
The Fibrotic Effects of TMAO on Human Renal Fibroblasts Is Mediated by NLRP3, Caspase-1 and the PERK/Akt/mTOR Pathway

Abstract
:1. Introduction
2. Results
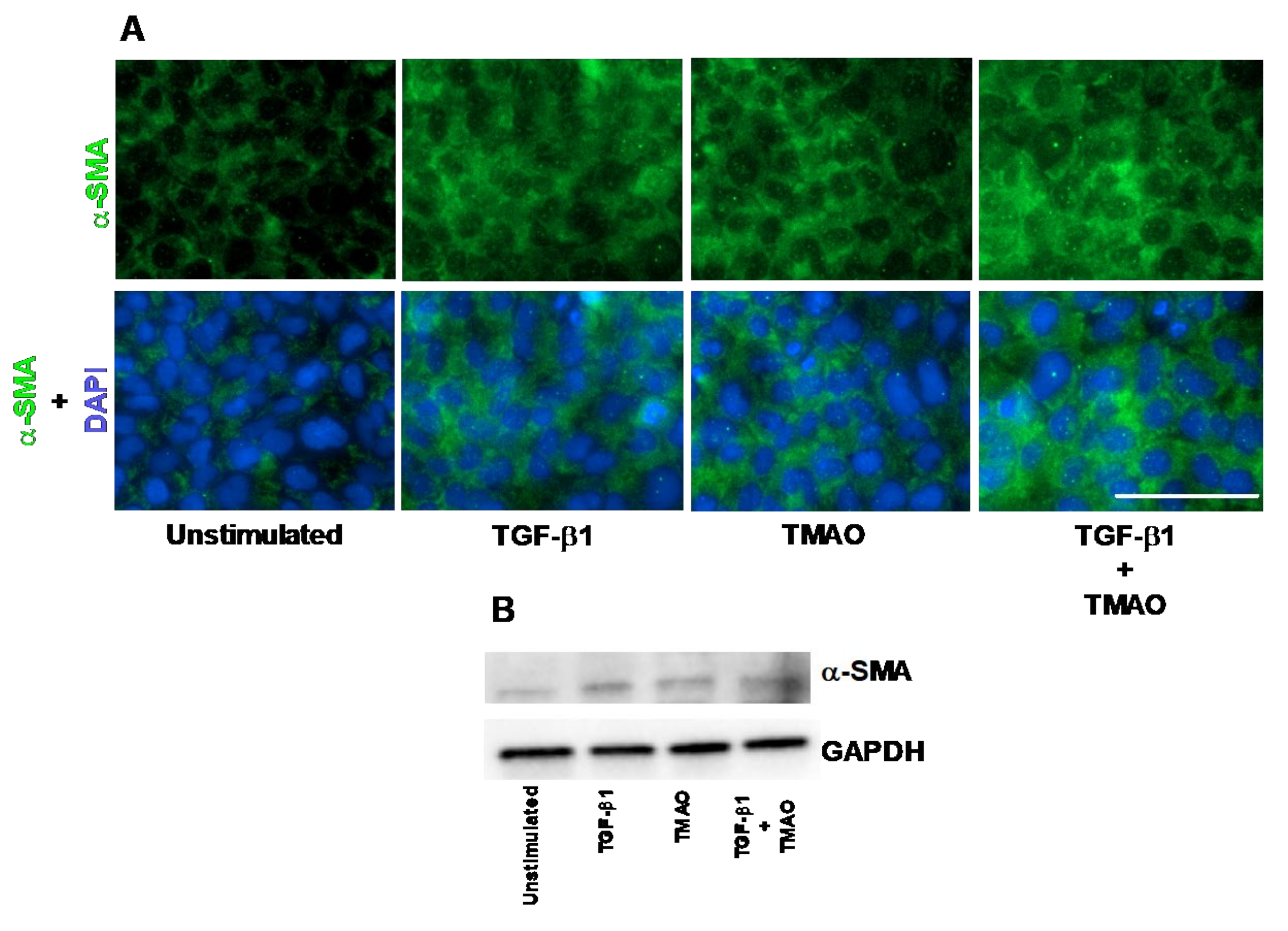
2.1. TMAO Induces Renal Fibroblast Activation
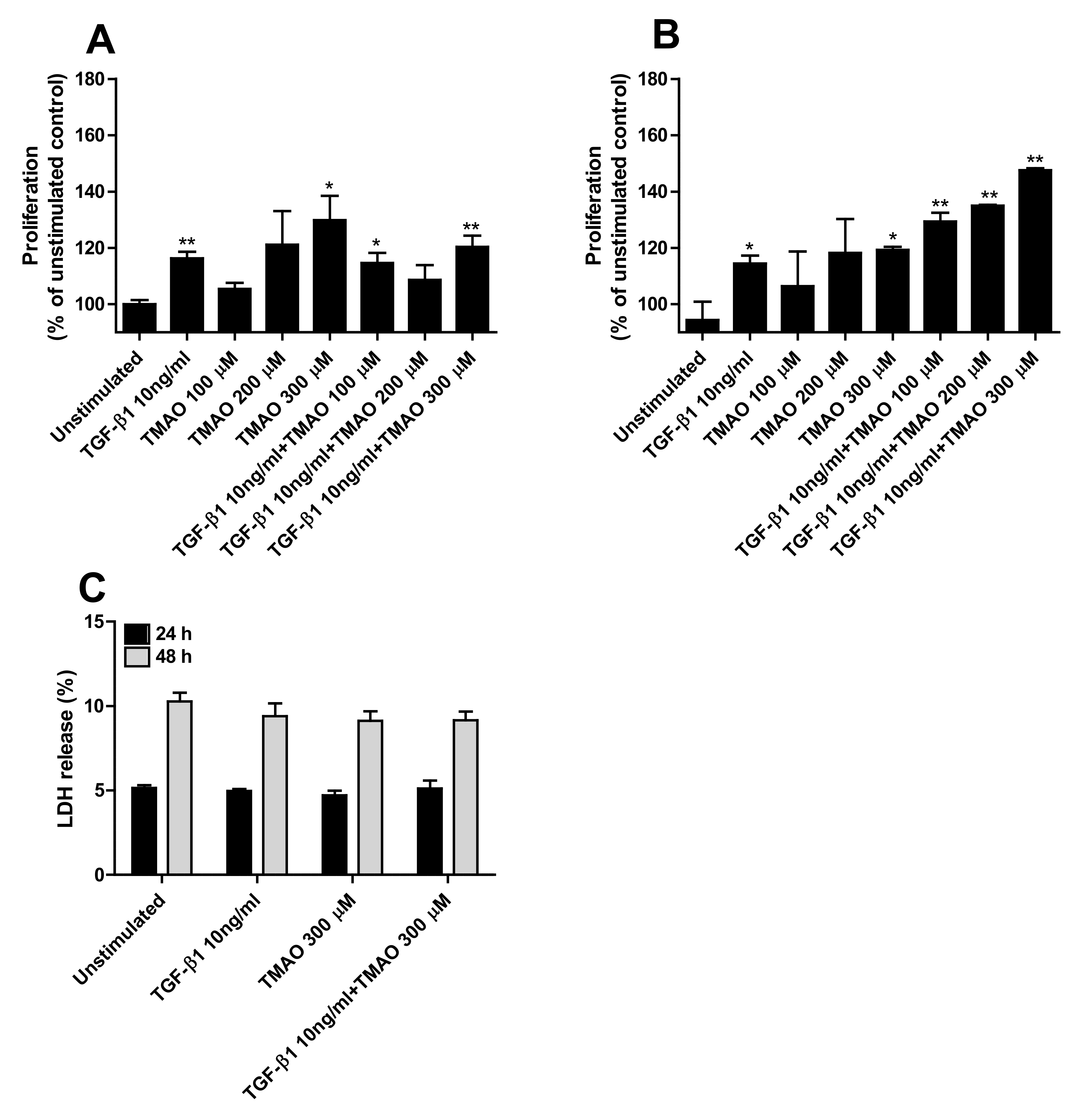
2.2. TMAO Promotes Renal Fibroblast Proliferation
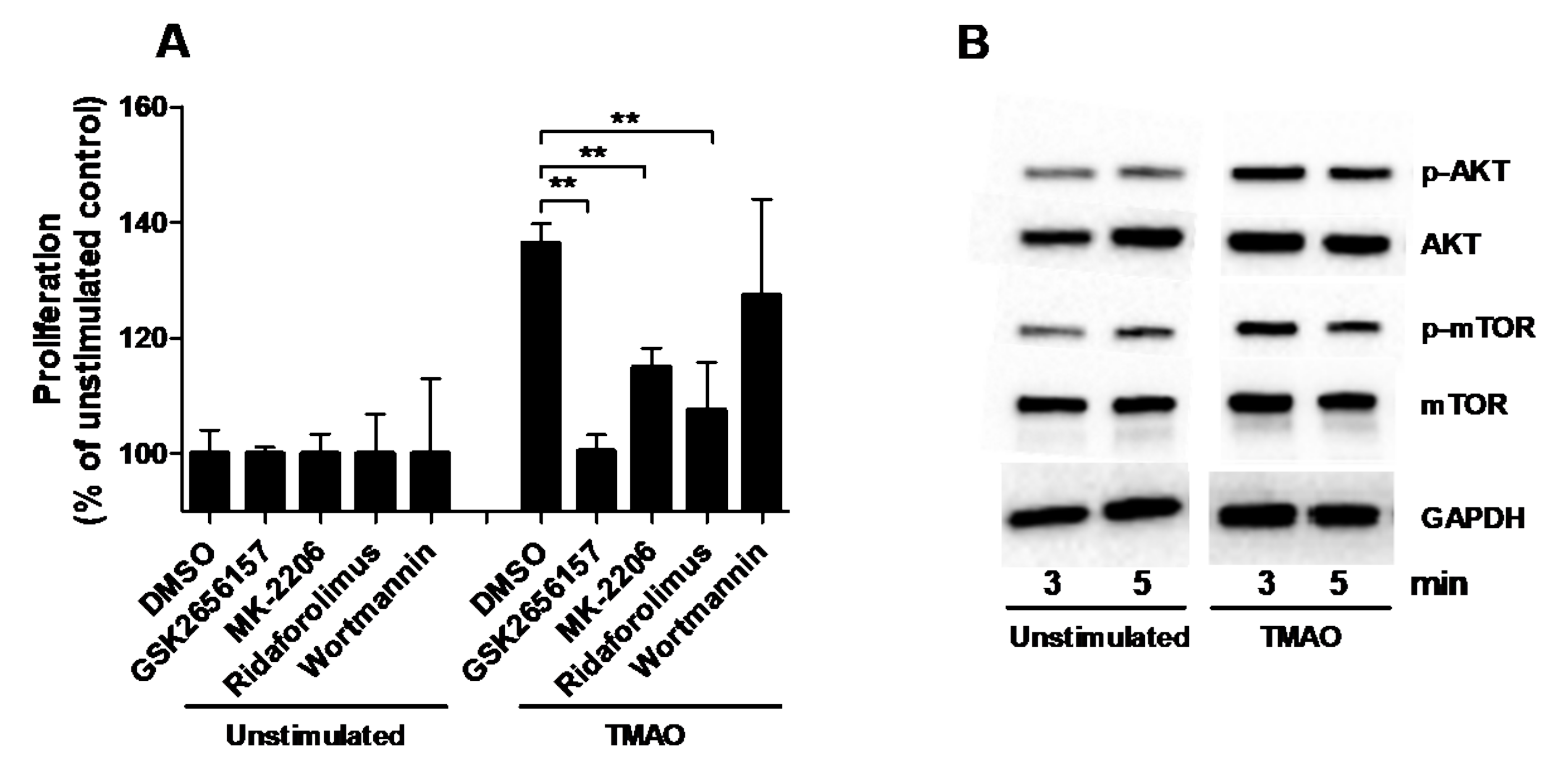
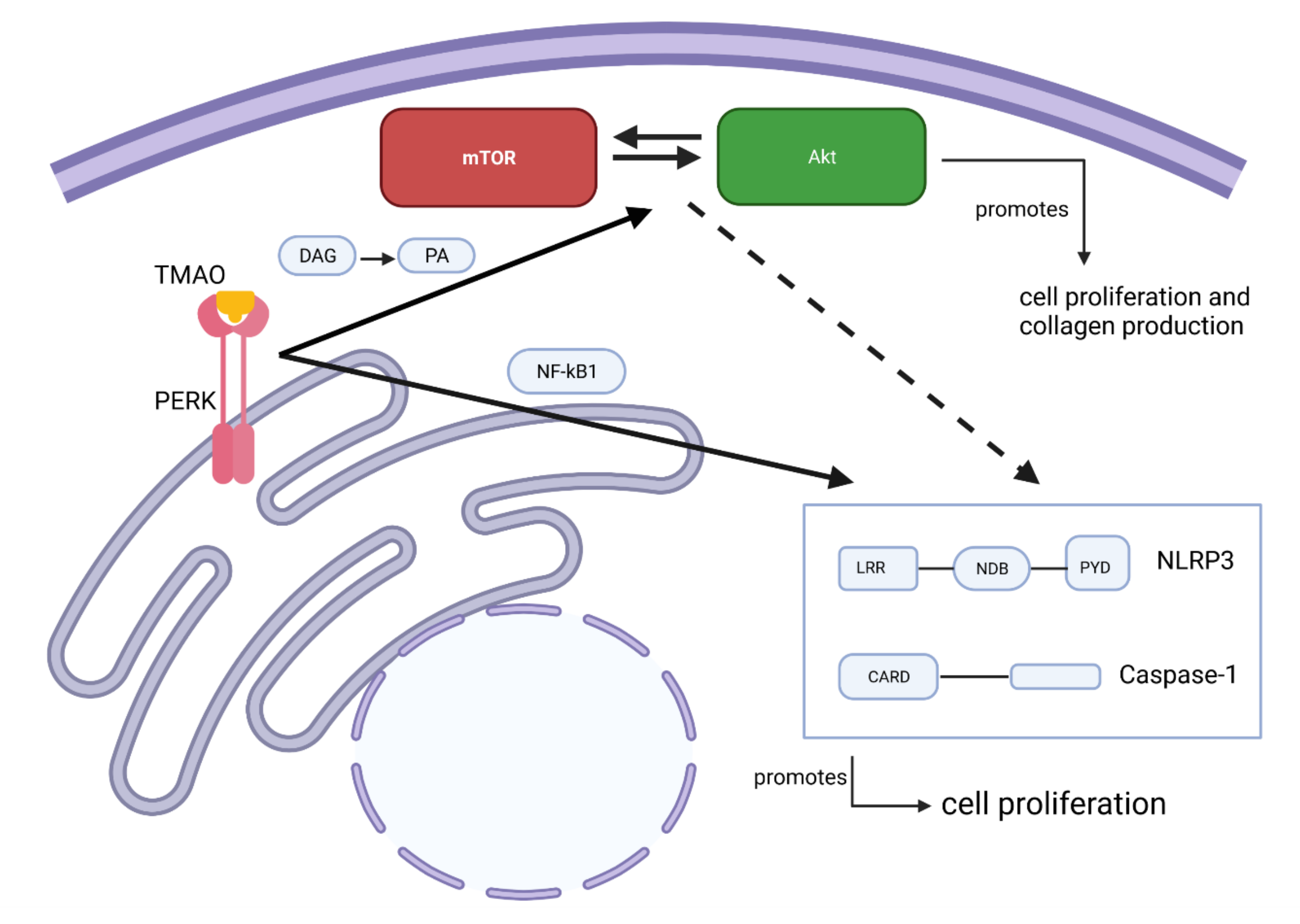
2.3. TMAO Increases the Proliferation of Renal Fibroblasts via the PERK/Akt/mTOR Pathway
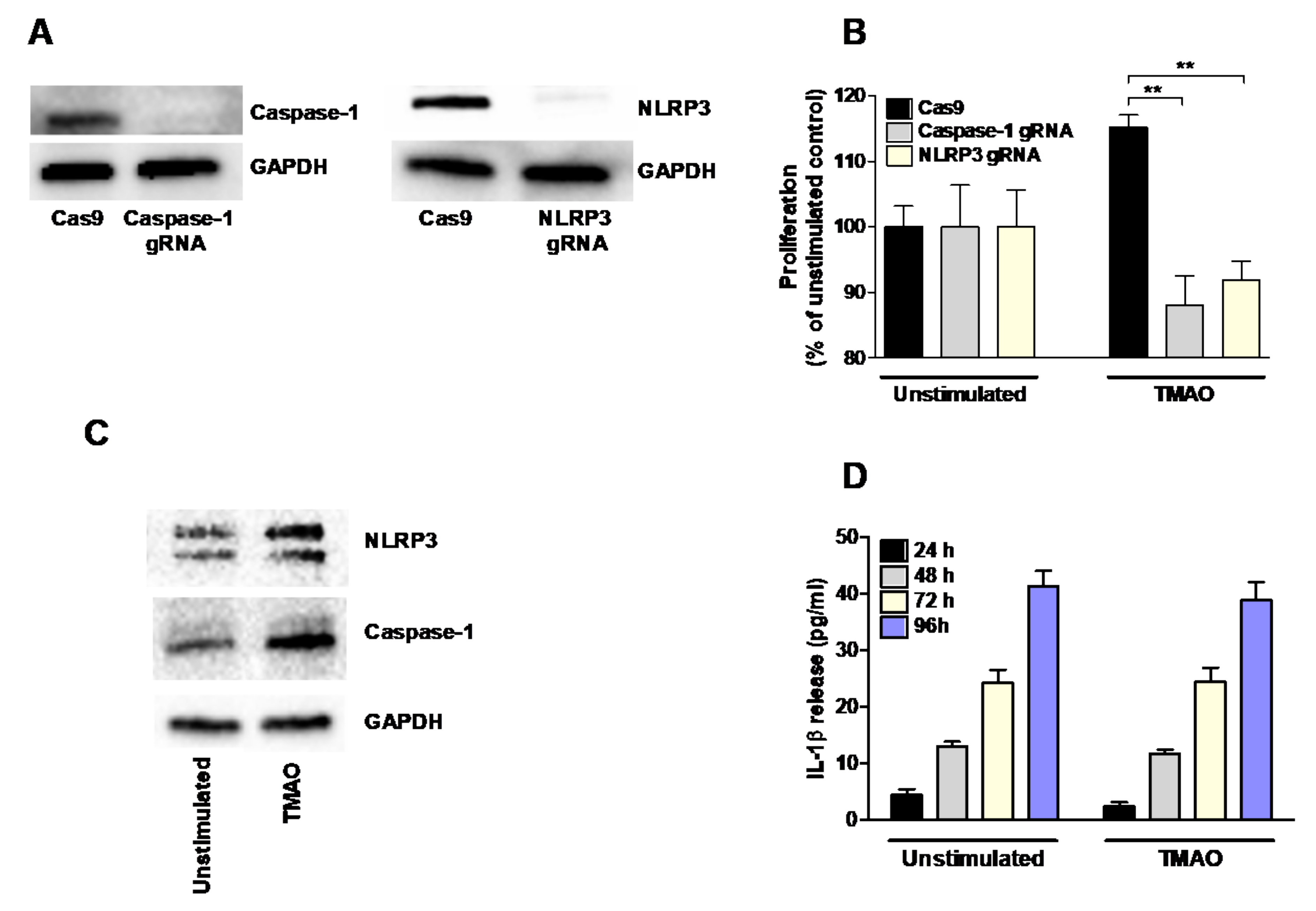
2.4. TMAO-Induced Proliferation of Renal Fibroblasts Is Mediated by NLRP3 and Caspase-1
2.5. TMAO Has no Effect on the Production of Fibronectin or TGF-β1 from Renal Fibroblasts
2.6. TMAO Increases Total Collagen Production via the Akt/mTOR Pathway
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. CRISPR/Cas9 Genome Editing of Renal Fibroblasts
4.3. Stimulation of Renal Fibroblasts
4.4. Immunofluorescence
4.5. Crystal Violet Proliferation Assay
4.6. Western Blot Analysis
4.7. Measurement of IL-1β, Fibronectin, TGF-β1 Release and Cell Viability
4.8. Quantification of Total Collagen Production
4.9. RNA Isolation and Real-Time RT-PCR
4.10. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janeiro, M.H.; Ramírez, M.J.; Milagro, F.I.; Martínez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018, 10, 1398. [Google Scholar] [CrossRef] [Green Version]
- Fennema, D.; Phillips, I.R.; Shephard, E.A. Trimethylamine and Trimethylamine N-Oxide, a Flavin-Containing Monooxygenase 3 (FMO3)-Mediated Host-Microbiome Metabolic Axis Implicated in Health and Disease. Drug Metab. Dispos. 2016, 44, 1839–1850. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Pazos, I.M.; Gai, F. Microscopic insights into the protein-stabilizing effect of trimethylamine N-oxide (TMAO). Proc. Natl. Acad. Sci. USA 2014, 111, 8476–8481. [Google Scholar] [CrossRef] [Green Version]
- Ufnal, M.; Zadlo, A.; Ostaszewski, R. TMAO: A small molecule of great expectations. Nutrition 2015, 31, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, Q.; Jiang, H. Gut microbiota in atherosclerosis: Focus on trimethylamine N-oxide. Apmis 2020, 128, 353–366. [Google Scholar] [CrossRef] [Green Version]
- Senthong, V.; Wang, Z.; Fan, Y.; Wu, Y.; Hazen, S.L.; Tang, W.H. Trimethylamine N-Oxide and Mortality Risk in Patients with Peripheral Artery Disease. J. Am. Heart Assoc. 2016, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.B.; Morse, B.L.; Djurdjev, O.; Tang, M.; Muirhead, N.; Barrett, B.; Holmes, D.T.; Madore, F.; Clase, C.M.; Rigatto, C.; et al. Advanced chronic kidney disease populations have elevated trimethylamine N-oxide levels associated with increased cardiovascular events. Kidney Int. 2016, 89, 1144–1152. [Google Scholar] [CrossRef]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. 2016, 27, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Manor, O.; Zubair, N.; Conomos, M.P.; Xu, X.; Rohwer, J.E.; Krafft, C.E.; Lovejoy, J.C.; Magis, A. A Multi-omic Association Study of Trimethylamine N-Oxide. Cell Rep. 2018, 24, 935–946. [Google Scholar] [CrossRef] [Green Version]
- Missailidis, C.; Hällqvist, J.; Qureshi, A.R.; Barany, P.; Heimbürger, O.; Lindholm, B.; Stenvinkel, P.; Bergman, P. Serum Trimethylamine-N-Oxide Is Strongly Related to Renal Function and Predicts Outcome in Chronic Kidney Disease. PLoS ONE 2016, 11, e0141738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, J.D.; Lee, J.A.; Lee, H.A.; Sadler, P.J.; Wilkie, D.R.; Woodham, R.H. Nuclear magnetic resonance studies of blood plasma and urine from subjects with chronic renal failure: Identification of trimethylamine-N-oxide. Biochim. Biophys. Acta 1991, 1096, 101–107. [Google Scholar] [CrossRef]
- Doucet, C.; Dutheil, D.; Petit, I.; Zhang, K.; Eugene, M.; Touchard, G.; Wahl, A.; Seguin, F.; Milinkevitch, S.; Hauet, T.; et al. Influence of colloid, preservation medium and trimetazidine on renal medulla injury. Biochim. Biophys. Acta 2004, 1673, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Hauet, T.; Baumert, H.; Gibelin, H.; Godart, C.; Carretier, M.; Eugene, M. Citrate, acetate and renal medullary osmolyte excretion in urine as predictor of renal changes after cold ischaemia and transplantation. Clin. Chem. Lab. Med. 2000, 38, 1093–1098. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Tang, Z.; You, L.; Wu, Y.; Liu, J.; Xue, J. Trimethylamine-N-oxide is an independent risk factor for hospitalization events in patients receiving maintenance hemodialysis. Ren. Fail. 2020, 42, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Miikeda, A.; Zuckerman, J.; Jia, X.; Charugundla, S.; Zhou, Z.; Kaczor-Urbanowicz, K.E.; Magyar, C.; Guo, F.; Wang, Z.; et al. Inhibition of microbiota-dependent TMAO production attenuates chronic kidney disease in mice. Sci. Rep. 2021, 11, 518. [Google Scholar] [CrossRef]
- Meran, S.; Steadman, R. Fibroblasts and myofibroblasts in renal fibrosis. Int. J. Exp. Pathol. 2011, 92, 158–167. [Google Scholar] [CrossRef]
- Nakagawa, S.; Nishihara, K.; Miyata, H.; Shinke, H.; Tomita, E.; Kajiwara, M.; Matsubara, T.; Iehara, N.; Igarashi, Y.; Yamada, H.; et al. Molecular Markers of Tubulointerstitial Fibrosis and Tubular Cell Damage in Patients with Chronic Kidney Disease. PLoS ONE 2015, 10, e0136994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artlett, C.M. The Role of the NLRP3 Inflammasome in Fibrosis. Open Rheumatol. J. 2012, 6, 80–86. [Google Scholar] [CrossRef]
- Artlett, C.M.; Thacker, J.D. Molecular activation of the NLRP3 Inflammasome in fibrosis: Common threads linking divergent fibrogenic diseases. Antioxid. Redox Signal. 2015, 22, 1162–1175. [Google Scholar] [CrossRef]
- Coca, S.G.; Yalavarthy, R.; Concato, J.; Parikh, C.R. Biomarkers for the diagnosis and risk stratification of acute kidney injury: A systematic review. Kidney Int. 2008, 73, 1008–1016. [Google Scholar] [CrossRef] [Green Version]
- Akcay, A.; Nguyen, Q.; Edelstein, C.L. Mediators of inflammation in acute kidney injury. Mediat. Inflamm. 2009, 2009, 137072. [Google Scholar] [CrossRef]
- Ram, C.; Jha, A.K.; Ghosh, A.; Gairola, S.; Syed, A.M.; Murty, U.S.; Naidu, V.; Sahu, B.D. Targeting NLRP3 inflammasome as a promising approach for treatment of diabetic nephropathy: Preclinical evidences with therapeutic approaches. Eur. J. Pharmacol. 2020, 885, 173503. [Google Scholar] [CrossRef]
- Qiu, Y.Y.; Tang, L.Q. Roles of the NLRP3 inflammasome in the pathogenesis of diabetic nephropathy. Pharmacol. Res. 2016, 114, 251–264. [Google Scholar] [CrossRef]
- Gao, M.; Sotomayor, M.; Villa, E.; Lee, E.H.; Schulten, K. Molecular mechanisms of cellular mechanics. Phys. Chem. Chem. Phys. 2006, 8, 3692–3706. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Zheng, B.; Liu, N.; Liu, J.; Liu, W.; Huang, X.; Zeng, X.; Chen, L.; Li, Z.; Ouyang, D. Trimethylamine N-Oxide Exacerbates Renal Inflammation and Fibrosis in Rats With Diabetic Kidney Disease. Front. Physiol. 2021, 12, 682482. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Buffa, J.A.; Roberts, A.B.; Sangwan, N.; Skye, S.M.; Li, L.; Ho, K.J.; Varga, J.; DiDonato, J.A.; Tang, W.W.; et al. Targeted Inhibition of Gut Microbial Trimethylamine N-Oxide Production Reduces Renal Tubulointerstitial Fibrosis and Functional Impairment in a Murine Model of Chronic Kidney Disease. Arter. Thromb. Vasc. Biol. 2020, 40, 1239–1255. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Geng, J.; Zhao, J.; Ni, Q.; Zhao, C.; Zheng, Y.; Chen, X.; Wang, L. Trimethylamine N-Oxide Exacerbates Cardiac Fibrosis via Activating the NLRP3 Inflammasome. Front. Physiol. 2019, 10, 866. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Hartupee, J.; Mann, D.L. Role of inflammatory cells in fibroblast activation. J. Mol. Cell Cardiol. 2016, 93, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. TGF-beta/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Desmouliere, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, D.G.K.; Boesby, L.; Nielsen, S.H.; Tepel, M.; Birot, S.; Karsdal, M.A.; Kamper, A.-L.; Genovese, F. Collagen turnover profiles in chronic kidney disease. Sci. Rep. 2019, 9, 16062. [Google Scholar] [CrossRef] [PubMed]
- Genovese, F.; Rasmussen, D.G.K.; Karsdal, M.A.; Jesky, M.; Ferro, C.; Fenton, A.; Cockwell, P. Imbalanced turnover of collagen type III is associated with disease progression and mortality in high-risk chronic kidney disease patients. Clin. Kidney J. 2021, 14, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Na, L.; Li, Y.; Chen, L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020, 10, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafari, M.; Ghadami, E.; Dadkhah, T.; Akhavan-Niaki, H. PI3k/AKT signaling pathway: Erythropoiesis and beyond. J. Cell Physiol. 2019, 234, 2373–2385. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.S.; Ramasamy, T.S.; Murphy, N.; Holt, M.K.; Czapiewski, R.; Wei, S.K.; Cui, W. PI3K/mTORC2 regulates TGF-β/Activin signalling by modulating Smad2/3 activity via linker phosphorylation. Nat. Commun. 2015, 6, 7212. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Su, P.; Wang, L.; Chen, J.; Zimmermann, M.; Genbacev, O.; Afonja, O.; Horne, M.C.; Tanaka, T.; Duan, E.; et al. mTOR supports long-term self-renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 7840–7845. [Google Scholar] [CrossRef] [Green Version]
- Paling, N.R.; Wheadon, H.; Bone, H.K.; Welham, M.J. Regulation of embryonic stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J. Biol. Chem. 2004, 279, 48063–48070. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.M.; Reynolds, D.; Cliff, T.; Ohtsuka, S.; Mattheyses, A.L.; Sun, Y.; Menendez, L.; Kulik, M.; Dalton, S. Signaling network crosstalk in human pluripotent cells: A Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem Cell 2012, 10, 312–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.; Silveira, G.G.; Soave, D.F.; Costa, J.P.O.; Silva, A.R. The Role of the LY294002—A Non-Selective Inhibitor of Phosphatidylinositol 3-Kinase (PI3K) Pathway- in Cell Survival and Proliferation in Cell Line SCC-25. Asian Pac. J. Cancer Prev. 2019, 20, 3377–3383. [Google Scholar] [CrossRef]
- Bavelloni, A.; Focaccia, E.; Piazzi, M.; Orsini, A.; Ramazzotti, G.; Cocco, L.; Blalock, W.; Faenza, I. Therapeutic potential of nvp-bkm120 in human osteosarcomas cells. J. Cell Physiol. 2019, 234, 10907–10917. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Becker, K.P.; Mekhail, T.; Chowdhary, S.A.; Eakle, J.F.; Wright, D.; Langdon, R.M.; Yost, K.J.; Padula, G.D.A.; West-Osterfield, K.; et al. Phase I/II study of bevacizumab with BKM120, an oral PI3K inhibitor, in patients with refractory solid tumors (phase I) and relapsed/refractory glioblastoma (phase II). J. Neurooncol. 2019, 144, 303–311. [Google Scholar] [CrossRef]
- Weinberg, M.A. RES-529: A PI3K/AKT/mTOR pathway inhibitor that dissociates the mTORC1 and mTORC2 complexes. Anticancer Drugs 2016, 27, 475–487. [Google Scholar] [CrossRef]
- Kaley, T.J.; Panageas, S.K.; Mellinghoff, I.K.; Nolan, C.; Gavrilovic, I.T.; DeAngelis, L.M.; Abrey, L.E.; Holland, E.C.; Lassman, A.B. Phase II trial of an AKT inhibitor (perifosine) for recurrent glioblastoma. J. Neurooncol. 2019, 144, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Fan, X.; Li, Q.; Wang, Y.; Chen, X.; Xiao, P.; Passerini, A.; Simon, S.I.; Sun, C. IRF-1 mediates the suppressive effects of mTOR inhibition on arterial endothelium. J. Mol. Cell Cardiol. 2020, 140, 30–41. [Google Scholar] [CrossRef]
- Méndez-Gómez, M.; Castro-Mercado, E.; Peña-Uribe, C.A.; Reyes-de la Cruz, H.; López-Bucio, J.; García-Pineda, E. Target Of Rapamycin signaling plays a role in Arabidopsis growth promotion by Azospirillum brasilense Sp245. Plant Sci. 2020, 293, 110416. [Google Scholar] [CrossRef] [PubMed]
- Brakemeier, S.; Arns, W.; Lehner, F.; Witzke, O.; Vonend, O.; Sommerer, C.; Mühlfeld, A.; Rath, T.; Schuhmann, R.; Zukunft, B.; et al. Everolimus in de novo kidney transplant recipients participating in the Eurotransplant senior program: Results of a prospective randomized multicenter study (SENATOR). PLoS ONE 2019, 14, e0222730. [Google Scholar] [CrossRef] [PubMed]
- Schötz, U.; Balzer, V.; Brandt, F.W.; Ziemann, F.; Subtil, F.S.B.; Rieckmann, T.; Köcher, S.; Engenhart-Cabillic, R.; Dikomey, E.; Wittig, A.; et al. Dual PI3K/mTOR Inhibitor NVP-BEZ235 Enhances Radiosensitivity of Head and Neck Squamous Cell Carcinoma (HNSCC) Cell Lines Due to Suppressed Double-Strand Break (DSB) Repair by Non-Homologous End Joining. Cancers 2020, 12, 467. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Sun, Y.; Chen, D.; Zhang, Y.; Yan, L.; Li, X.; Wang, J. Negative regulation of PI3K/AKT/mTOR axis regulates fibroblast proliferation, apoptosis and autophagy play a vital role in triptolide-induced epidural fibrosis reduction. Eur. J. Pharmacol. 2019, 864, 172724. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Dai, J.; Jiao, R.; Jiang, Q.; Wang, J. Homoharringtonine inhibits fibroblasts proliferation, extracellular matrix production and reduces surgery-induced knee arthrofibrosis via PI3K/AKT/mTOR pathway-mediated apoptosis. J. Orthop. Surg. Res. 2021, 16, 9. [Google Scholar] [CrossRef]
- Xing, W.; Guo, W.; Zou, C.H.; Fu, T.T.; Li, X.Y.; Zhu, M.; Qi, J.-H.; Song, J.; Dong, C.-H.; Li, Z.; et al. Acemannan accelerates cell proliferation and skin wound healing through AKT/mTOR signaling pathway. J. Dermatol. Sci. 2015, 79, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Henderson, A.; Petriello, M.C.; Romano, K.A.; Gearing, M.; Miao, J.; Schell, M.; Sandoval-Espinola, W.J.; Tao, J.; Sha, B.; et al. Trimethylamine N-oxide binds and activates PERK to promote metabolic dysfunction. Cell Metab. 2019, 30, 1141–1151.e5. [Google Scholar] [CrossRef]
- McQuiston, A.; Diehl, J.A. Recent insights into PERK-dependent signaling from the stressed endoplasmic reticulum. F1000Research 2017, 6, 1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anjum, R.; Blenis, J. The RSK family of kinases: Emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 2008, 9, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Toschi, A.; Lee, E.; Xu, L.; Garcia, A.; Gadir, N.; Foster, D.A. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: Competition with rapamycin. Mol. Cell Biol. 2009, 29, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Gallego, P.; Castejón-Vega, B.; Del Campo, J.A.; Cordero, M.D. The Absence of NLRP3-inflammasome Modulates Hepatic Fibrosis Progression, Lipid Metabolism, and Inflammation in KO NLRP3 Mice during Aging. Cells 2020, 9, 2148. [Google Scholar] [CrossRef]
- Boini, K.M.; Hussain, T.; Li, P.L.; Koka, S. Trimethylamine-N-Oxide Instigates NLRP3 Inflammasome Activation and Endothelial Dysfunction. Cell Physiol. Biochem. 2017, 44, 152–162. [Google Scholar] [CrossRef]
- Zhao, W.; Shi, C.S.; Harrison, K.; Hwang, I.Y.; Nabar, N.R.; Wang, M.; Kehrl, J.H. AKT Regulates NLRP3 Inflammasome Activation by Phosphorylating NLRP3 Serine 5. J. Immunol. 2020, 205, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, X.; Pan, Y.; Shi, G.; Ren, J.; Fan, H.; Dou, H.; Hou, Y. mTOR regulates NLRP3 inflammasome activation via reactive oxygen species in murine lupus. Acta Biochim. Biophys. Sin. 2018, 50, 888–896. [Google Scholar] [CrossRef] [Green Version]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 2010, 21, 1732–1744. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, Z. Effect and Regulation of the NLRP3 Inflammasome During Renal Fibrosis. Front. Cell Dev. Biol. 2019, 7, 379. [Google Scholar] [CrossRef]
- Zhou, Y.; Tong, Z.; Jiang, S.; Zheng, W.; Zhao, J.; Zhou, X. The Roles of Endoplasmic Reticulum in NLRP3 Inflammasome Activation. Cells 2020, 9, 1219. [Google Scholar] [CrossRef]
- Kim, S.; Joe, Y.; Jeong, S.O.; Zheng, M.; Back, S.H.; Park, S.W.; Ryter, S.W.; Chung, H.T. Endoplasmic reticulum stress is sufficient for the induction of IL-1β production via activation of the NF-κB and inflammasome pathways. Innate Immun. 2014, 20, 799–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.Y.; Rho, H.S.; Kim, A.; Kim, T.H.; Jang, K.; Jun, D.W.; Kim, J.W.; Kim, B.; Kim, S.G. FXR Inhibits Endoplasmic Reticulum Stress-Induced NLRP3 Inflammasome in Hepatocytes and Ameliorates Liver Injury. Cell Rep. 2018, 24, 2985–2999. [Google Scholar] [CrossRef] [Green Version]
- Muller, G.A.; Frank, J.; Rodemann, H.P.; Engler-Blum, G. Human renal fibroblast cell lines (tFKIF and tNKF) are new tools to investigate pathophysiologic mechanisms of renal interstitial fibrosis. Exp. Nephrol. 1995, 3, 127–133. [Google Scholar] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Demirel, I.; Persson, A.; Brauner, A.; Särndahl, E.; Kruse, R.; Persson, K. Activation of the NLRP3 Inflammasome Pathway by Uropathogenic Escherichia coli Is Virulence Factor-Dependent and Influences Colonization of Bladder Epithelial Cells. Front. Cell Infect. Microbiol. 2018, 8, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Oligonucleotide Sequences (5′–3′) |
|---|---|
| Fibronectin | F: ACAACACCGAGGTGACTGAGAC R: GGACACAACGATGCTTCCTGAG |
| Collagen 1 | F: GATTCCCTGGACCTAAAGGTGC R: AGCCTCTCCATCTTTGCCAGCA |
| Collagen 3 | F: TGGTCTGCAAGGAATGCCTGGA R: TCTTTCCCTGGGACACCATCAG |
| Collagen 4 | F: TGTTGACGGCTTACCTGGAGAC R: GGTAGACCAACTCCAGGCTCTC |
| GAPDH | F: GTCTCCTCTGACTTCAACAGCG R: ACCACCCTGTTGCTGTAGCCAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapetanaki, S.; Kumawat, A.K.; Persson, K.; Demirel, I. The Fibrotic Effects of TMAO on Human Renal Fibroblasts Is Mediated by NLRP3, Caspase-1 and the PERK/Akt/mTOR Pathway. Int. J. Mol. Sci. 2021, 22, 11864. https://doi.org/10.3390/ijms222111864
Kapetanaki S, Kumawat AK, Persson K, Demirel I. The Fibrotic Effects of TMAO on Human Renal Fibroblasts Is Mediated by NLRP3, Caspase-1 and the PERK/Akt/mTOR Pathway. International Journal of Molecular Sciences. 2021; 22(21):11864. https://doi.org/10.3390/ijms222111864
Chicago/Turabian StyleKapetanaki, Stefania, Ashok Kumar Kumawat, Katarina Persson, and Isak Demirel. 2021. "The Fibrotic Effects of TMAO on Human Renal Fibroblasts Is Mediated by NLRP3, Caspase-1 and the PERK/Akt/mTOR Pathway" International Journal of Molecular Sciences 22, no. 21: 11864. https://doi.org/10.3390/ijms222111864