastic Reaction in Breast Cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar] [CrossRef] [Green Version]
- Jotzu, C.; Alt, E.; Welte, G.; Li, J.; Hennessy, B.T.; Devarajan, E.; Krishnappa, S.; Pinilla, S.; Droll, L.; Song, Y.-H. Adipose Tissue Derived Stem Cells Differentiate into Carcinoma-Associated Fibroblast-like Cells under the Influence of Tumor Derived Factors. Cell Oncol. 2011, 34, 55–67. [Google Scholar] [CrossRef]
- Peiris-Pagès, M.; Sotgia, F.; Lisanti, M.P. Chemotherapy Induces the Cancer-Associated Fibroblast Phenotype, Activating Paracrine Hedgehog-GLI Signalling in Breast Cancer Cells. Oncotarget 2015, 6, 10728–10745. [Google Scholar] [CrossRef] [Green Version]
- Sappino, A.P.; Skalli, O.; Jackson, B.; Schürch, W.; Gabbiani, G. Smooth-Muscle Differentiation in Stromal Cells of Malignant and Non-Malignant Breast Tissues. Int. J. Cancer 1988, 41, 707–712. [Google Scholar] [CrossRef]
- Bernard, V.; Semaan, A.; Huang, J.; San Lucas, F.A.; Mulu, F.C.; Stephens, B.M.; Guerrero, P.A.; Huang, Y.; Zhao, J.; Kamyabi, N.; et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin. Cancer Res. 2019, 25, 2194–2205. [Google Scholar] [CrossRef] [Green Version]
- Cremasco, V.; Astarita, J.L.; Grauel, A.L.; Keerthivasan, S.; MacIsaac, K.; Woodruff, M.C.; Wu, M.; Spel, L.; Santoro, S.; Amoozgar, Z.; et al. FAP Delineates Heterogeneous and Functionally Divergent Stromal Cells in Immune-Excluded Breast Tumors. Cancer Immunol. Res. 2018, 6, 1472–1485. [Google Scholar] [CrossRef] [Green Version]
- Maris, P.; Blomme, A.; Palacios, A.P.; Costanza, B.; Bellahcène, A.; Bianchi, E.; Gofflot, S.; Drion, P.; Trombino, G.E.; Valentin, E.D.; et al. Asporin Is a Fibroblast-Derived TGF-Β1 Inhibitor and a Tumor Suppressor Associated with Good Prognosis in Breast Cancer. PLOS Med. 2015, 12, e1001871. [Google Scholar] [CrossRef]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.M.; Gillen, A.E.; Cittelly, D.M.; Tan, A.-C.; Sams, S.B.; Pillai, M.M.; Elias, A.D.; Robinson, W.A.; et al. Fibroblast Subtypes Regulate Responsiveness of Luminal Breast Cancer to Estrogen. Clin. Cancer Res. 2017, 23, 1710–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of Fibroblast Heterogeneity in the Tumor Microenvironment. Cancer Biol. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [Green Version]
- Friedman, G.; Levi-Galibov, O.; David, E.; Bornstein, C.; Giladi, A.; Dadiani, M.; Mayo, A.; Halperin, C.; Pevsner-Fischer, M.; Lavon, H.; et al. Cancer-Associated Fibroblast Compositions Change with Breast Cancer Progression Linking the Ratio of S100A4 + and PDPN + CAFs to Clinical Outcome. Nat. Cancer 2020, 1, 692–708. [Google Scholar] [CrossRef]
- Kauppila, S.; Stenbäck, F.; Risteli, J.; Jukkola, A.; Risteli, L. Aberrant Type I and Type III Collagen Gene Expression in Human Breast Cancer in Vivo. J. Pathol. 1998, 186, 262–268. [Google Scholar] [CrossRef]
- Eble, J.A.; Niland, S. The Extracellular Matrix in Tumor Progression and Metastasis. Clin. Exp. Metast. 2019, 36, 171–198. [Google Scholar] [CrossRef]
- Kuchnio, A.; Moens, S.; Bruning, U.; Kuchnio, K.; Cruys, B.; Thienpont, B.; Broux, M.; Ungureanu, A.A.; Leite de Oliveira, R.; Bruyère, F.; et al. The Cancer Cell Oxygen Sensor PHD2 Promotes Metastasis via Activation of Cancer-Associated Fibroblasts. Cell Rep. 2015, 12, 992–1005. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Chen, S.; Yuan, W.; Fan, Q.; Tian, J.; Wang, X.; Chen, L.; Zhang, X.; Wei, W.; Liu, R.; et al. Oriented Collagen Fibers Direct Tumor Cell Intravasation. PNAS 2016, 113, 11208–11213. [Google Scholar] [CrossRef] [Green Version]
- Pickup, M.W.; Laklai, H.; Acerbi, I.; Owens, P.; Gorska, A.E.; Chytil, A.; Aakre, M.; Weaver, V.M.; Moses, H.L. Stromally Derived Lysyl Oxidase Promotes Metastasis of Transforming Growth Factor-β-Deficient Mouse Mammary Carcinomas. Cancer Res. 2013, 73, 5336–5346. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Santhanam, A.N.; Baker, A.R.; Hegamyer, G.; Kirschmann, D.A.; Colburn, N.H. Pdcd4 Repression of Lysyl Oxidase Inhibits Hypoxia-Induced Breast Cancer Cell Invasion. Oncogene 2010, 29, 3921–3932. [Google Scholar] [CrossRef] [Green Version]
- Wolf, K.; Te Lindert, M.; Krause, M.; Alexander, S.; Te Riet, J.; Willis, A.L.; Hoffman, R.M.; Figdor, C.G.; Weiss, S.J.; Friedl, P. Physical Limits of Cell Migration: Control by ECM Space and Nuclear Deformation and Tuning by Proteolysis and Traction Force. J. Cell Biol. 2013, 201, 1069–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human Breast Cancer Invasion and Aggression Correlates with ECM Stiffening and Immune Cell Infiltration. Integr. Biol. 2015, 7, 1120–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kai, F.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef]
- Oudin, M.J.; Jonas, O.; Kosciuk, T.; Broye, L.C.; Guido, B.C.; Wyckoff, J.; Riquelme, D.; Lamar, J.M.; Asokan, S.B.; Whittaker, C.; et al. Tumor Cell–Driven Extracellular Matrix Remodeling Drives Haptotaxis during Metastatic Progression. Cancer Discov. 2016, 6, 516–531. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in Mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-Dependent Matrix Remodelling Is Required for the Generation and Maintenance of Cancer-Associated Fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Miao, L.; Liu, Q.; Lin, C.M.; Luo, C.; Wang, Y.; Liu, L.; Yin, W.; Hu, S.; Kim, W.Y.; Huang, L. Targeting Tumor-Associated Fibroblasts for Therapeutic Delivery in Desmoplastic Tumors. Cancer Res. 2017, 77, 719–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Bruni, D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity 2020, 52, 55–81. [Google Scholar] [CrossRef]
- Olkhanud, P.B.; Baatar, D.; Bodogai, M.; Hakim, F.; Gress, R.; Anderson, R.L.; Deng, J.; Xu, M.; Briest, S.; Biragyn, A. Breast Cancer Lung Metastasis Requires Expression of Chemokine Receptor CCR4 and Regulatory T Cells. Cancer Res. 2009, 69, 5996–6004. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Shaffer, A.L.; Emre, N.C.T.; Ceribelli, M.; Zhang, M.; Wright, G.; Xiao, W.; Powell, J.; Platig, J.; Kohlhammer, H.; et al. Exploiting Synthetic Lethality for the Therapy of ABC Diffuse Large B Cell Lymphoma. Cancer Cell 2012, 21, 723–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.C.B.; Goedegebuure, P.S.; Belt, B.A.; Flaherty, B.; Sankpal, N.; Gillanders, W.E.; Eberlein, T.J.; Hsieh, C.-S.; Linehan, D.C. Disruption of CCR5-Dependent Homing of Regulatory T Cells Inhibits Tumor Growth in a Murine Model of Pancreatic Cancer. J. Immunol. 2009, 182, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.; Shani, O.; Raz, Y.; Sharon, Y.; Hoffman, D.; Abramovitz, L.; Erez, N. Fibroblasts Drive an Immunosuppressive and Growth-Promoting Microenvironment in Breast Cancer via Secretion of Chitinase 3-like 1. Oncogene 2017, 36, 4457–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augsten, M.; Sjoberg, E.; Frings, O.; Vorrink, S.U.; Frijhoff, J.; Olsson, E.; Borg, A.; Ostman, A. Cancer-Associated Fibroblasts Expressing CXCL14 Rely upon Nos1-Derived Nitric Oxide Signaling for Their Tumor Supporting Properties. Cancer Res. 2014, 74, 2999–3010. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Deng, J.; Rychahou, P.G.; Qiu, S.; Evers, B.M.; Zhou, B.P. Stabilization of Snail by NF-KappaB Is Required for Inflammation-Induced Cell Migration and Invasion. Cancer Cell 2009, 15, 416–428. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Kugeratski, F.G.; Atkinson, S.J.; Neilson, L.J.; Lilla, S.; Knight, J.R.P.; Serneels, J.; Juin, A.; Ismail, S.; Bryant, D.M.; Markert, E.K.; et al. Hypoxic Cancer-Associated Fibroblasts Increase NCBP2-AS2/HIAR to Promote Endothelial Sprouting through Enhanced VEGF Signaling. Sci Signal. 2019, 12, eaan8247. [Google Scholar] [CrossRef] [Green Version]
- Koyama, H.; Kobayashi, N.; Harada, M.; Takeoka, M.; Kawai, Y.; Sano, K.; Fujimori, M.; Amano, J.; Ohhashi, T.; Kannagi, R.; et al. Significance of Tumor-Associated Stroma in Promotion of Intratumoral Lymphangiogenesis. Am. J. Pathol. 2008, 172, 179–193. [Google Scholar] [CrossRef] [Green Version]
- Liao, D.; Luo, Y.; Markowitz, D.; Xiang, R.; Reisfeld, R.A. Cancer Associated Fibroblasts Promote Tumor Growth and Metastasis by Modulating the Tumor Immune Microenvironment in a 4T1 Murine Breast Cancer Model. PLoS ONE 2009, 4, e7965. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The Reverse Warburg Effect: Aerobic Glycolysis in Cancer Associated Fibroblasts and the Tumor Stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor Cells Induce the Cancer Associated Fibroblast Phenotype via Caveolin-1 Degradation: Implications for Breast Cancer and DCIS Therapy with Autophagy Inhibitors. Cell Cycle 2010, 9, 2423–2433. [Google Scholar] [CrossRef] [Green Version]
- Pavlides, S.; Tsirigos, A.; Vera, I.; Flomenberg, N.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; Pestell, R.G.; et al. Loss of Stromal Caveolin-1 Leads to Oxidative Stress, Mimics Hypoxia and Drives Inflammation in the Tumor Microenvironment, Conferring the “Reverse Warburg Effect”: A Transcriptional Informatics Analysis with Validation. Cell Cycle 2010, 9, 2201–2219. [Google Scholar] [CrossRef] [Green Version]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Andò, S.; Martinez-Outschoorn, U.; et al. Metabolic Reprogramming of Cancer-Associated Fibroblasts by TGF-β Drives Tumor Growth: Connecting TGF-β Signaling with “Warburg-like” Ca.a.a.ancer Metabolism and L-Lactate Production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic Reprogramming of Cancer-Associated Fibroblasts by IDH3α Downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Yang, G.; Hou, Y.; Tang, X.; Wu, C.; Wu, X.-A.; Guo, L.; Zhu, Q.; Luo, H.; Du, Y.-E.; et al. Cytoplasmic GPER Translocation in Cancer-Associated Fibroblasts Mediates CAMP/PKA/CREB/Glycolytic Axis to Confer Tumor Cells with Multidrug Resistance. Oncogene 2017, 36, 2131–2145. [Google Scholar] [CrossRef]
- Demircioglu, F.; Wang, J.; Candido, J.; Costa, A.S.H.; Casado, P.; de Luxan Delgado, B.; Reynolds, L.E.; Gomez-Escudero, J.; Newport, E.; Rajeeve, V.; et al. Cancer Associated Fibroblast FAK Regulates Malignant Cell Metabolism. Nat. Commun. 2020, 11, 1290. [Google Scholar] [CrossRef] [Green Version]
- Codogno, P.; Meijer, A.J. Autophagy and Signaling: Their Role in Cell Survival and Cell Death. Cell Death Differ. 2005, 12 (Suppl. 2), 1509–1518. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy Is an HIF-1-Dependent Adaptive Metabolic Response to Hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.-L.; Mo, E.-P.; Yang, L.; Du, J.; Wang, H.-S.; Zhang, H.; Kurihara, H.; Xu, J.; Cai, S.-H. Autophagy Is Involved in TGF-Β1-Induced Protective Mechanisms and Formation of Cancer-Associated Fibroblasts Phenotype in Tumor Microenvironment. Oncotarget 2016, 7, 4122–4141. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, J.; Huang, Y.; Ji, S.; Shao, G.; Feng, S.; Chen, D.; Zhao, K.; Wang, Z.; Wu, A. Cancer-Associated Fibroblasts Autophagy Enhances Progression of Triple-Negative Breast Cancer Cells. Med. Sci. Monit. 2017, 23, 3904–3912. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Chen, X.; Wang, X.; Zhao, Z.; Hu, W.; Zeng, S.; Wei, J.; Yang, X.; Qian, L.; Zhou, S.; et al. The Effects and the Mechanisms of Autophagy on the Cancer-Associated Fibroblasts in Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 171. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. Let-7 Regulates Self Renewal and Tumorigenicity of Breast Cancer Cells. Cell 2007, 131, 1109–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenos, K.J.; Miedema, D.M.; Lodestijn, S.C.; Nijman, L.E.; van den Bosch, T.; Romero Ros, X.; Lourenço, F.C.; Lecca, M.C.; van der Heijden, M.; van Neerven, S.M.; et al. Stem Cell Functionality Is Microenvironmentally Defined during Tumour Expansion and Therapy Response in Colon Cancer. Nat. Cell Biol. 2018, 20, 1193–1202. [Google Scholar] [CrossRef]
- Du, Y.; Shao, H.; Moller, M.; Prokupets, R.; Tse, Y.T.; Liu, Z.-J. Intracellular Notch1 Signaling in Cancer-Associated Fibroblasts Dictates the Plasticity and Stemness of Melanoma Stem/Initiating Cells. Stem Cells 2019, 37, 865–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, P.-J.; Rama, N.; Imbach, J.; Fiore, S.; Ducarouge, B.; Neves, D.; Chen, H.-W.; Bernard, D.; Yang, P.-C.; Bernet, A.; et al. Cancer-Associated Fibroblasts Produce Netrin-1 to Control Cancer Cell Plasticity. Cancer Res. 2019, 79, 3651–3661. [Google Scholar] [CrossRef]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.L.; Wu, S.Z.; Roden, D.; Chan, C.-L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting Stromal Remodeling and Cancer Stem Cell Plasticity Overcomes Chemoresistance in Triple Negative Breast Cancer. Nat. Commun. 2018, 9, 2897. [Google Scholar] [CrossRef] [Green Version]
- Malanchi, I.; Santamaria-Martínez, A.; Susanto, E.; Peng, H.; Lehr, H.-A.; Delaloye, J.-F.; Huelsken, J. Interactions between Cancer Stem Cells and Their Niche Govern Metastatic Colonization. Nature 2011, 481, 85–89. [Google Scholar] [CrossRef]
- Pontiggia, O.; Sampayo, R.; Raffo, D.; Motter, A.; Xu, R.; Bissell, M.J.; de Joffé, E.B.K.; Simian, M. The Tumor Microenvironment Modulates Tamoxifen Resistance in Breast Cancer: A Role for Soluble Stromal Factors and Fibronectin through Β1 Integrin. Breast Cancer Res. Treat. 2012, 133, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Shekhar, M.P.V.; Santner, S.; Carolin, K.A.; Tait, L. Direct Involvement of Breast Tumor Fibroblasts in the Modulation of Tamoxifen Sensitivity. Am. J. Pathol. 2007, 170, 1546–1560. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Zhao, J.; Xing, Y.; Zhang, X.; Liu, J.; Ouyang, Q.; Chen, J.; Su, F.; Liu, Q.; Song, E. Immune Checkpoint Inhibition Overcomes ADCP-Induced Immunosuppression by Macrophages. Cell 2018, 175, 442–457.e23. [Google Scholar] [CrossRef] [Green Version]
- Boesch, M.; Onder, L.; Cheng, H.-W.; Novkovic, M.; Mörbe, U.; Sopper, S.; Gastl, G.; Jochum, W.; Ruhstaller, T.; Knauer, M.; et al. Interleukin 7-Expressing Fibroblasts Promote Breast Cancer Growth through Sustenance of Tumor Cell Stemness. Oncoimmunology 2018, 7, e1414129. [Google Scholar] [CrossRef] [Green Version]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W.; et al. CCL2 Mediates Cross-Talk between Cancer Cells and Stromal Fibroblasts That Regulates Breast Cancer Stem Cells. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef] [Green Version]
- Amornsupak, K.; Insawang, T.; Thuwajit, P.; O-Charoenrat, P.; Eccles, S.A.; Thuwajit, C. Cancer-Associated Fibroblasts Induce High Mobility Group Box 1 and Contribute to Resistance to Doxorubicin in Breast Cancer Cells. BMC Cancer 2014, 14, 955. [Google Scholar] [CrossRef]
- Zhao, X.-L.; Lin, Y.; Jiang, J.; Tang, Z.; Yang, S.; Lu, L.; Liang, Y.; Liu, X.; Tan, J.; Hu, X.-G.; et al. High-Mobility Group Box 1 Released by Autophagic Cancer-Associated Fibroblasts Maintains the Stemness of Luminal Breast Cancer Cells. J. Pathol. 2017, 243, 376–389. [Google Scholar] [CrossRef]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual Breast Cancers after Conventional Therapy Display Mesenchymal as Well as Tumor-Initiating Features. PNAS 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.-F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic Resistance of Tumorigenic Breast Cancer Cells to Chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef]
- Chan, T.-S.; Hsu, C.-C.; Pai, V.C.; Liao, W.-Y.; Huang, S.-S.; Tan, K.-T.; Yen, C.-J.; Hsu, S.-C.; Chen, W.-Y.; Shan, Y.-S.; et al. Metronomic Chemotherapy Prevents Therapy-Induced Stromal Activation and Induction of Tumor-Initiating Cells. J. Exp. Med. 2016, 213, 2967–2988. [Google Scholar] [CrossRef]
- Luo, H.; Liu, M.; Luo, S.; Yu, T.; Wu, C.; Yang, G.; Tu, G. Dynamic Monitoring of GPER-Mediated Estrogenic Effects in Breast Cancer Associated Fibroblasts: An Alternative Role of Estrogen in Mammary Carcinoma Development. Steroids 2016, 112, 1–11. [Google Scholar] [CrossRef]
- Ribeiro, M.P.C.; Santos, A.E.; Custódio, J.B.A. The Activation of the G Protein-Coupled Estrogen Receptor (GPER) Inhibits the Proliferation of Mouse Melanoma K1735-M2 Cells. Chem. Biol. Interact. 2017, 277, 176–184. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Goldberg, A.F.; Lin, Z.; Ko, Y.-H.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; et al. Anti-Estrogen Resistance in Breast Cancer Is Induced by the Tumor Microenvironment and Can Be Overcome by Inhibiting Mitochondrial Function in Epithelial Cancer Cells. Cancer Biol. Ther. 2011, 12, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.A.; Pollard, J.W. Microenvironmental Regulation of Metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Science, A.A. for the A. of Erratum for the Report “Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition” by M. Yu, A. Bardia, B. S. Wittner, S. L. Stott, M. E. Smas, D. T. Ting, S. J. Isakoff, J. C. Ciciliano, M. N. Wells, A. M. Shah, K. F. Concannon, M. C. Donaldson, L. V. Sequist, E. Brachtel, D. Sgroi, J. Baselga, S. Ramaswamy, M. Toner, D. A. Haber, S. Maheswaran. Science 2019, 363. [Google Scholar] [CrossRef] [Green Version]
- Micalizzi, D.S.; Maheswaran, S.; Haber, D.A. A Conduit to Metastasis: Circulating Tumor Cell Biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-Metastatic Niches: Organ-Specific Homes for Metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Goddard, E.T.; Bozic, I.; Riddell, S.R.; Ghajar, C.M. Dormant Tumour Cells, Their Niches and the Influence of Immunity. Nat. Cell Biol. 2018, 20, 1240–1249. [Google Scholar] [CrossRef]
- Yu, Y.; Xiao, C.-H.; Tan, L.-D.; Wang, Q.-S.; Li, X.-Q.; Feng, Y.-M. Cancer-Associated Fibroblasts Induce Epithelial-Mesenchymal Transition of Breast Cancer Cells through Paracrine TGF-β Signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [Green Version]
- Al-Ansari, M.M.; Hendrayani, S.F.; Shehata, A.I.; Aboussekhra, A. Erratum: P16 INK4A Represses the Paracrine Tumor-Promoting Effects of Breast Stromal Fibroblasts. Oncogene 2013, 32, 2356–2364. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xu, K.; Chase, M.; Ji, Y.; Logan, J.K.; Buchsbaum, R.J. Tiam1-Regulated Osteopontin in Senescent Fibroblasts Contributes to the Migration and Invasion of Associated Epithelial Cells. J. Cell Sci. 2012, 125, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Nguyen-Ngoc, K.-V.; Cheung, K.J.; Brenot, A.; Shamir, E.R.; Gray, R.S.; Hines, W.C.; Yaswen, P.; Werb, Z.; Ewald, A.J. ECM Microenvironment Regulates Collective Migration and Local Dissemination in Normal and Malignant Mammary Epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, E2595–E2604. [Google Scholar] [CrossRef] [Green Version]
- Buchsbaum, R.J.; Oh, S.Y. Breast Cancer-Associated Fibroblasts: Where We Are and Where We Need to Go. Cancers 2016, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Duda, D.G.; Duyverman, A.M.M.J.; Kohno, M.; Snuderl, M.; Steller, E.J.A.; Fukumura, D.; Jain, R.K. Malignant Cells Facilitate Lung Metastasis by Bringing Their Own Soil. PNAS 2010, 107, 21677–21682. [Google Scholar] [CrossRef] [Green Version]
- Labernadie, A.; Kato, T.; Brugués, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; González-Tarragó, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A Mechanically Active Heterotypic E-Cadherin/N-Cadherin Adhesion Enables Fibroblasts to Drive Cancer Cell Invasion. Nat. Cell Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef]
- Gao, M.-Q.; Kim, B.G.; Kang, S.; Choi, Y.P.; Park, H.; Kang, K.S.; Cho, N.H. Stromal Fibroblasts from the Interface Zone of Human Breast Carcinomas Induce an Epithelial-Mesenchymal Transition-like State in Breast Cancer Cells in Vitro. J. Cell Sci. 2010, 123, 3507–3514. [Google Scholar] [CrossRef] [Green Version]
- Rummel, S.; Valente, A.L.; Kane, J.L.; Shriver, C.D.; Ellsworth, R.E. Genomic (In)Stability of the Breast Tumor Microenvironment. Mol. Cancer Res. 2012, 10, 1526–1531. [Google Scholar] [CrossRef] [Green Version]
- Togo, S.; Polanska, U.M.; Horimoto, Y.; Orimo, A. Carcinoma-Associated Fibroblasts Are a Promising Therapeutic Target. Cancers 2013, 5, 149–169. [Google Scholar] [CrossRef] [Green Version]
- Gascard, P.; Tlsty, T.D. Carcinoma-Associated Fibroblasts: Orchestrating the Composition of Malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Hosein, A.N.; Wu, M.; Arcand, S.L.; Lavallée, S.; Hébert, J.; Tonin, P.N.; Basik, M. Breast Carcinoma–Associated Fibroblasts Rarely Contain P53 Mutations or Chromosomal Aberrations. Cancer Res. 2010, 70, 5770–5777. [Google Scholar] [CrossRef] [Green Version]
- Allinen, M.; Beroukhim, R.; Cai, L.; Brennan, C.; Lahti-Domenici, J.; Huang, H.; Porter, D.; Hu, M.; Chin, L.; Richardson, A.; et al. Molecular Characterization of the Tumor Microenvironment in Breast Cancer. Cancer Cell 2004, 6, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Pelham, R.J.; Rodgers, L.; Hall, I.; Lucito, R.; Nguyen, K.C.Q.; Navin, N.; Hicks, J.; Mu, D.; Powers, S.; Wigler, M.; et al. Identification of Alterations in DNA Copy Number in Host Stromal Cells during Tumor Progression. PNAS 2006, 103, 19848–19853. [Google Scholar] [CrossRef] [Green Version]
- Moinfar, F.; Man, Y.G.; Arnould, L.; Bratthauer, G.L.; Ratschek, M.; Tavassoli, F.A. Concurrent and Independent Genetic Alterations in the Stromal and Epithelial Cells of Mammary Carcinoma: Implications for Tumorigenesis. Cancer Res. 2000, 60, 2562–2566. [Google Scholar]
- Hawsawi, N.M.; Ghebeh, H.; Hendrayani, S.-F.; Tulbah, A.; Al-Eid, M.; Al-Tweigeri, T.; Ajarim, D.; Alaiya, A.; Dermime, S.; Aboussekhra, A. Breast Carcinoma-Associated Fibroblasts and Their Counterparts Display Neoplastic-Specific Changes. Cancer Res. 2008, 68, 2717–2725. [Google Scholar] [CrossRef] [Green Version]
- Patocs, A.; Zhang, L.; Xu, Y.; Weber, F.; Caldes, T.; Mutter, G.L.; Platzer, P.; Eng, C. Breast-Cancer Stromal Cells with TP53 Mutations and Nodal Metastases. N. Engl. J. Med. 2007, 357, 2543–2551. [Google Scholar] [CrossRef]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic Switch Drives the Conversion of Fibroblasts into Proinvasive Cancer-Associated Fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef] [Green Version]
- Fiegl, H.; Millinger, S.; Goebel, G.; Müller-Holzner, E.; Marth, C.; Laird, P.W.; Widschwendter, M. Breast Cancer DNA Methylation Profiles in Cancer Cells and Tumor Stroma: Association with HER-2/Neu Status in Primary Breast Cancer. Cancer Res. 2006, 66, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Yao, J.; Cai, L.; Bachman, K.E.; van den Brûle, F.; Velculescu, V.; Polyak, K. Distinct Epigenetic Changes in the Stromal Cells of Breast Cancers. Nat. Genet. 2005, 37, 899–905. [Google Scholar] [CrossRef]
- Tomasetti, M.; Gaetani, S.; Monaco, F.; Neuzil, J.; Santarelli, L. Epigenetic Regulation of MiRNA Expression in Malignant Mesothelioma: MiRNAs as Biomarkers of Early Diagnosis and Therapy. Front. Oncol. 2019, 9, 1293. [Google Scholar] [CrossRef]
- Silva, M.; Melo, S.A. Non-Coding RNAs in Exosomes: New Players in Cancer Biology. Curr. Genom. 2015, 16, 295–303. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.H.; Miller, P.; Garcia-Contreras, M.; Ao, Z.; Machlin, L.; Issa, E.; El-Ashry, D. Hierarchical Paracrine Interaction of Breast Cancer Associated Fibroblasts with Cancer Cells via HMAPK-MicroRNAs to Drive ER-Negative Breast Cancer Phenotype. Cancer Biol. 2015, 16, 1671–1681. [Google Scholar] [CrossRef] [Green Version]
- Verghese, E.T.; Drury, R.; Green, C.A.; Holliday, D.L.; Lu, X.; Nash, C.; Speirs, V.; Thorne, J.L.; Thygesen, H.H.; Zougman, A.; et al. MiR-26b Is down-Regulated in Carcinoma-Associated Fibroblasts from ER-Positive Breast Cancers Leading to Enhanced Cell Migration and Invasion. J. Pathol. 2013, 231, 388–399. [Google Scholar] [CrossRef]
- Humphries, B.; Yang, C. The MicroRNA-200 Family: Small Molecules with Novel Roles in Cancer Development, Progression and Therapy. Oncotarget 2015, 6, 6472–6498. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Tu, G.; Yang, G.; Li, G.; Yang, D.; Lang, L.; Xi, L.; Sun, K.; Chen, Y.; Shu, K.; et al. MiR-205/YAP1 in Activated Fibroblasts of Breast Tumor Promotes VEGF-Independent Angiogenesis through STAT3 Signaling. Theranostics 2017, 7, 3972–3988. [Google Scholar] [CrossRef]
- Bronisz, A.; Godlewski, J.; Wallace, J.A.; Merchant, A.S.; Nowicki, M.O.; Mathsyaraja, H.; Srinivasan, R.; Trimboli, A.J.; Martin, C.K.; Li, F.; et al. Reprogramming of the Tumour Microenvironment by Stromal PTEN-Regulated MiR-320. Nat. Cell Biol. 2012, 14, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Baroni, S.; Romero-Cordoba, S.; Plantamura, I.; Dugo, M.; D’Ippolito, E.; Cataldo, A.; Cosentino, G.; Angeloni, V.; Rossini, A.; Daidone, M.G.; et al. Exosome-Mediated Delivery of MiR-9 Induces Cancer-Associated Fibroblast-like Properties in Human Breast Fibroblasts. Cell Death Dis. 2016, 7, e2312. [Google Scholar] [CrossRef]
- Finak, G.; Bertos, N.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal Gene Expression Predicts Clinical Outcome in Breast Cancer. Nat. Med. 2008, 14, 518–527. [Google Scholar] [CrossRef]
- Conklin, M.W.; Eickhoff, J.C.; Riching, K.M.; Pehlke, C.A.; Eliceiri, K.W.; Provenzano, P.P.; Friedl, A.; Keely, P.J. Aligned Collagen Is a Prognostic Signature for Survival in Human Breast Carcinoma. Am. J. Pathol. 2011, 178, 1221–1232. [Google Scholar] [CrossRef]
- Maller, O.; Drain, A.P.; Barrett, A.S.; Borgquist, S.; Ruffell, B.; Zakharevich, I.; Pham, T.T.; Gruosso, T.; Kuasne, H.; Lakins, J.N.; et al. Tumour-Associated Macrophages Drive Stromal Cell-Dependent Collagen Crosslinking and Stiffening to Promote Breast Cancer Aggression. Nat. Mater. 2021, 20, 548–559. [Google Scholar] [CrossRef]
- Gjaltema, R.A.F.; de Rond, S.; Rots, M.G.; Bank, R.A. Procollagen Lysyl Hydroxylase 2 Expression Is Regulated by an Alternative Downstream Transforming Growth Factor β-1 Activation Mechanism. J. Biol. Chem. 2015, 290, 28465–28476. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.-H.; Hwang-Verslues, W.W.; Chang, Y.-C.; Chen, C.-C.; Hsiao, M.; Jeng, Y.-M.; Chang, K.-J.; Lee, E.Y.-H.P.; Shew, J.-Y.; Lee, W.-H. Activation of Robo1 Signaling of Breast Cancer Cells by Slit2 from Stromal Fibroblast Restrains Tumorigenesis via Blocking PI3K/Akt/β-Catenin Pathway. Cancer Res. 2012, 72, 4652–4661. [Google Scholar] [CrossRef] [Green Version]
- Marlow, R.; Strickland, P.; Lee, J.S.; Wu, X.; PeBenito, M.; Binnewies, M.; Le, E.K.; Moran, A.; Macias, H.; Cardiff, R.D.; et al. SLITs Suppress Tumor Growth In Vivo by Silencing Sdf1/Cxcr4 within Breast Epithelium. Cancer Res. 2008, 68, 7819–7827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, P.; Bonnefoi, H.; Anderle, P.; Cameron, D.; Wirapati, P.; Wirapati, P.; Becette, V.; André, S.; Piccart, M.; Campone, M.; et al. A Stroma-Related Gene Signature Predicts Resistance to Neoadjuvant Chemotherapy in Breast Cancer. Nat. Med. 2009, 15, 68–74. [Google Scholar] [CrossRef]
- Gieniec, K.A.; Butler, L.M.; Worthley, D.L.; Woods, S.L. Cancer-Associated Fibroblasts—Heroes or Villains? Br. J. Cancer 2019, 121, 293–302. [Google Scholar] [CrossRef]
- Ao, Z.; Shah, S.H.; Machlin, L.M.; Parajuli, R.; Miller, P.C.; Rawal, S.; Williams, A.J.; Cote, R.J.; Lippman, M.E.; Datar, R.H.; et al. Identification of Cancer-Associated Fibroblasts in Circulating Blood from Patients with Metastatic Breast Cancer. Cancer Res. 2015, 75, 4681–4687. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Ernsting, M.J.; Undzys, E.; Holwell, N.; Foltz, W.D.; Li, S.-D. Docetaxel Conjugate Nanoparticles That Target α-Smooth Muscle Actin–Expressing Stromal Cells Suppress Breast Cancer Metastasis. Cancer Res. 2013, 73, 4862–4871. [Google Scholar] [CrossRef] [Green Version]
- Pant, S.; Saleh, M.; Bendell, J.; Infante, J.R.; Jones, S.; Kurkjian, C.D.; Moore, K.M.; Kazakin, J.; Abbadessa, G.; Wang, Y.; et al. A Phase I Dose Escalation Study of Oral C-MET Inhibitor Tivantinib (ARQ 197) in Combination with Gemcitabine in Patients with Solid Tumors. Ann. Oncol. 2014, 25, 1416–1421. [Google Scholar] [CrossRef]
- Espina, V.A.; Liotta, L.; Rassulova, S.; Gallimore, H.; Grant-Wisdom, T.; Menezes, G.; Nayer, H.; Edmiston, K. Abstract CT140: PINC Trial: Preventing Invasive Breast Neoplasia with Chloroquine. Cancer Res. 2017, 77, CT140. [Google Scholar]
- Monti, D.; Sotgia, F.; Whitaker-Menezes, D.; Tuluc, M.; Birbe, R.; Berger, A.; Lazar, M.; Cotzia, P.; Draganova-Tacheva, R.; Lin, Z.; et al. Pilot Study Demonstrating Metabolic and Anti-Proliferative Effects of in Vivo Anti-Oxidant Supplementation with N-Acetylcysteine in Breast Cancer. Semin. Oncol. 2017, 44, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.; von Pawel, J.; Novello, S.; Ramlau, R.; Favaretto, A.; Barlesi, F.; Akerley, W.; Orlov, S.; Santoro, A.; Spigel, D.; et al. Phase III Multinational, Randomized, Double-Blind, Placebo-Controlled Study of Tivantinib (ARQ 197) Plus Erlotinib Versus Erlotinib Alone in Previously Treated Patients With Locally Advanced or Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2015, 33, 2667–2674. [Google Scholar] [CrossRef]
- Gerber, D.E.; Socinski, M.A.; Neal, J.W.; Wakelee, H.A.; Shirai, K.; Sequist, L.V.; Rosovsky, R.P.; Lilenbaum, R.C.; Bastos, B.R.; Huang, C.; et al. Randomized Phase 2 Study of Tivantinib plus Erlotinib versus Single-Agent Chemotherapy in Previously Treated KRAS Mutant Advanced Non-Small Cell Lung Cancer. Lung Cancer 2018, 117, 44–49. [Google Scholar] [CrossRef]
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan Inhibits Collagen I Synthesis and Improves the Distribution and Efficacy of Nanotherapeutics in Tumors. PNAS 2011, 108, 2909–2914. [Google Scholar] [CrossRef] [Green Version]
- Gilkes, D.M.; Chaturvedi, P.; Bajpai, S.; Wong, C.C.; Wei, H.; Pitcairn, S.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Collagen Prolyl Hydroxylases Are Essential for Breast Cancer Metastasis. Cancer Res. 2013, 73, 3285–3296. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.-H.; Hsia, S.-M.; Shieh, T.-M. Lysyl Oxidase and the Tumor Microenvironment. Int. J. Mol. Sci. 2016, 18, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankowski, K.; Kucia, M.; Wysoczynski, M.; Reca, R.; Zhao, D.; Trzyna, E.; Trent, J.; Peiper, S.; Zembala, M.; Ratajczak, J.; et al. Both Hepatocyte Growth Factor (HGF) and Stromal-Derived Factor-1 Regulate the Metastatic Behavior of Human Rhabdomyosarcoma Cells, but Only HGF Enhances Their Resistance to Radiochemotherapy. Cancer Res. 2003, 63, 7926–7935. [Google Scholar]
- Purcell, J.W.; Tanlimco, S.G.; Hickson, J.; Fox, M.; Sho, M.; Durkin, L.; Uziel, T.; Powers, R.; Foster, K.; McGonigal, T.; et al. LRRC15 Is a Novel Mesenchymal Protein and Stromal Target for Antibody-Drug Conjugates. Cancer Res. 2018, 78, 4059–4072. [Google Scholar] [CrossRef] [Green Version]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Belgodere, J.A.; King, C.T.; Bursavich, J.B.; Burow, M.E.; Martin, E.C.; Jung, J.P. Engineering Breast Cancer Microenvironments and 3D Bioprinting. Front. Bioeng. Biotechnol. 2018, 6, 66. [Google Scholar] [CrossRef]
- Estrada, M.F.; Rebelo, S.P.; Davies, E.J.; Pinto, M.T.; Pereira, H.; Santo, V.E.; Smalley, M.J.; Barry, S.T.; Gualda, E.J.; Alves, P.M.; et al. Modelling the Tumour Microenvironment in Long-Term Microencapsulated 3D Co-Cultures Recapitulates Phenotypic Features of Disease Progression. Biomaterials 2016, 78, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Mejia, D.L.; Chiang, B.; Luker, K.E.; Luker, G.D. Hybrid Collagen Alginate Hydrogel as a Platform for 3D Tumor Spheroid Invasion. Acta Biomater. 2018, 75, 213–225. [Google Scholar] [CrossRef]
- Truong, D.D.; Kratz, A.; Park, J.G.; Barrientos, E.S.; Saini, H.; Nguyen, T.; Pockaj, B.; Mouneimne, G.; LaBaer, J.; Nikkhah, M. A Human Organotypic Microfluidic Tumor Model Permits Investigation of the Interplay between Patient-Derived Fibroblasts and Breast Cancer Cells. Cancer Res. 2019, 79, 3139–3151. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elwakeel, E.; Weigert, A. Breast Cancer CAFs: Spectrum of Phenotypes and Promising Targeting Avenues. Int. J. Mol. Sci. 2021, 22, 11636. https://doi.org/10.3390/ijms222111636
Elwakeel E, Weigert A. Breast Cancer CAFs: Spectrum of Phenotypes and Promising Targeting Avenues. International Journal of Molecular Sciences. 2021; 22(21):11636. https://doi.org/10.3390/ijms222111636
Chicago/Turabian StyleElwakeel, Eiman, and Andreas Weigert. 2021. "Breast Cancer CAFs: Spectrum of Phenotypes and Promising Targeting Avenues" International Journal of Molecular Sciences 22, no. 21: 11636. https://doi.org/10.3390/ijms222111636
APA StyleElwakeel, E., & Weigert, A. (2021). Breast Cancer CAFs: Spectrum of Phenotypes and Promising Targeting Avenues. International Journal of Molecular Sciences, 22(21), 11636. https://doi.org/10.3390/ijms222111636