
CRISPRi-Guided Metabolic Flux Engineering for Enhanced Protopanaxadiol Production in Saccharomyces cerevisiae

 , ,
, ,
Abstract
:1. Introduction
2. Results
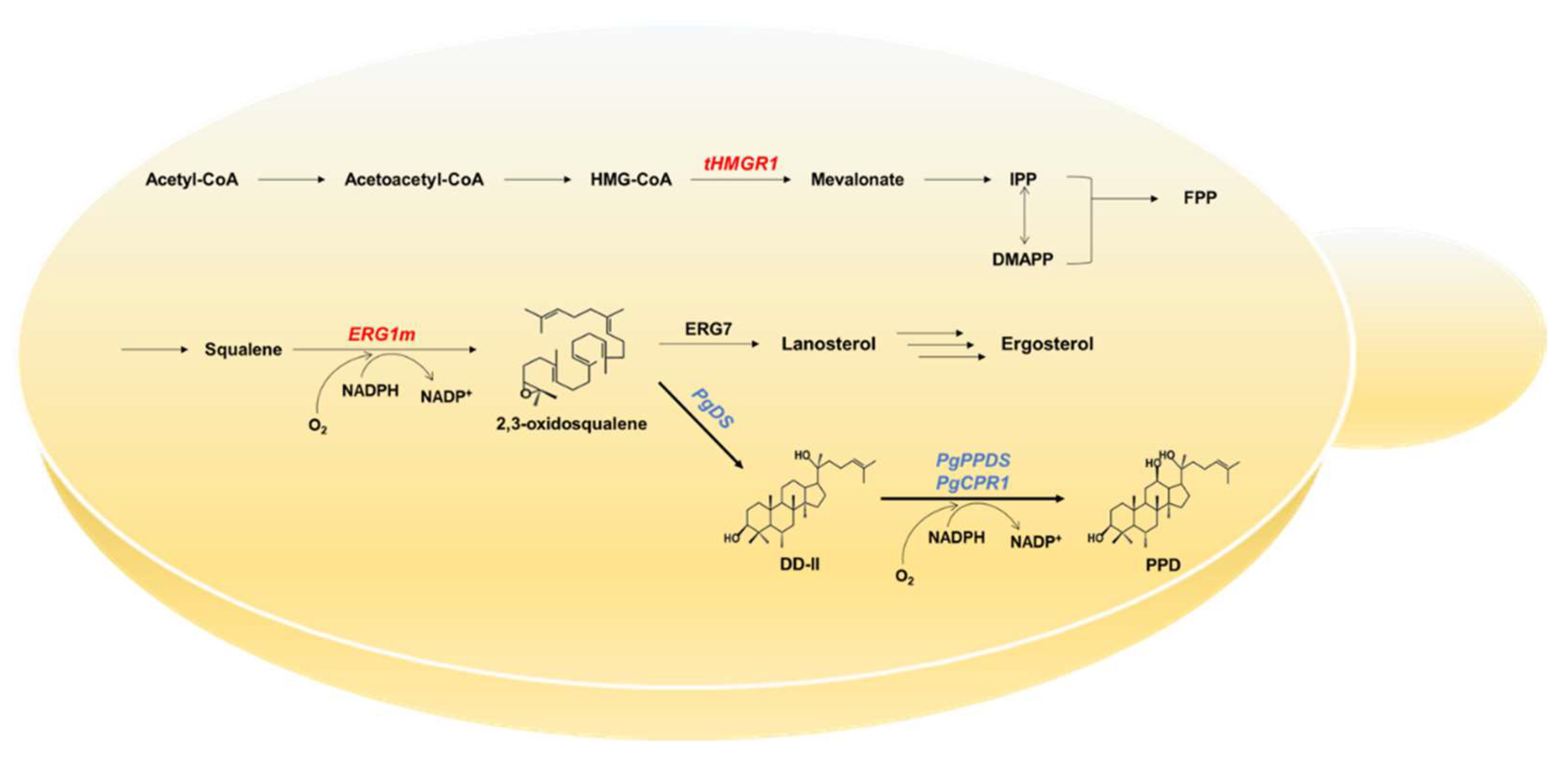
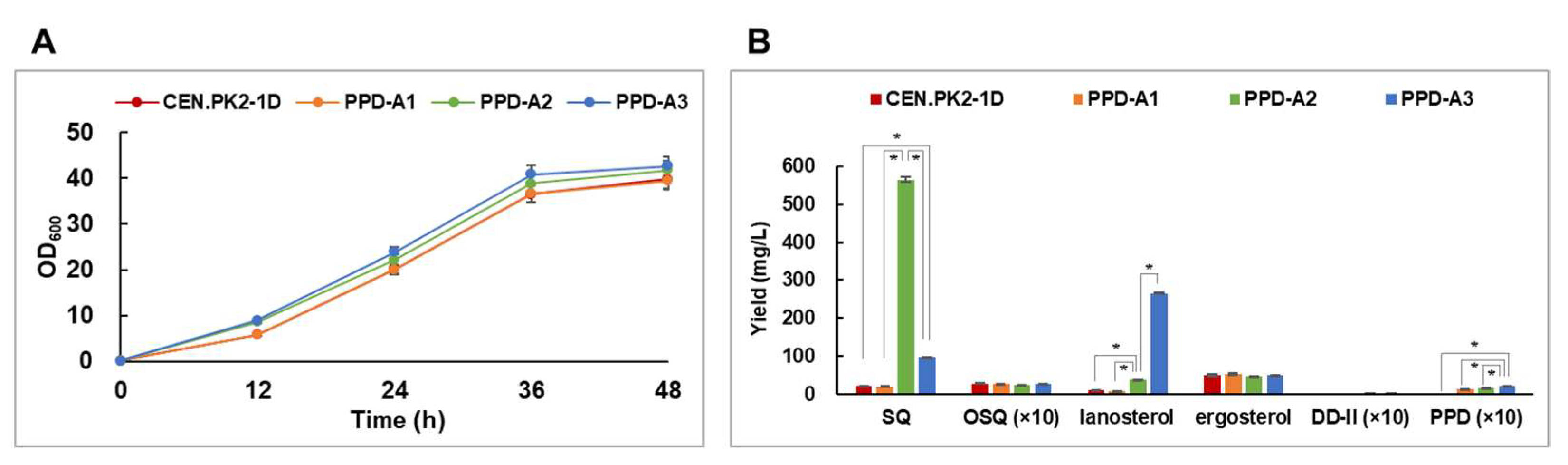
2.1. Construction of the PPD-Producing Yeast Strain
2.2. Enhancing the MVA Pathway and Squalene Monooxygenase Stability Upregulated the Triterpene Biosynthetic Pathway
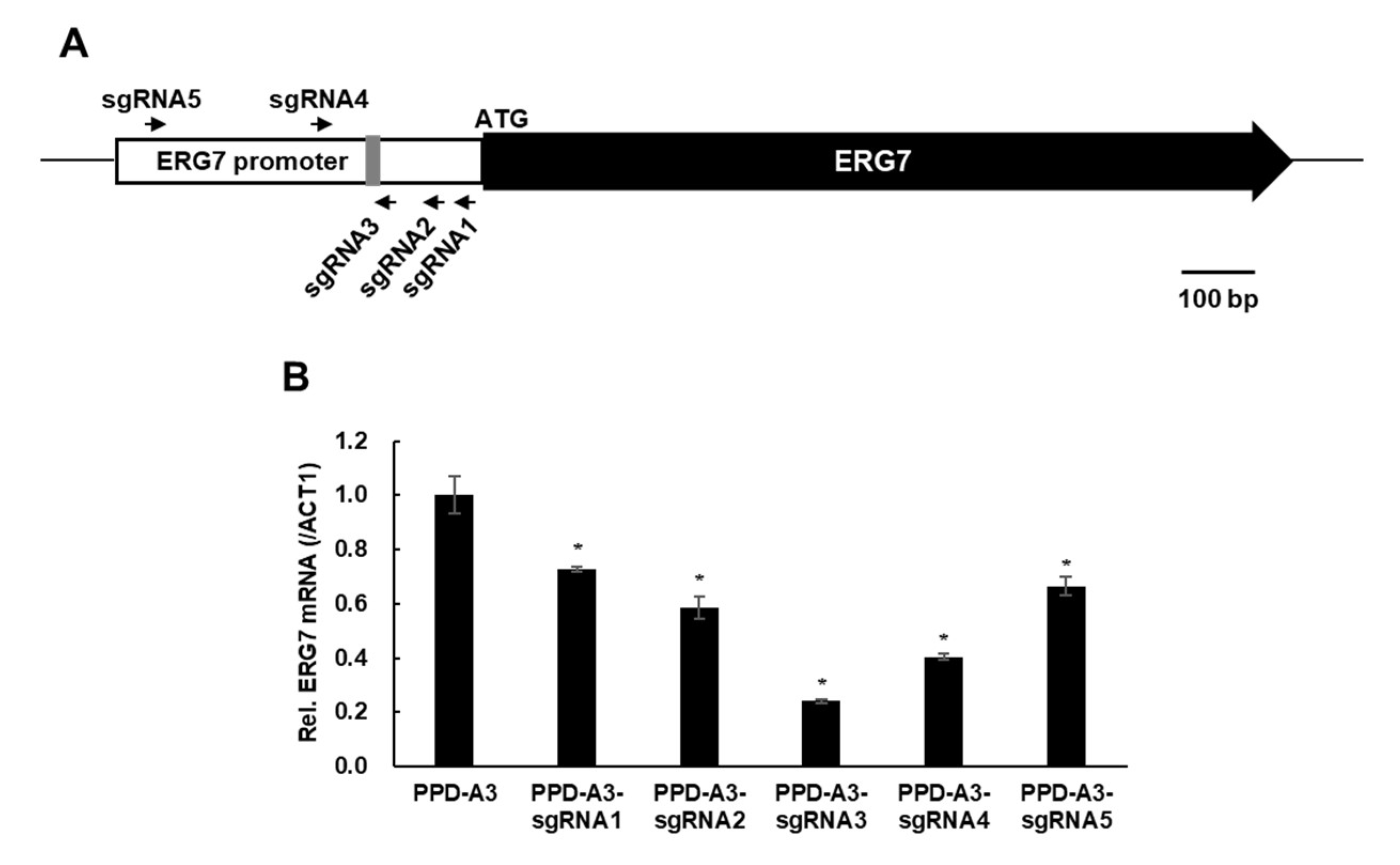
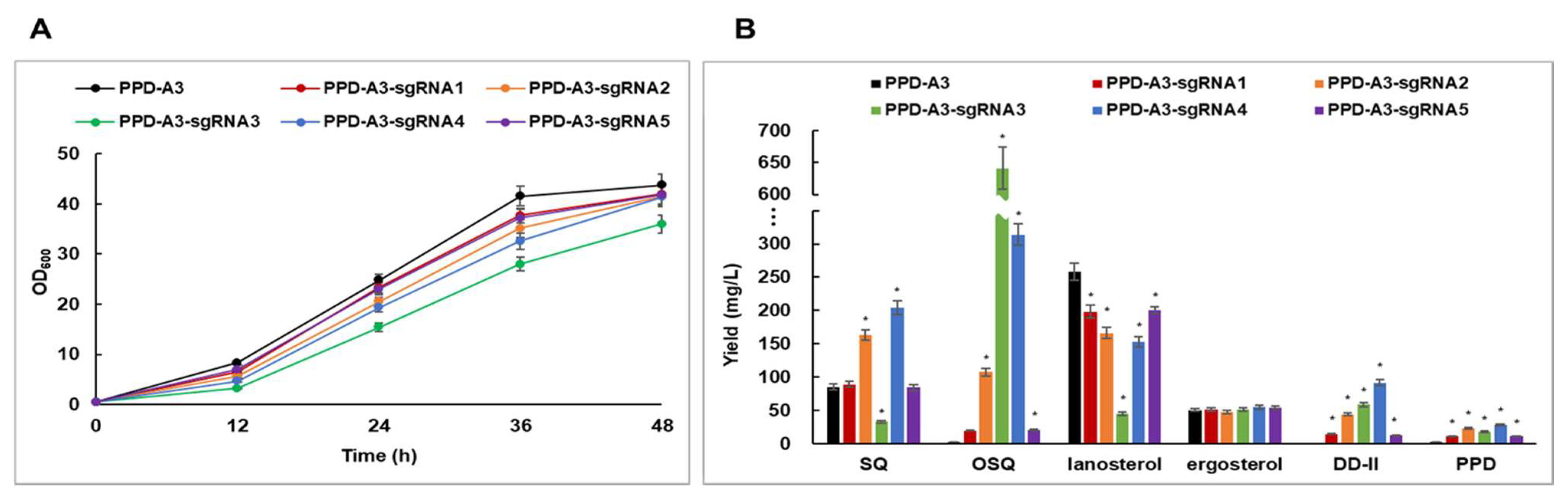
2.3. CRISPRi-Mediated ERG7 Suppression Improved PPD Production
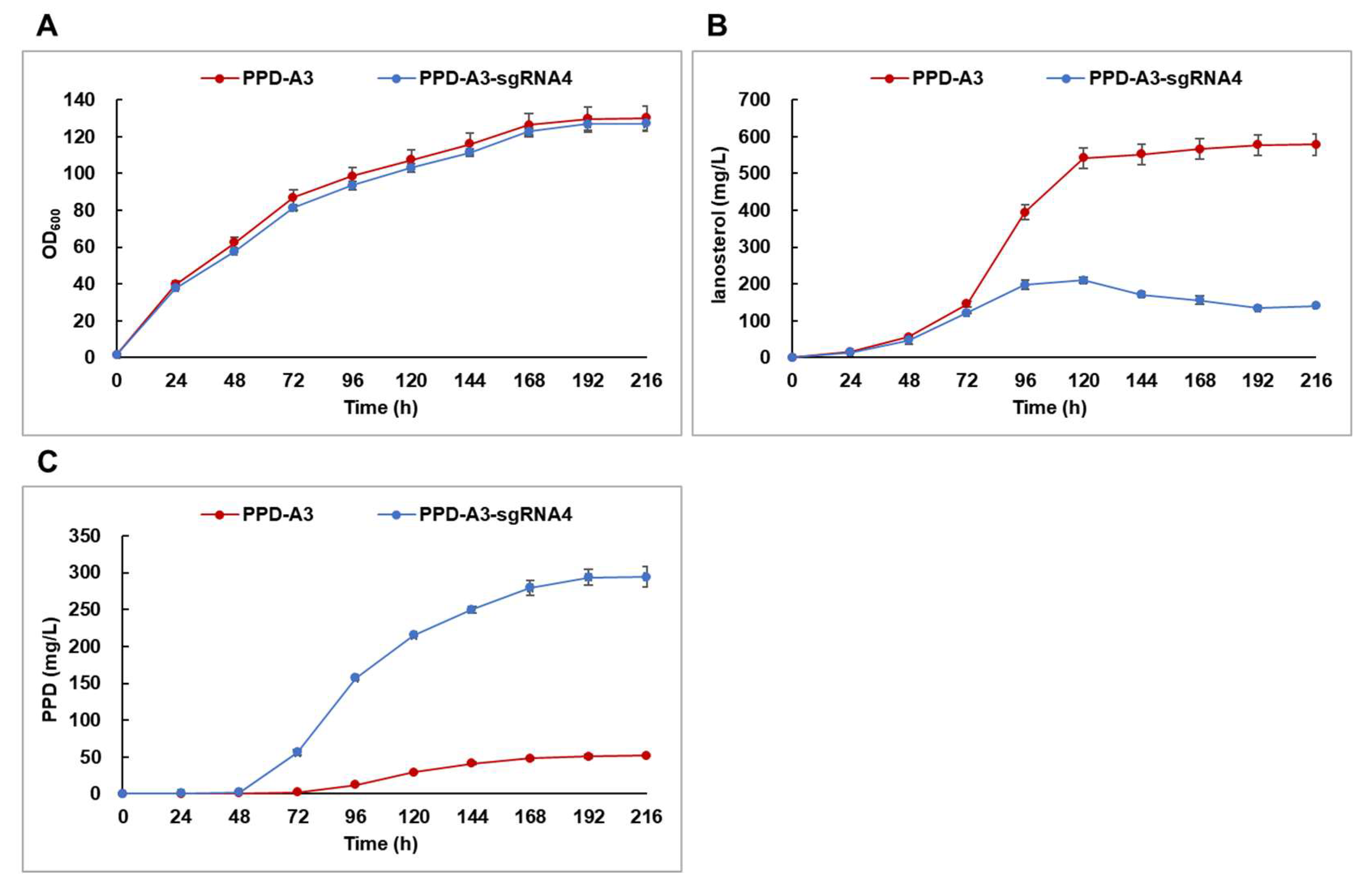
2.4. PPD Production via Batch-Fed Fermentation
3. Discussion
4. Materials and Methods
4.1. Strains and Medium
4.2. Plasmid Construction
4.3. Engineering S. cerevisiae for PPD Production
4.4. Yeast Cultivation and Batch-Fed Fermentation
4.5. Metabolite Extraction and HPLC Analysis
4.6. RNA Extraction and qRT-PCR
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siraj, F.M.; SathishKumar, N.; Kim, Y.J.; Kim, S.Y.; Yang, D.C. Ginsenoside F2 possesses anti-obesity activity via binding with PPARgamma and inhibiting adipocyte differentiation in the 3T3-L1 cell line. J. Enzyme Inhib. Med. Chem. 2015, 30, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.E.; Chang, B.Y.; Jeon, B.M.; Baek, J.I.; Kim, S.C.; Kim, S.Y. SGL 121 Attenuates Nonalcoholic Fatty Liver Disease through Adjusting Lipid Metabolism Through AMPK Signaling Pathway. Int. J. Mol. Sci. 2020, 21, 4534. [Google Scholar] [CrossRef]
- Park, S.; Ko, E.; Lee, J.H.; Song, Y.; Cui, C.-H.; Hou, J.; Jeon, B.M.; Kim, H.S.; Kim, S.C. Gypenoside LXXV Promotes Cutaneous Wound Healing In Vivo by Enhancing Connective Tissue Growth Factor Levels Via the Glucocorticoid Receptor Pathway. Molecules 2019, 24, 1595. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chu, S.; Lin, M.; Gao, Y.; Liu, Y.; Yang, S.; Zhou, X.; Zhang, Y.; Hu, Y.; Wang, H.; et al. Anticancer property of ginsenoside Rh2 from ginseng. Eur. J. Med. Chem. 2020, 203, 112627. [Google Scholar] [CrossRef]
- Gao, H.; Kang, N.; Hu, C.; Zhang, Z.; Xu, Q.; Liu, Y.; Yang, S. Ginsenoside Rb1 exerts anti-inflammatory effects in vitro and in vivo by modulating toll-like receptor 4 dimerization and NF-kB/MAPKs signaling pathways. Phytomedicine 2020, 69, 153197. [Google Scholar] [CrossRef]
- Chen, W.; Balan, P.; Popovich, D.G. Analysis of Ginsenoside Content (Panax ginseng) from Different Regions. Molecules 2019, 24, 3491. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.; Liu, Y.; Zhang, X.; Shi, M.; Wang, B.; Wang, D.; Huang, L.; Zhang, X. Metabolic engineering of Saccharomyces cerevisiae for production of ginsenosides. Metab. Eng. 2013, 20, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Jang, I.S.; Son, S.H.; Ko, Y.J.; Cho, B.K.; Kim, S.C.; Lee, J.Y. Tailoring the Saccharomyces cerevisiae endoplasmic reticulum for functional assembly of terpene synthesis pathway. Metab. Eng. 2019, 56, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Jang, I.S.; Sung, B.H.; Kim, S.C.; Lee, J.Y. Rerouting of NADPH synthetic pathways for increased protopanaxadiol production in Saccharomyces cerevisiae. Sci. Rep. 2018, 8, 15820. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Bai, P.; Liu, T.; Li, D.; Zhang, X.; Lu, W.; Yuan, Y. Optimization of a cytochrome P450 oxidation system for enhancing protopanaxadiol production in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2016, 113, 1787–1795. [Google Scholar] [CrossRef]
- Zhao, F.; Bai, P.; Nan, W.; Li, D.; Zhang, C.; Lu, C.; Qi, H.; Lu, W. A modular engineering strategy for high-level production of protopanaxadiol from ethanol by Saccharomyces cerevisiae. AIChE J. 2018, 65, 866–874. [Google Scholar] [CrossRef]
- Zhao, F.; Du, Y.; Bai, P.; Liu, J.; Lu, W.; Yuan, Y. Enhancing Saccharomyces cerevisiae reactive oxygen species and ethanol stress tolerance for high-level production of protopanoxadiol. Bioresour. Technol. 2017, 227, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.L.; Montecillo, J.A.V.; Bae, H. Recent Advances in the Metabolic Engineering of Yeasts for Ginsenoside Biosynthesis. Front. Bioeng. Biotechnol. 2020, 8, 139. [Google Scholar] [CrossRef]
- Vranová, E.; Coman, D.; Gruissem, W. Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 2013, 64, 665–700. [Google Scholar] [CrossRef] [PubMed]
- Tansakul, P.; Shibuya, M.; Kushiro, T.; Ebizuka, Y. Dammarenediol-II synthase, the first dedicated enzyme for ginsenoside biosynthesis, in Panax ginseng. FEBS Lett. 2006, 580, 5143–5149. [Google Scholar] [CrossRef] [Green Version]
- Han, J.Y.; Hwang, H.S.; Choi, S.W.; Kim, H.J.; Choi, Y.E. Cytochrome P450 CYP716A53v2 catalyzes the formation of protopanaxatriol from protopanaxadiol during ginsenoside biosynthesis in Panax ginseng. Plant Cell Physiol. 2012, 53, 1535–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Fan, Y.; Wei, W.; Wang, P.; Liu, Q.; Wei, Y.; Zhang, L.; Zhao, G.; Yue, J.; Zhou, Z. Production of bioactive ginsenoside compound K in metabolically engineered yeast. Cell Res. 2014, 24, 770–773. [Google Scholar] [CrossRef]
- Hu, Z.; He, B.; Ma, L.; Sun, Y.; Niu, Y.; Zeng, B. Recent Advances in Ergosterol Biosynthesis and Regulation Mechanisms in Saccharomyces cerevisiae. Indian J. Microbiol. 2017, 57, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ching, C.B. Dynamic control of ERG9 expression for improved amorpha-4,11-diene production in Saccharomyces cerevisiae. Microb. Cell Fact. 2015, 14, 38. [Google Scholar] [CrossRef] [Green Version]
- Paddon, C.J.; Westfall, P.J.; Pitera, D.J.; Benjamin, K.; Fisher, K.; McPhee, D.J.; Leavell, M.D.; Tai, A.; Main, A.; Eng, D.; et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Ozaydın, B.; Burd, H.; Lee, T.S.; Keasling, J.D. Carotenoid-based phenotypic screen of the yeast deletion collection reveals new genes with roles in isoprenoid production. Metab. Eng. 2013, 15, 174–183. [Google Scholar] [CrossRef]
- Rodriguez, S.; Denby, C.M.; Van Vu, T.; Baidoo, E.E.; Wang, G.; Keasling, J.D. ATP citrate lyase mediated cytosolic acetyl-CoA biosynthesis increases mevalonate production in Saccharomyces cerevisiae. Microb. Cell Fact. 2016, 15, 48. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.C.; Kim, W.; Park, S.C.; Jeong, J.; Park, M.K.; Lim, S.; Lee, Y.; Im, W.T.; Lee, J.H.; Choi, G.; et al. Two ginseng UDP-glycosyltransferases synthesize ginsenoside Rg3 and Rd. Plant Cell Physiol. 2014, 55, 2177–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, J.; Romanini, D.W.; Paradise, E.M.; Keasling, J.D. Engineering triterpene production in Saccharomyces cerevisiae-beta-amyrin synthase from Artemisia annua. FEBS J. 2008, 275, 1852–1859. [Google Scholar] [CrossRef]
- Broker, J.N.; Muller, B.; van Deenen, N.; Prufer, D.; Gronover, C.S. Upregulating the mevalonate pathway and repressing sterol synthesis in Saccharomyces cerevisiae enhances the production of triterpenes. Appl. Microbiol. Biotechnol. 2018, 102, 6923–6934. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Plan, M.R.; Chrysanthopoulos, P.; Hodson, M.; Nielsen, L.; Vickers, C.E. A squalene synthase protein degradation method for improved sesquiterpene production in Saccharomyces cerevisiae. Metab. Eng. 2016, 39, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Scalcinati, G.; Knuf, C.; Partow, S.; Chen, Y.; Maury, J.; Schalk, M.; Daviet, L.; Nielsen, J.; Siewers, V. Dynamic control of gene expression in Saccharomyces cerevisiae engineered for the production of plant sesquitepene alpha-santalene in a fed-batch mode. Metab. Eng. 2012, 14, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ho, J.; Shis, D.L.; Gupta, C.; Long, J.; Wagner, D.S.; Ott, W.; Josić, K.; Bennett, M.R. Tuning the dynamic range of bacterial promoters regulated by ligand-inducible transcription factors. Nat. Commun. 2018, 9, 64. [Google Scholar] [CrossRef]
- Lv, L.; Ren, Y.; Chen, J.-C.; Wu, Q.; Chen, G.-Q. Application of CRISPRi for prokaryotic metabolic engineering involving multiple genes, a case study: Controllable P(3HB-co-4HB) biosynthesis. Metab. Eng. 2015, 29, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Yasokawa, D.; Murata, S.; Kitagawa, E.; Iwahashi, Y.; Nakagawa, R.; Hashido, T.; Iwahashi, H. Mechanisms of copper toxicity in Saccharomyces cerevisiae determined by microarray analysis. Environ. Toxicol. 2008, 23, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Hu, Z.; Zhang, T.; Gong, T.; Chen, J.; Zhu, P.; Li, Y.; Yang, J. Production of a bioactive unnatural ginsenoside by metabolically engineered yeasts based on a new UDP-glycosyltransferase from Bacillus subtilis. Metab. Eng. 2017, 44, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wei, W.; Ye, W.; Li, X.; Zhao, W.; Yang, C.; Li, C.; Yan, X.; Zhou, Z. Synthesizing ginsenoside Rh2 in Saccharomyces cerevisiae cell factory at high-efficiency. Cell Discov. 2019, 5, 5. [Google Scholar] [CrossRef]
- Wang, P.; Wei, Y.; Fan, Y.; Liu, Q.; Wei, W.; Yang, C.; Zhang, L.; Zhao, G.; Yue, J.; Yan, X.; et al. Production of bioactive ginsenosides Rh2 and Rg3 by metabolically engineered yeasts. Metab. Eng. 2015, 29, 97–105. [Google Scholar] [CrossRef]
- Foresti, O.; Ruggiano, A.; Hannibal-Bach, H.K.; Ejsing, C.S.; Carvalho, P. Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. Elife 2013, 2, e00953. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-F.; Gu, A.-D.; Liang, L.; Li, Y.; Gong, T.; Chen, J.-J.; Chen, T.-J.; Yang, J.L.; Ping Zhu, P. Construction and optimization of microbial cell factories for sustainable production of bioactive dammarenediol-II glucosides. Green Chem. 2019, 21, 3286–3299. [Google Scholar] [CrossRef] [Green Version]
- Jensen, E.D.; Ferreira, R.; Jakociunas, T.; Arsovska, D.; Zhang, J.; Ding, L.; Smith, J.D.; David, F.; Nielsen, J.; Jensen, M.K.; et al. Transcriptional reprogramming in yeast using dCas9 and combinatorial gRNA strategies. Microb. Cell Fact. 2017, 16, 46. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.; Suresh, S.; Schlecht, U.; Wu, M.; Wagih, O.; Peltz, G.; Davis, R.W.; Steinmetz, L.M.; Parts, L.; Onge, R.P.S. Quantitative CRISPR interference screens in yeast identify chemical-genetic interactions and new rules for guide RNA design. Genome Biol. 2016, 17, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanegas, K.G.; Lehka, B.J.; Mortensen, U.H. SWITCH: A dynamic CRISPR tool for genome engineering and metabolic pathway control for cell factory construction in Saccharomyces cerevisiae. Microb. Cell Fact. 2017, 16, 25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.L.; Peng, Y.Z.; Liu, D.; Liu, H.; Cao, Y.X.; Li, B.Z.; Li, C.; Yuan, Y.J. Gene repression via multiplex gRNA strategy in Y. lipolytica. Microb. Cell Fact. 2018, 17, 62. [Google Scholar] [CrossRef]
- Polakowski, T.; Stahl, U.; Lang, C. Overexpression of a cytosolic hydroxymethylglutaryl-CoA reductase leads to squalene accumulation in yeast. Appl. Microbiol. Biotechnol. 1998, 49, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Thoma, R.; Schulz-Gasch, T.; D’Arcy, B.; Benz, J.; Aebi, J.D.; Dehmlow, H.; Hennig, M.; Stihle, M.; Ruf, A. Insight into steroid scaffold formation from the structure of human oxidosqualene cyclase. Nature 2004, 432, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.-L.; Zhao, S.-J.; Xu, L.-X.; Zhang, X.-Y. Heterologous expression of dammarenediol synthase gene in an engineered Saccharomyces cerevisiae. Lett. Appl. Microbiol. 2012, 55, 323–329. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Source | |
|---|---|---|
| CEN.PK2-1D | MATα, ura3-52, trp1-289, leu2-3,112, his3-Δ1, MAL2-8c, SUC2 | EUROSCARF |
| PPD-A1 | GPDpro-DS-CYC1ter, GPDpro-PgPPDS-CYC1ter, PGK1pro-PgCPR1-CYC1ter cassettes were inserted into CEN.PK2-1D | This study |
| PPD-A2 | GPDpro-tHMGR1-CYC1ter cassette was inserted into PPD-A1 | This study |
| PPD-A3 | TEF1pro-ERG1m-CYC1ter cassette was inserted into PPD-A2 | This study |
| PPD-A3-sgRNA1 | [GPDpro-dCas9-ADH1ter]-[SNR52pro-sgRNA1ERG7pro-SUP4ter] cassette was inserted into PPD-A3 | This study |
| PPD-A3-sgRNA2 | [GPDpro-dCas9-ADH1ter]-[SNR52pro-sgRNA2ERG7pro-SUP4ter] cassette was inserted into PPD-A3 | This study |
| PPD-A3-sgRNA3 | [GPDpro-dCas9-ADH1ter]-[SNR52pro-sgRNA3ERG7pro-SUP4ter] cassette was inserted into PPD-A3 | This study |
| PPD-A3-sgRNA4 | [GPDpro-dCas9-ADH1ter]-[SNR52pro-sgRNA4ERG7pro-SUP4ter] cassette was inserted into PPD-A3 | This study |
| PPD-A3-sgRNA5 | [GPDpro-dCas9-ADH1ter]-[SNR52pro-sgRNA5ERG7pro-SUP4ter] cassette was inserted into PPD-A3 | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, S.-H.; Baek, J.-I.; Jeon, B.-M.; Seo, J.-W.; Kim, M.-S.; Byun, J.-Y.; Park, S.-H.; Kim, S.-J.; Lee, J.-Y.; Lee, J.-H.; et al. CRISPRi-Guided Metabolic Flux Engineering for Enhanced Protopanaxadiol Production in Saccharomyces cerevisiae. Int. J. Mol. Sci. 2021, 22, 11836. https://doi.org/10.3390/ijms222111836
Lim S-H, Baek J-I, Jeon B-M, Seo J-W, Kim M-S, Byun J-Y, Park S-H, Kim S-J, Lee J-Y, Lee J-H, et al. CRISPRi-Guided Metabolic Flux Engineering for Enhanced Protopanaxadiol Production in Saccharomyces cerevisiae. International Journal of Molecular Sciences. 2021; 22(21):11836. https://doi.org/10.3390/ijms222111836
Chicago/Turabian StyleLim, Soo-Hwan, Jong-In Baek, Byeong-Min Jeon, Jung-Woo Seo, Min-Sung Kim, Ji-Young Byun, Soo-Hoon Park, Su-Jin Kim, Ju-Young Lee, Jun-Hyoung Lee, and et al. 2021. "CRISPRi-Guided Metabolic Flux Engineering for Enhanced Protopanaxadiol Production in Saccharomyces cerevisiae" International Journal of Molecular Sciences 22, no. 21: 11836. https://doi.org/10.3390/ijms222111836
APA StyleLim, S.-H., Baek, J.-I., Jeon, B.-M., Seo, J.-W., Kim, M.-S., Byun, J.-Y., Park, S.-H., Kim, S.-J., Lee, J.-Y., Lee, J.-H., & Kim, S.-C. (2021). CRISPRi-Guided Metabolic Flux Engineering for Enhanced Protopanaxadiol Production in Saccharomyces cerevisiae. International Journal of Molecular Sciences, 22(21), 11836. https://doi.org/10.3390/ijms222111836