
Cerium Oxide Nanoparticles Alleviate Hepatic Fibrosis Phenotypes In Vitro



Abstract
:1. Introduction
2. Results
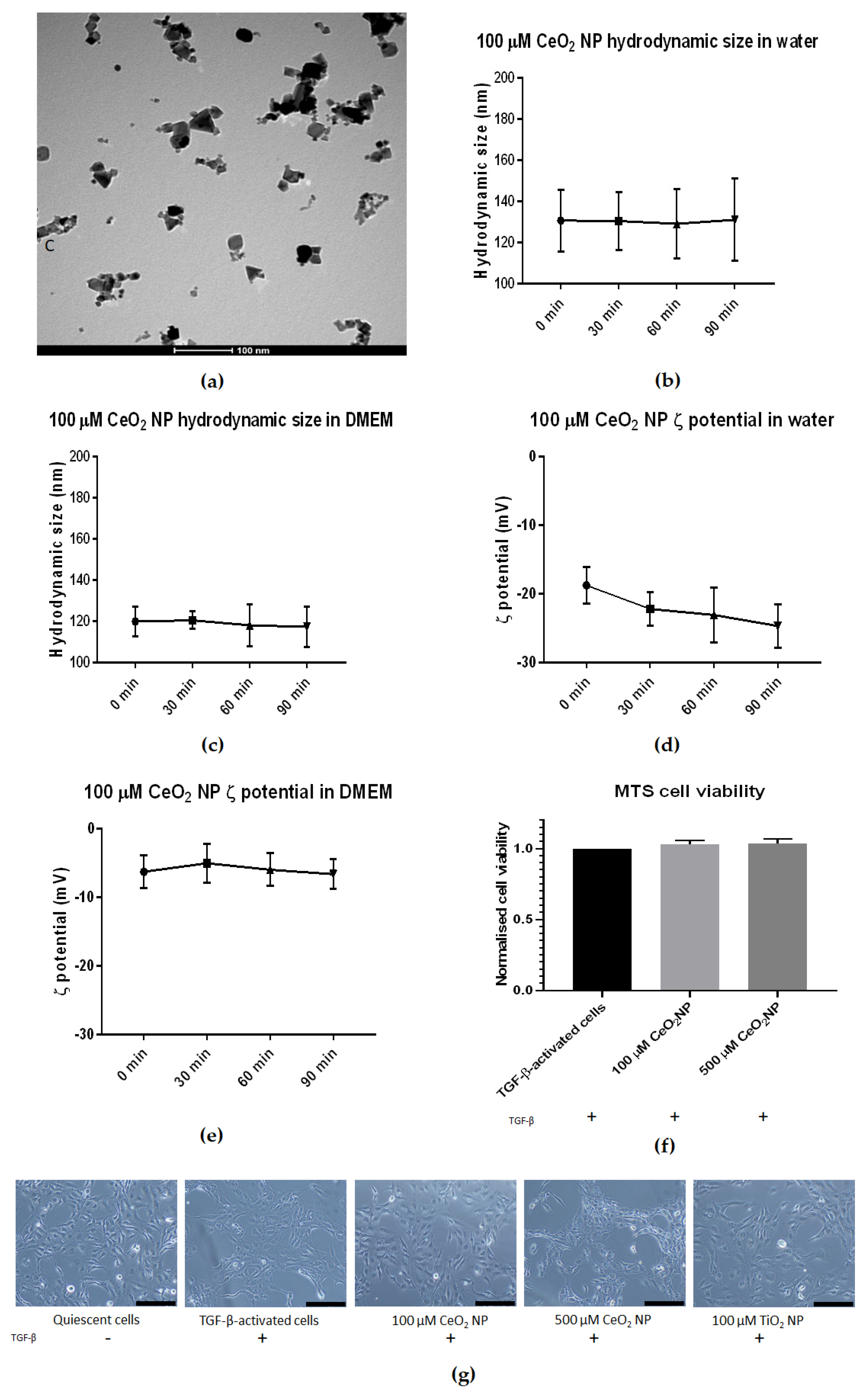
2.1. Characterizing Cerium Oxide Nanoparticles Used in This Study
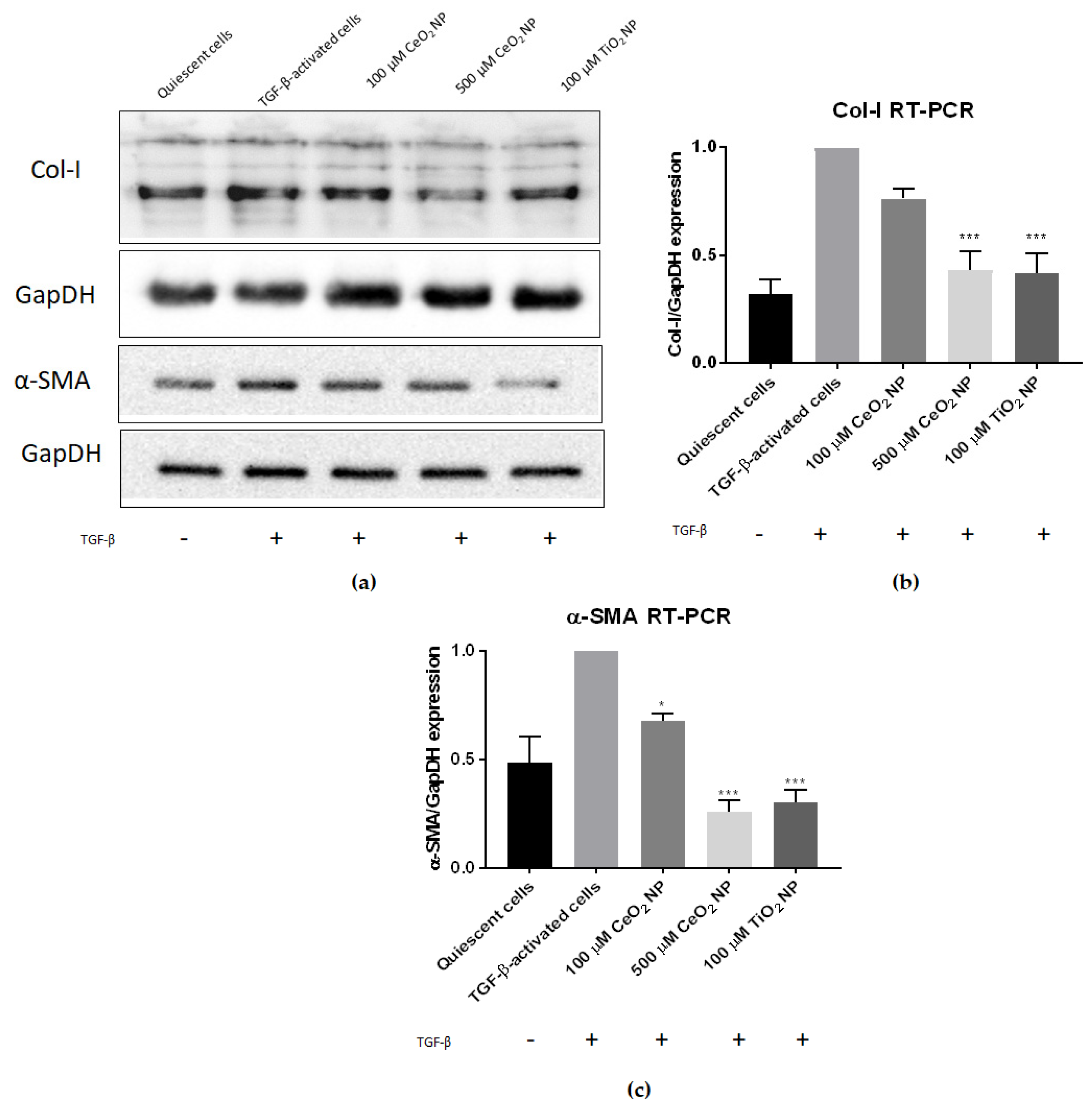
2.2. Cerium Oxide Nanoparticles Are Able to Reduce Fibrogenesis Marker Expression
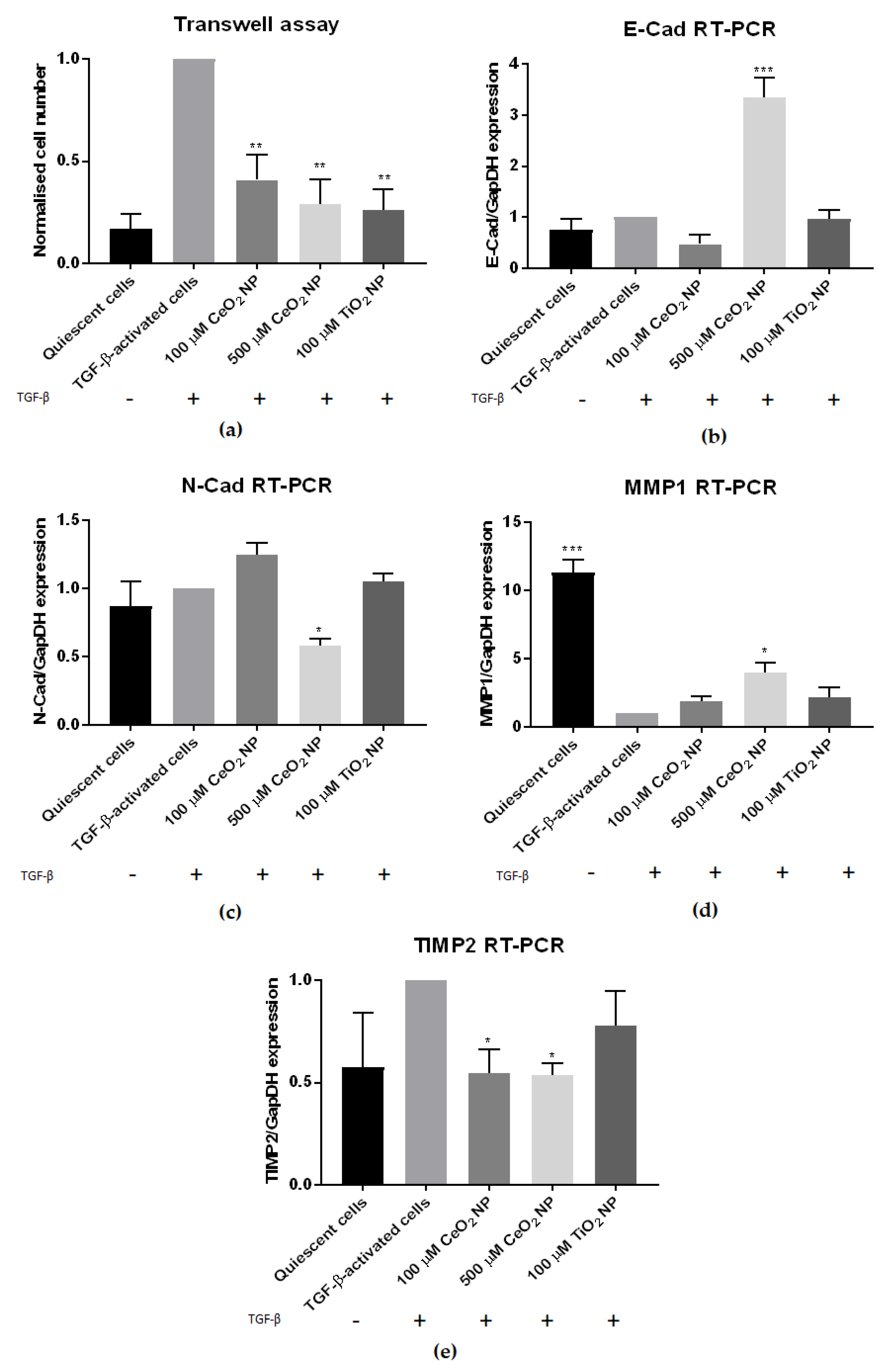
2.3. CeO2 NP Affect Other Classical Hallmarks of Hepatic Fibrosis
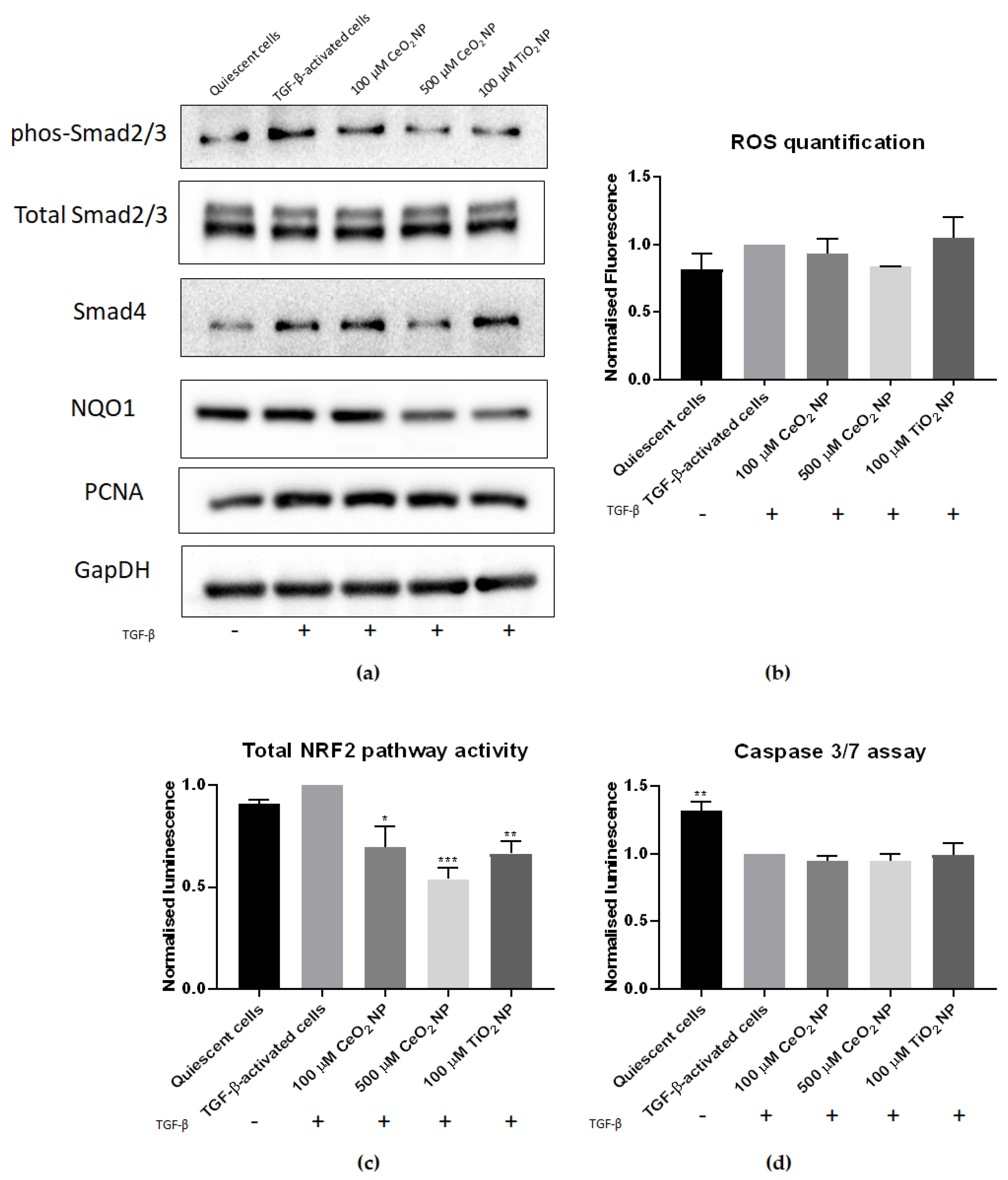
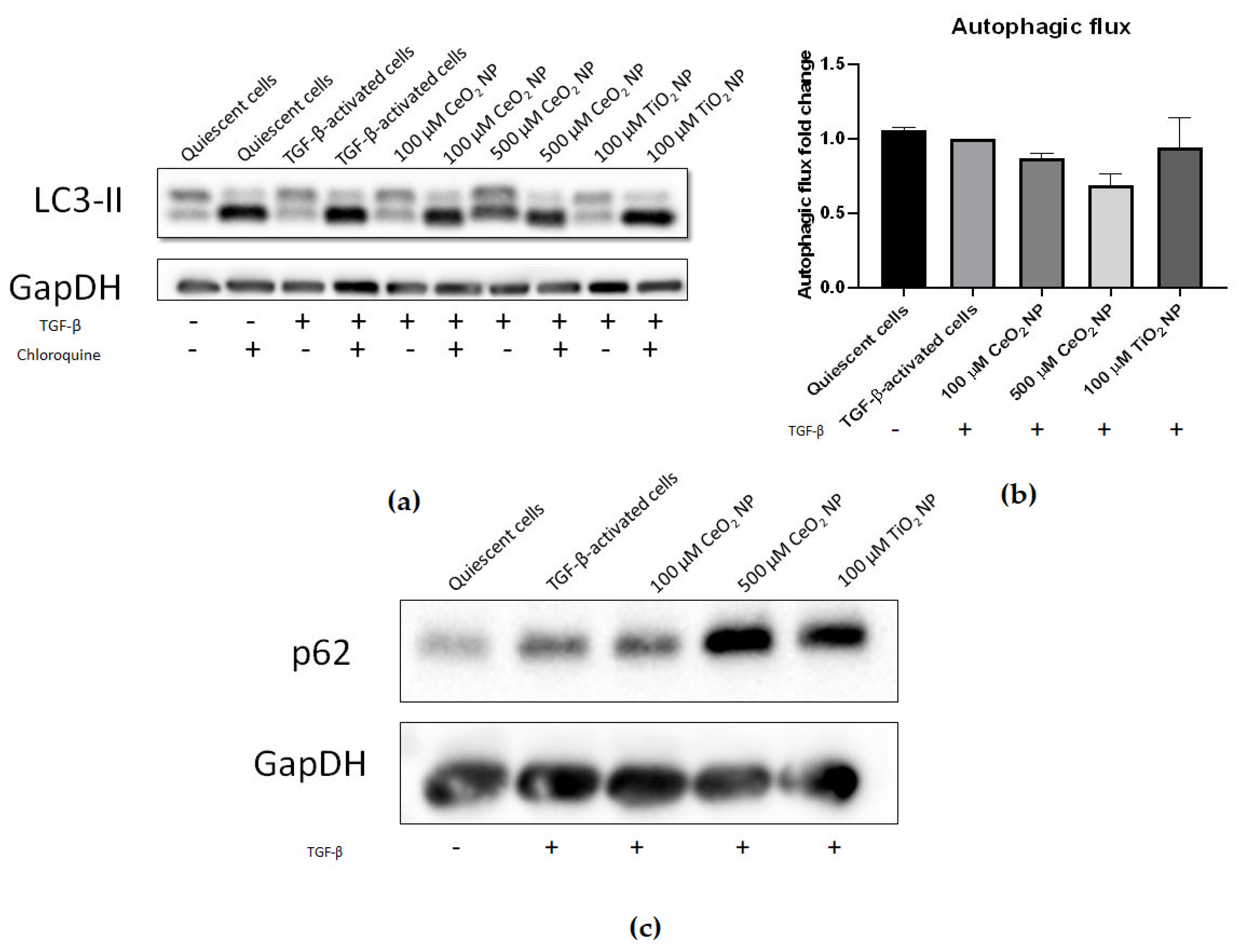
2.4. HSC Activation Phenotypes Are Reduced in CeO2 NP-Treated Cells
3. Discussion
4. Materials and Methods
4.1. Nanoparticles Used in This Study
4.2. Cell Lines Used
4.3. Dynamic Light Scattering
4.4. MTS Assay
4.5. Cell Imaging with Light Microscopy
4.6. Cell Migration Assay
4.7. Oxidative Stress Measurement
4.8. Nrf2 Activity Assay
4.9. Immunoblots
4.10. RNA Extraction, cDNA Synthesis and Real-Time PCR
4.11. Caspase 3/7 Activity Assay
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Bansal, R.; Nagórniewicz, B.; Prakash, J. Clinical advancements in the targeted therapies against liver fibrosis. Mediat. Inflamm. 2016, 2016, 1–16. [Google Scholar] [CrossRef]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68–69, 435–451. [Google Scholar] [CrossRef]
- Boey, A.; Ho, H.K. All roads lead to the liver: Metal nanoparticles and their implications for liver health. Small 2020, 16, e2000153. [Google Scholar] [CrossRef]
- Elsaesser, A.; Howard, C.V. Toxicology of nanoparticles. Adv. Drug Deliv. Rev. 2012, 64, 129–137. [Google Scholar] [CrossRef]
- Das, S.; Dowding, J.M.; Klump, K.E.; McGinnis, J.F.; Self, W.; Seal, S. Cerium oxide nanoparticles: Applications and prospects in nanomedicine. Nanomedicine 2013, 8, 1483–1508. [Google Scholar] [CrossRef]
- Korsvik, C.; Patil, S.; Seal, S.; Self, W.T. Superoxide dismutase mimetic properties exhibited by vacancy engineered ceria nanoparticles. Chem. Commun. 2007, 1056–1058. [Google Scholar] [CrossRef]
- Baldim, V.; Bedioui, F.; Mignet, N.; Margaill, I.; Berret, J.-F. The enzyme-like catalytic activity of cerium oxide nanoparticles and its dependency on Ce3+ surface area concentration. Nanoscale 2018, 10, 6971–6980. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Qu, X. Cerium oxide nanoparticle: A remarkably versatile rare earth nanomaterial for biological applications. NPG Asia Mater. 2014, 6, e90. [Google Scholar] [CrossRef]
- Estevez, A.Y.; Erlichman, J.S. The potential of cerium oxide nanoparticles (nanoceria) for neurodegenerative disease therapy. Nanomedicine 2014, 9, 1437–1440. [Google Scholar] [CrossRef] [Green Version]
- Rzigalinski, B.A.; Carfagna, C.S.; Ehrich, M. Cerium oxide nanoparticles in neuroprotection and considerations for efficacy and safety. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1444. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.K.; Kim, T.; Choi, I.-Y.; Soh, M.; Kim, D.; Kim, Y.-J.; Jang, H.; Yang, H.-S.; Kim, J.Y.; Park, H.-K.; et al. Ceria nanoparticles that can protect against ischemic stroke. Angew. Chem. Int. Ed. 2012, 51, 11039–11043. [Google Scholar] [CrossRef]
- Kyosseva, S.V.; Kyosseva, S.V. Cerium oxide nanoparticles as promising ophthalmic therapeutics for the treatment of retinal diseases. World J. Ophthalmol. 2015, 5, 23. [Google Scholar] [CrossRef]
- Casals, E.; Zeng, M.; Parra-Robert, M.; Fernández-Varo, G.; Morales-Ruiz, M.; Jiménez, W.; Puntes, V.; Casals, G. Cerium oxide nanoparticles: Advances in biodistribution, toxicity, and preclinical exploration. Small 2020, 16, e1907322. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Morishita, M.; Wagner, J.G.; Fatouraie, M.; Wooldridge, M.; Eagle, W.E.; Barres, J.; Carlander, U.; Emond, C.; Jolliet, O. In vivo biodistribution and physiologically based pharmacokinetic modeling of inhaled fresh and aged cerium oxide nanoparticles in rats. Part. Fibre Toxicol. 2015, 13, 45. [Google Scholar] [CrossRef] [Green Version]
- Yokel, R.A.; Tseng, M.T.; Dan, M.; Unrine, J.; Graham, U.M.; Wu, P.; Grulke, E.A. Biodistribution and biopersistence of ceria engineered nanomaterials: Size dependence. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 398–407. [Google Scholar] [CrossRef]
- Geraets, L.; Oomen, A.G.; Schroeter, J.D.; Coleman, V.; Cassee, F.R. Tissue Distribution of inhaled micro- and nano-sized cerium oxide particles in rats: Results from a 28-day exposure study. Toxicol. Sci. 2012, 127, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Hirst, S.M.; Karakoti, A.; Singh, S.; Self, W.; Tyler, R.; Seal, S.; Reilly, C.M. Bio-distribution and in vivo antioxidant effects of cerium oxide nanoparticles in mice. Environ. Toxicol. 2013, 28, 107–118. [Google Scholar] [CrossRef]
- Molina, R.M.; Konduru, N.V.; Jimenez, R.J.; Pyrgiotakis, G.; Demokritou, P.; Wohlleben, W.; Brain, J.D. Bioavailability, distribution and clearance of tracheally instilled, gavaged or injected cerium dioxide nanoparticles and ionic cerium. Environ. Sci. Nano 2014, 1, 561–573. [Google Scholar] [CrossRef]
- Tseng, M.T.; Lu, X.; Duan, X.; Hardas, S.S.; Sultana, R.; Wu, P.; Unrine, J.M.; Graham, U.; Butterfield, D.A.; Grulke, E.A.; et al. Alteration of hepatic structure and oxidative stress induced by intravenous nanoceria. Toxicol. Appl. Pharmacol. 2012, 260, 173–182. [Google Scholar] [CrossRef]
- Tseng, M.T.; Fu, Q.; Lor, K.; Fernandez-Botran, G.R.; Deng, Z.-B.; Graham, U.; Butterfield, D.A.; Grulke, E.A.; Yokel, R. Persistent hepatic structural alterations following nanoceria vascular infusion in the rat. Toxicol. Pathol. 2014, 42, 984–996. [Google Scholar] [CrossRef] [PubMed]
- Adebayo, O.A.; Akinloye, O.; Adaramoye, O.A. Cerium oxide nanoparticles attenuate oxidative stress and inflammation in the liver of diethylnitrosamine-treated mice. Biol. Trace Elem. Res. 2020, 193, 214–225. [Google Scholar] [CrossRef]
- Córdoba-Jover, B.; Arce-Cerezo, A.; Ribera, J.; Pauta, M.; Oró, D.; Casals, G.; Fernández-Varo, G.; Casals, E.; Puntes, V.; Jiménez, W.; et al. Cerium oxide nanoparticles improve liver regeneration after acetaminophen-induced liver injury and partial hepatectomy in rats. J. Nanobiotechnol. 2019, 17, 1–12. [Google Scholar] [CrossRef]
- Ibrahim, H.G.; Attia, N.; Hashem, F.E.Z.A.; El Heneidy, M.A. Cerium oxide nanoparticles: In pursuit of liver protection against doxorubicin-induced injury in rats. Biomed. Pharmacother. 2018, 103, 773–781. [Google Scholar] [CrossRef]
- Sangomla, S.; Saifi, M.A.; Khurana, A.; Godugu, C. Nanoceria ameliorates doxorubicin induced cardiotoxicity: Possible mitigation via reduction of oxidative stress and inflammation. J. Trace Elements Med. Biol. 2018, 47, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Oró, D.; Yudina, T.; Fernández-Varo, G.; Casals, E.; Reichenbach, V.; Casals, G.; de la Presa, B.G.; Sandalinas, S.; Carvajal, S.; Puntes, V.; et al. Cerium oxide nanoparticles reduce steatosis, portal hypertension and display anti-inflammatory properties in rats with liver fibrosis. J. Hepatol. 2016, 64, 691–698. [Google Scholar] [CrossRef]
- Kobyliak, N.; Virchenko, O.; Falalyeyeva, T.; Kondro, M.; Beregova, T.; Bodnar, P.; Shcherbakov, O.; Bubnov, R.; Caprnda, M.; Delev, D.; et al. Cerium dioxide nanoparticles possess anti-inflammatory properties in the conditions of the obesity-associated NAFLD in rats. Biomed. Pharmacother. 2017, 90, 608–614. [Google Scholar] [CrossRef]
- Carvajal, S.; Perramón, M.; Oró, D.; Casals, E.; Fernández-Varo, G.; Casals, G.; Parra, M.; de la Presa, B.G.; Ribera, J.; Pastor, Ó.; et al. Cerium oxide nanoparticles display antilipogenic effect in rats with non-alcoholic fatty liver disease. Sci. Rep. 2019, 9, 12848. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Varo, G.; Perramón, M.; Carvajal, S.; Oró, D.; Casals, E.; Boix, L.; Oller, L.; Macías-Muñoz, L.; Marfà, S.; Casals, G.; et al. Bespoken nanoceria: An effective treatment in experimental hepatocellular carcinoma. Hepatology 2020, 72, 1267–1282. [Google Scholar] [CrossRef] [Green Version]
- Peng, F.; Tee, J.K.; Setyawati, M.I.; Ding, X.; Yeo, H.L.A.; Tan, Y.L.; Leong, D.T.; Ho, H.K. Inorganic nanomaterials as highly efficient inhibitors of cellular hepatic fibrosis. ACS Appl. Mater. Interfaces 2018, 10, 31938–31946. [Google Scholar] [CrossRef]
- Bashandy, S.A.; Alaamer, A.; Moussa, S.; Omara, E.A. Role of zinc oxide nanoparticles in alleviating hepatic fibrosis and nephrotoxicity induced by thioacetamide in rats. Can. J. Physiol. Pharmacol. 2018, 96, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Shalby, A.; El-Maksoud, M.; Moneim, A.; Ahmed, H. Antifibrotic candidates of Selenium nanoparticles and selenium in the experimental model. J. Appl. Pharm. Sci. 2017, 7, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.; Sandberg, A.; Heckert, E.; Self, W.; Seal, S. Protein adsorption and cellular uptake of cerium oxide nanoparticles as a function of zeta potential. Biomaterials 2007, 28, 4600–4607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iimuro, Y.; Nishio, T.; Morimoto, T.; Nitta, T.; Stefanovic, B.; Choi, S.K.; Brenner, D.; Yamaoka, Y. Delivery of matrix metalloproteinase-1 attenuates established liver fibrosis in the rat. Gastroenterology 2003, 124, 445–458. [Google Scholar] [CrossRef]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44–46, 247–254. [Google Scholar] [CrossRef]
- Thoen, L.F.; Guimarães, E.L.; Dollé, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef]
- Lippai, M.; Lőw, P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. BioMed Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Tovar, E.; Muriel, P. Molecular mechanisms that link oxidative stress, inflammation, and fibrosis in the liver. Antioxidants 2020, 9, 1279. [Google Scholar] [CrossRef]
- Galicia-Moreno, M.; Favari, L.; Muriel, P. Antifibrotic and antioxidant effects of N-acetylcysteine in an experimental cholestatic model. Eur. J. Gastroenterol. Hepatol. 2012, 24, 179–185. [Google Scholar] [CrossRef]
- Mata-Santos, H.A.; Dutra, F.F.; Rocha, C.C.; Lino, F.G.; Xavier, F.R.; Chinalia, L.A.; Hossy, B.H.; Castelo-Branco, M.T.L.; Teodoro, A.J.; Paiva, C.N.; et al. Silymarin reduces profibrogenic cytokines and reverses hepatic fibrosis in chronic murine schistosomiasis. Antimicrob. Agents Chemother. 2014, 58, 2076–2083. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.-A.; Chen, G.-M.; Liu, Y.; Chen, Y.-X.; Wu, H.-Y.; Chen, J.; Xiong, Y.-L.; Tian, C.; Wang, G.-Y.; Jia, B.; et al. Inhibitory effect of silymarin on CCl4-induced liver fibrosis by reducing Ly6Chi monocytes infiltration. Int. J. Clin. Exp. Pathol. 2017, 10, 11941–11951. [Google Scholar]
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.; Siegel, D. Functions of NQO1 in cellular protection and CoQ10 metabolism and its potential role as a redox sensitive molecular switch. Front. Physiol. 2017, 8, 595. [Google Scholar] [CrossRef]
- Hashem, R.; Rashd, L.A.; Hashem, K.S.; Soliman, H.M. Cerium oxide nanoparticles alleviate oxidative stress and decreases Nrf-2/HO-1 in D-GALN/LPS induced hepatotoxicity. Biomed. Pharmacother. 2015, 73, 80–86. [Google Scholar] [CrossRef]
- Rubio, L.; Annangi, B.; Vila, L.; Hernandez, A.; Marcos, R. Antioxidant and anti-genotoxic properties of cerium oxide nanoparticles in a pulmonary-like cell system. Arch. Toxicol. 2016, 90, 269–278. [Google Scholar] [CrossRef]
- Carvajal, S.; Perramón, M.; Casals, G.; Oró, D.; Ribera, J.; Morales-Ruiz, M.; Casals, E.; Casado, P.; Melgar-Lesmes, P.; Fernández-Varo, G.; et al. Cerium oxide nanoparticles protect against oxidant injury and interfere with oxidative mediated kinase signaling in human-derived hepatocytes. Int. J. Mol. Sci. 2019, 20, 5959. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.-M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Liu, G.-S.; Dusting, G.J.; Chan, E.C. NADPH oxidase-dependent redox signaling in TGF-β-mediated fibrotic responses. Redox Biol. 2014, 2, 267–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samarakoon, R.; Overstreet, J.M.; Higgins, P.J. TGF-β signaling in tissue fibrosis: Redox controls, target genes and therapeutic opportunities. Cell. Signal. 2013, 25, 264–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsi, F.; Caputo, F.; Traversa, E.; Ghibelli, L. Not only redox: The multifaceted activity of cerium oxide nanoparticles in cancer prevention and therapy. Front. Oncol. 2018, 8, 309. [Google Scholar] [CrossRef] [Green Version]
- Graham, U.M.; Yokel, R.A.; Dozier, A.K.; Drummy, L.; Mahalingam, K.; Tseng, M.T.; Birch, E.; Fernback, J. Analytical high-resolution electron microscopy reveals organ-specific nanoceria bioprocessing. Toxicol. Pathol. 2017, 46, 47–61. [Google Scholar] [CrossRef]
- Bartneck, M.; Warzecha, K.T.; Tacke, F. Therapeutic targeting of liver inflammation and fibrosis by nanomedicine. Hepatobiliary Surg. Nutr. 2014, 3, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Sulthana, S.; Banerjee, T.; Kallu, J.; Vuppala, S.R.; Heckert, B.; Naz, S.; Shelby, T.; Yambem, O.; Santra, S. Combination therapy of NSCLC using Hsp90 inhibitor and doxorubicin carrying functional nanoceria. Mol. Pharm. 2017, 14, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M.; Sciences, H.; Sciences, M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qie, Y.; Yuan, H.; von Roemeling, C.; Chen, Y.; Liu, X.; Shih, K.D.; Knight, J.A.; Tun, H.W.; Wharen, R.E.; Jiang, W.; et al. Surface modification of nanoparticles enables selective evasion of phagocytic clearance by distinct macrophage phenotypes. Sci. Rep. 2016, 6, 26269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, K.; Arya, A.; Gangwar, A.; Singh, S.K.; Roy, M.; Das, M.; Sethy, N.K. Cerium oxide nanoparticles promote neurogenesis and abrogate hypoxia-induced memory impairment through AMPK–PKC–CBP signaling cascade. Int. J. Nanomed. 2016, 11, 1159–1173. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.Y.; Mercer, R.R.; Barger, M.; Schwegler-Berry, D.; Scabilloni, J.; Ma, J.K.; Castranova, V. Induction of Pulmonary Fibrosis by Cerium Oxide Nanoparticles. Toxicol. Appl. Pharmacol. 2012, 262, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Bishoff, B.; Mercer, R.R.; Barger, M.; Schwegler-Berry, D.; Castranova, V. Role of Epithelial Mesenchymal Transition (EMT) and Fibroblast Function in Cerium Oxide Nanoparticles-Induced Lung Fibrosis. Toxicol. Appl. Pharmacol. 2017, 323, 16–25. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, L.; Pytel, D.; Mucha, B.; Szymanek, K.; Szaflik, J.; Szaflik, J.P.; Majsterek, I. Altered expression levels of MMP1, MMP9, MMP12, TIMP1, and IL-1β as a risk factor for the elevated IOP and optic nerve head damage in the primary open-angle glaucoma patients. BioMed Res. Int. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahlin, L.; Stjernholm-Vladic, Y.; Roos, N.; Masironi, B.; Ekman-Ordeberg, G. Impaired leukocyte influx in cervix of postterm women not responding to prostaglandin priming. Reprod. Biol. Endocrinol. 2008, 6, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mean ROS Level | Difference from TGF-β-activated Cell Mean * | Percentage Reduction from Quiescent Cell Mean # |
|---|---|---|---|
| Quiescent cell | 0.817 | −0.183 | N.A. |
| TGF-β-activated cell | 1.00 | N.A. | N.A. |
| 100 µM CeO2 NP | 0.936 | −0.064 | 35.03% |
| 500 µM CeO2 NP | 0.793 | −0.207 | 113.0% |
| 100 µM TiO2 NP | 1.04 | +0.40 | −26.20% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boey, A.; Leong, S.Q.; Bhave, S.; Ho, H.K. Cerium Oxide Nanoparticles Alleviate Hepatic Fibrosis Phenotypes In Vitro. Int. J. Mol. Sci. 2021, 22, 11777. https://doi.org/10.3390/ijms222111777
Boey A, Leong SQ, Bhave S, Ho HK. Cerium Oxide Nanoparticles Alleviate Hepatic Fibrosis Phenotypes In Vitro. International Journal of Molecular Sciences. 2021; 22(21):11777. https://doi.org/10.3390/ijms222111777
Chicago/Turabian StyleBoey, Adrian, Shu Qing Leong, Sayali Bhave, and Han Kiat Ho. 2021. "Cerium Oxide Nanoparticles Alleviate Hepatic Fibrosis Phenotypes In Vitro" International Journal of Molecular Sciences 22, no. 21: 11777. https://doi.org/10.3390/ijms222111777
APA StyleBoey, A., Leong, S. Q., Bhave, S., & Ho, H. K. (2021). Cerium Oxide Nanoparticles Alleviate Hepatic Fibrosis Phenotypes In Vitro. International Journal of Molecular Sciences, 22(21), 11777. https://doi.org/10.3390/ijms222111777