
Spike Formation Is a Turning Point Determining Wheat Root Microbiome Abundance, Structures and Functions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
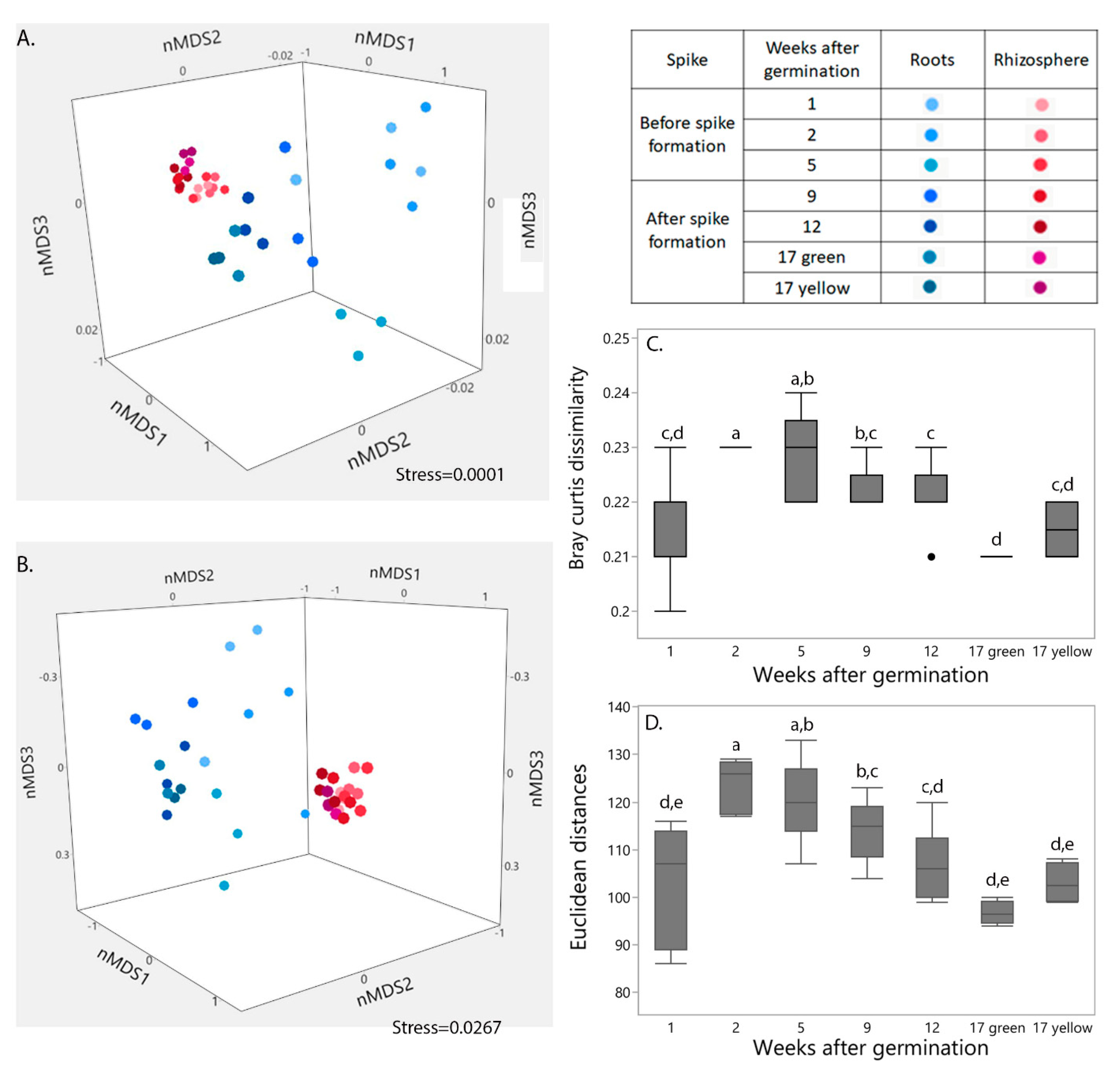
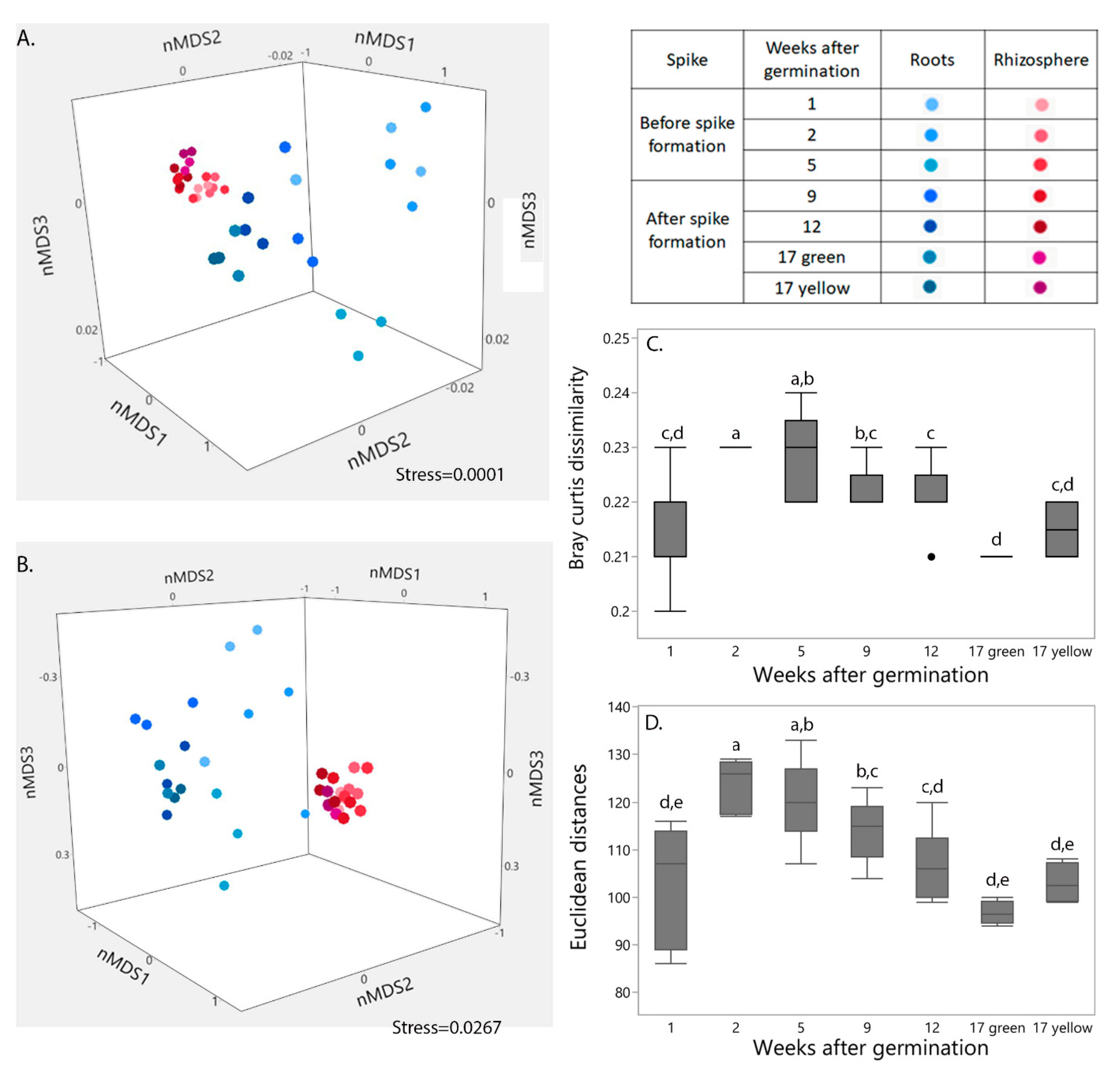
2.1. Influence of Niche and Plant Developmental Stage on Bacterial Community Structure and Functional Profile
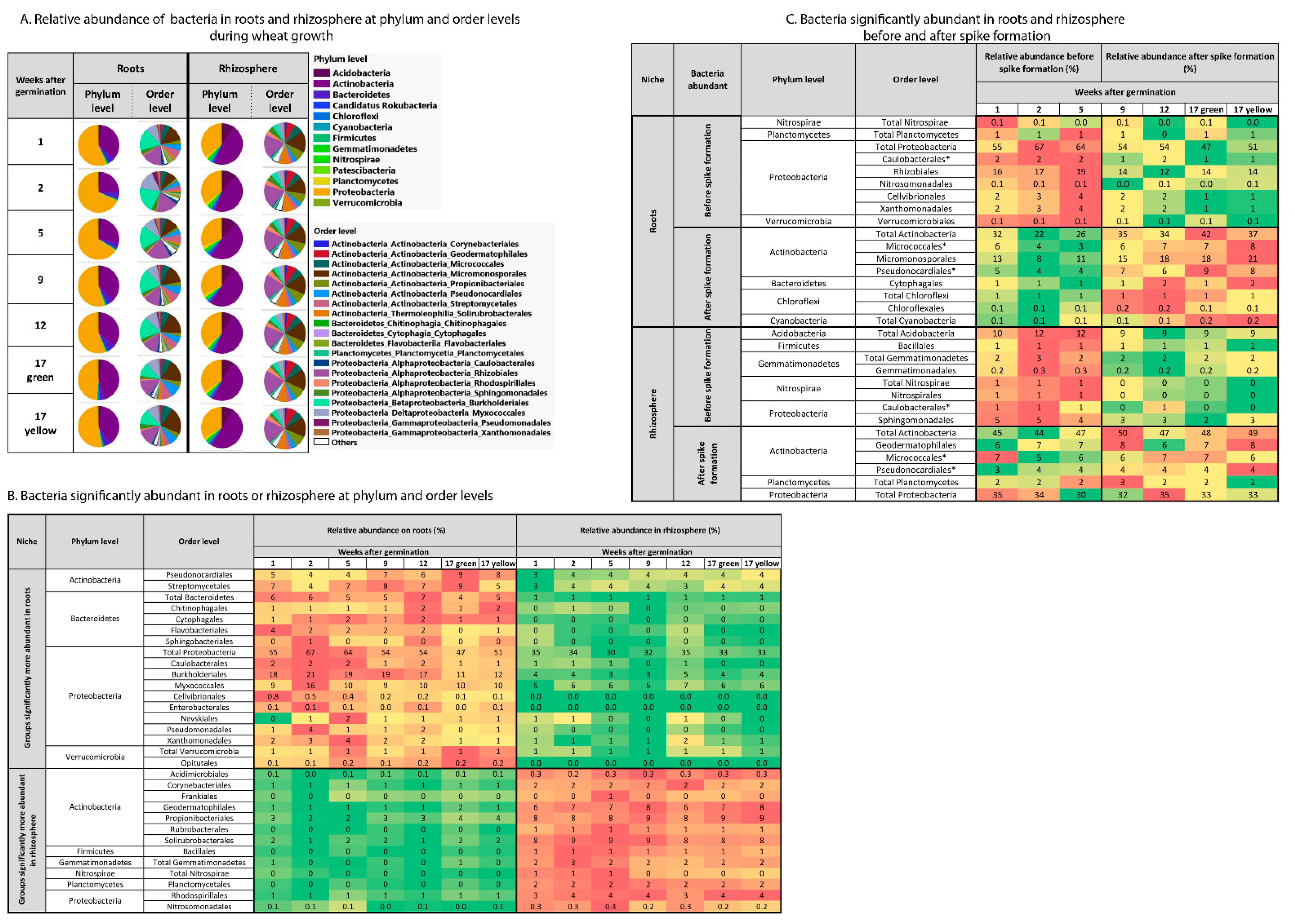
2.2. Influence of Niche and Spike Formation on Bacterial Community Composition
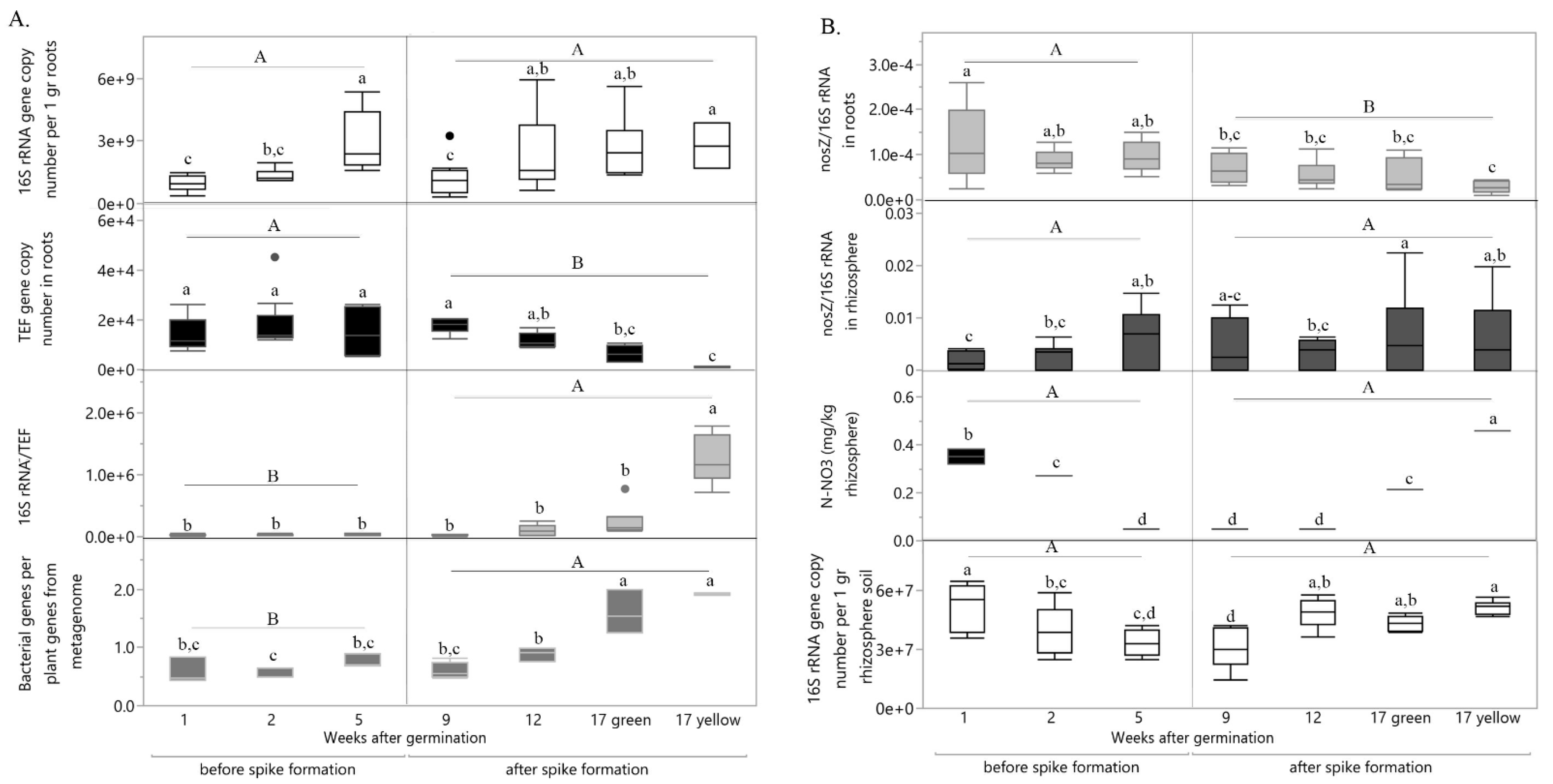
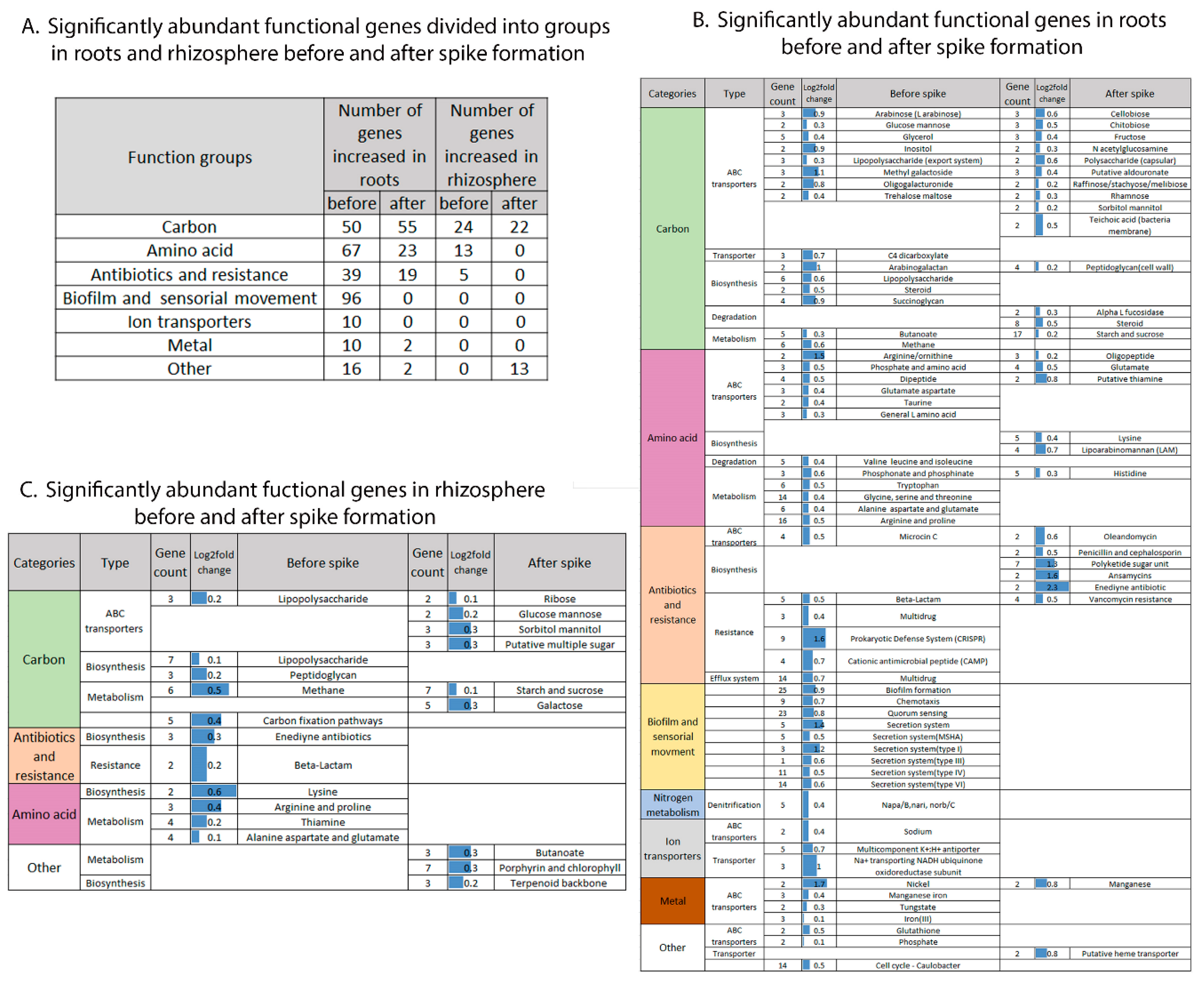
2.3. The Effect of Niche and Spike Formation on Specific Root-Associated and Rhizosphere Gene Functions
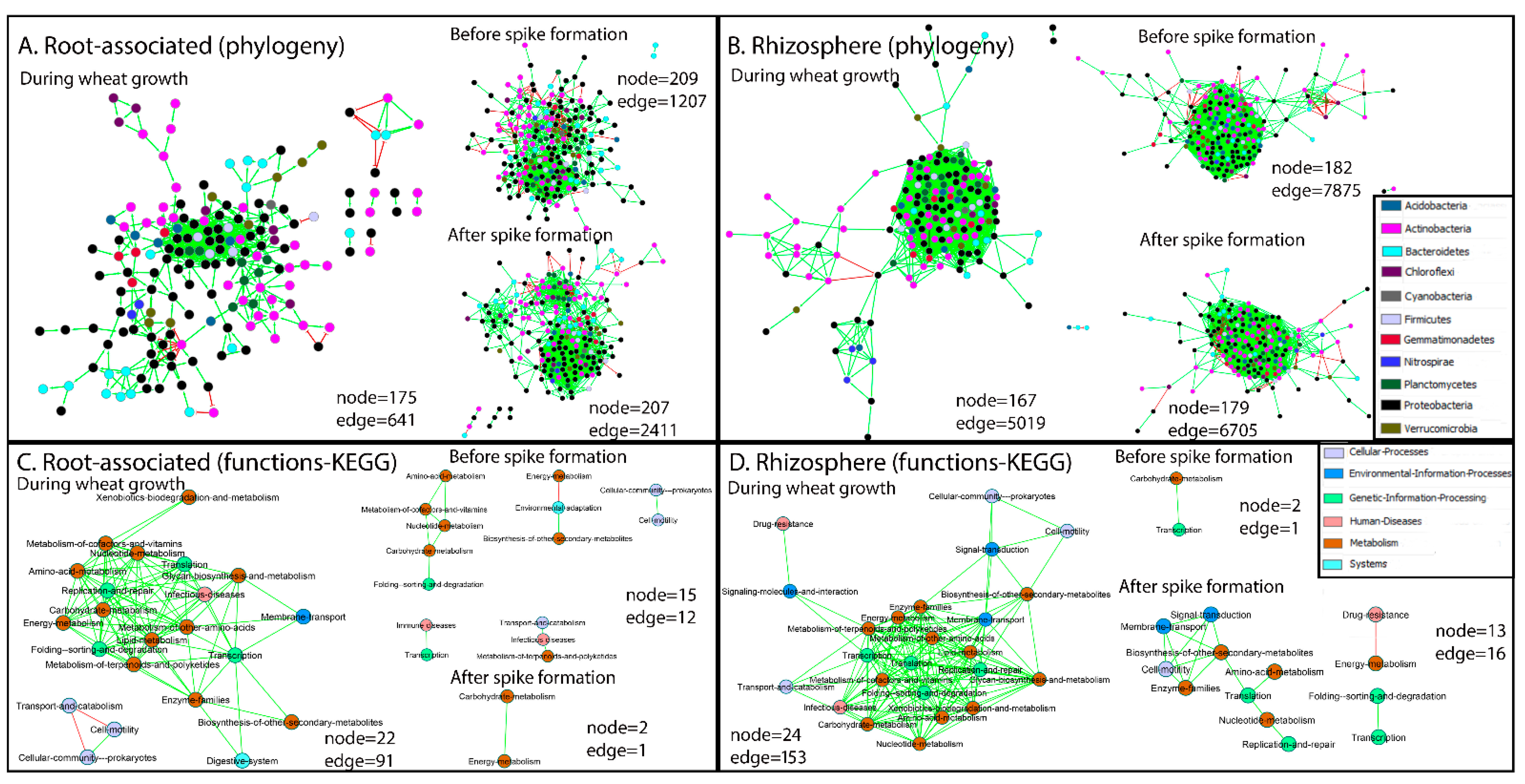
2.4. Links between Taxonomy and Significantly Changed Functions
3. Discussion
4. Materials and Methods
4.1. Soil and Root Sampling
4.2. Rhizosphere and Root DNA Extraction for Sequencing and qPCR
4.3. Generation of qPCR Plasmid Standards
4.4. Assessment of Gene Copy Numbers by qPCR
4.5. Shotgun Sequencing
4.6. Data Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berg, G.; Smalla, K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol. Ecol. 2009, 68, 1–13. [Google Scholar] [CrossRef] [Green Version]
- İnceoğlu, Ö.; Falcão Salles, J.; van Elsas, J.D. Soil and Cultivar Type Shape the Bacterial Community in the Potato Rhizosphere. Microb. Ecol. 2012, 63, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; Van Der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, D.; Garrido-Oter, R.; Münch, P.C.; Weiman, A.; Dröge, J.; Pan, Y.; McHardy, A.C.; Schulze-Lefert, P. Structure and Function of the Bacterial Root Microbiota in Wild and Domesticated Barley Resource Structure and Function of the Bacterial Root Microbiota in Wild and Domesticated Barley. Cell Host Microbe 2015, 17, 392–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.L.; Nguyen, C.; Finlay, R.D. Carbon flow in the rhizosphere: Carbon trading at the soil–root interface. Plant Soil 2009, 321, 5–33. [Google Scholar] [CrossRef]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. Oxford Academic. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef] [PubMed]
- Bertin, C.; Yang, X.; Weston, L.A. The role of root exudates and allelochemicals in the rhizosphere. Plant Soil. 2003, 256, 67–83. [Google Scholar] [CrossRef]
- Bais, H.P.; Weir, T.L.; Perry, L.G.; Gilroy, S.; Vivanco, J.M. The role of root exudates in rhizosphere interactions with plants and other organisms. Annu. Rev. Plant Biol. 2006, 57, 233–266. [Google Scholar] [CrossRef] [Green Version]
- Badri, D.V.; Vivanco, J.M. Regulation and function of root exudates. Plant Cell Environ. 2009, 32, 666–681. [Google Scholar] [CrossRef]
- Jaeger, C.H.; Lindow, S.E.; Miller, W.; Clark, E.; Firestone, M.K. Mapping of sugar and amino acid availability in soil around roots with bacterial sensors of sucrose and tryptophan. Appl. Environ. Microbiol. 1999, 65, 2685–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doan, T.H.; Doan, T.A.; Kangas, M.; Ernest, A.E.; Tran, D.; Wilson, C.L.; Holmes, A.E.; Doyle, E.L.; Brooks, T.L.D. A low-cost imaging method for the temporal and spatial colorimetric detection of free amines on maize root surfaces. Front. Plant Sci. 2017, 8, 1513. [Google Scholar] [CrossRef] [Green Version]
- Sasse, J.; Martinoia, E.; Northen, T. Feed your friends: Do plant exudates shape the root microbiome? Trends Plant Sci. 2018, 23, 25–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovira, A.D. Zones of exudation along plant roots and spatial distribution of micro-organisms in the rhizosphere. Pestic. Sci. 1973, 4, 361–366. [Google Scholar] [CrossRef]
- Sauer, D.; Kuzyakov, Y.; Stahr, K. Spatial distribution of root exudates of five plant species as assessed by14C labeling. J. Plant Nutr Soil Sci. 2006, 169, 360–362. [Google Scholar] [CrossRef]
- Vidal, A.; Hirte, J.; Bender, S.F.; Mayer, J.; Gattinger, A.; Höschen, C.; Schädler, S.; Iqbal, T.M.; Mueller, C.W. Linking 3d soil structure and plant-microbe-soil carbon transfer in the rhizosphere. Front. Environ. Sci. 2018, 6, 9. [Google Scholar] [CrossRef]
- Canarini, A.; Kaiser, C.; Merchant, A.; Richter, A.; Wanek, W. Root Exudation of Primary Metabolites: Mechanisms and Their Roles in Plant Responses to Environmental Stimuli. Front. Plant Sci. 2019, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Kuramae, E.E.; De Hollander, M.; Klinkhamer, P.G.L.; Van Veen, J.A. Functional traits dominate the diversity-related selection of bacterial communities in the rhizosphere. ISME J. 2017, 11, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamutando, C.N.; Vikram, S.; Kamgan-Nkuekam, G.; Makhalanyane, T.; Greve, M.; Le Roux, J.; Richardson, D.M.; Cowan, D.A.; Valverde, A. The functional potential of the rhizospheric microbiome of an invasive tree species. Acacia Dealbata. Microb. Ecol. 2019, 77, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badri, D.V.; Chaparro, J.M.; Zhang, R.; Shen, Q.; Vivanco, J.M. Application of natural blends of phytochemicals derived from the root exudates of arabidopsis to the soil reveal that phenolic-related compounds predominantly modulate the soil microbiome. J. Biol. Chem. 2013, 288, 4502–4512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaparro, J.M.; Badri, D.V.; Bakker, M.G.; Sugiyama, A.; Manter, D.K.; Vivanco, J.M. Root Exudation of Phytochemicals in Arabidopsis Follows Specific Patterns That Are Developmentally Programmed and Correlate with Soil Microbial Functions. PLoS ONE 2013, 8, e55731. [Google Scholar] [CrossRef]
- Pausch, J.; Kuzyakov, Y. Carbon input by roots into the soil: Quantification of rhizodeposition from root to ecosystem scale. Glob. Chang. Biol. 2018, 24, 1–12. [Google Scholar] [CrossRef]
- Na, X.; Cao, X.; Ma, C.; Ma, S.; Xu, P.; Liu, S.; Wang, J.; Wang, H.; Chen, L.; Qiao, Z. Plant stage, not drought stress, determines the effect of cultivars on bacterial community diversity in the rhizosphere of broomcorn millet (Panicum miliaceum L.). Front. Microbiol. 2019, 10, 828. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.A.; Santos-Medellín, C.M.; Liechty, Z.S.; Nguyen, B.; Lurie, E.; Eason, S.; Phillips, G.; Sundaresan, V. Compositional shifts in root-associated bacterial and archaeal microbiota track the plant life cycle in field-grown rice. PLoS Biol. 2018, 16, e2003862. [Google Scholar] [CrossRef] [Green Version]
- Haichar, F.E.Z.; Marol, C.; Berge, O.; Rangel-Castro, J.I.; Prosser, J.; Balesdent, J.; Heulin, T.; Achouak, W. Plant host habitat and root exudates shape soil bacterial community structure. ISME J. 2008, 2, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Lomond, J.S.; Tong, A.Z. Rapid analysis of dissolved methane, ethylene, acetylene and ethane using partition coefficients and headspace-gas chromatography. J. Chromatogr. Sci. 2011, 49, 469–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Li, Y.; Shi, Z.; Wang, S.; Wang, Z.; Liu, Y.; Wen, X.; Mo, F.; Han, J.; Liao, Y. Crop development has more influence on shaping rhizobacteria of wheat than tillage practice and crop rotation pattern in an arid agroecosystem. Appl. Soil Ecol. 2021, 165, 104016. [Google Scholar] [CrossRef]
- Betlach, M.R.; Tiedje, J.M. Kinetic explanation for accumulation of nitrite, nitric oxide, and nitrous oxide during bacterial denitrification. Appl. Environ. Microbiol. 1981, 42, 1074–1084. [Google Scholar] [CrossRef] [Green Version]
- Thomson, A.J.; Giannopoulos, G.; Pretty, J.; Baggs, E.M.; Richardson, D.J. Biological sources and sinks of nitrous oxide and strategies to mitigate emissions. Philos Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 1157–1168. [Google Scholar] [CrossRef] [Green Version]
- Bazin, M.J.; Markham, P.; Scott, E.M.; Lynch, J.M. Population dynamics and rhizosphere interactions. In The Rhizosphere; Lynch, J.M., Ed.; John Wiley and Sons Ltd.: Hoboken, NJ, USA, 1990; pp. 99–127. [Google Scholar]
- Reinhold-Hurek, B.; Bünger, W.; Burbano, C.S.; Sabale, M.; Hurek, T. Roots shaping their microbiome: Global hotspots for microbial activity. Annu. Rev. Phytopathol. 2015, 53, 403–424. [Google Scholar] [CrossRef]
- Kuzyakov, Y.; Razavi, B.S. Rhizosphere size and shape: Temporal dynamics and spatial stationarity. Soil Biol. Biochem. 2019, 135, 343–360. [Google Scholar] [CrossRef]
- Chaparro, J.M.; Badri, D.V.; Vivanco, J.M. Rhizosphere microbiome assemblage is affected by plant development. ISME J. 2014, 8, 790–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Weert, S.; Vermeiren, H.; Mulders, I.H.M.; Kuiper, I.; Hendrickx, N.; Bloemberg, G.V.; Vanderleyden, J.; De Mot, R.; Lugtenberg, B.J.J. Flagella-driven chemotaxis towards exudate components is an important trait for tomato root colonization by Pseudomonas fluorescens. Mol. Plant Microbe Interact. 2002, 15, 1173–1180. [Google Scholar] [CrossRef] [Green Version]
- Lareen, A.; Burton, F.; Schäfer, P. Plant root-microbe communication in shaping root microbiomes. Plant. Mol. Biol. 2016, 90, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Harshey, R.M. Bacterial motility on a surface: Many ways to a common goal. Annu. Rev. Microbiol. 2003, 57, 249–273. [Google Scholar] [CrossRef] [PubMed]
- Lugtenberg, B.J.J.; Dekkers, L.; Bloemberg, G.V. Molecular determinants of rhizosphere colonization by Pseudomonas. Annu. Rev. Phytopathol. 2001, 39, 461–490. [Google Scholar] [CrossRef] [PubMed]
- Khosla, C. Structures and mechanisms of polyketide synthases. American Chemical Society. J. Org. Chem. 2009, 74, 6416–6420. [Google Scholar] [CrossRef] [PubMed]
- Iannucci, A.; Fragasso, M.; Beleggia, R.; Nigro, F.; Papa, R. Evolution of the crop rhizosphere: Impact of domestication on root exudates in tetraploid wheat (Triticum turgidum L.). Front. Plant Sci. 2017, 8, 2124. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.W.; Mah, T.F. Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol. Rev. 2017, 41, 276–301. [Google Scholar] [CrossRef]
- Shah, S.A.; Vestergaard, G.; Garrett, R.A. CRISPR/Cas and CRISPR/Cmr immune systems of archaea. In Regulatory RNAs in Prokaryotes; Springer: Wien, Austria, 2012; pp. 163–182. [Google Scholar]
- Allard-Massicotte, R.; Tessier, L.; Lécuyer, F.; Lakshmanan, V.; Lucier, J.-F.; Garneau, D.; Caudwell, L.; Vlamakis, H.; Bais, H.P.; Beauregard, P.B. Bacillus subtilis early colonization of Arabidopsis thaliana roots involves multiple chemotaxis receptors. MBio 2016, 7, e01664-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oburger, E.; Jones, D.L. Sampling root exudates—Mission impossible? Rhizosphere 2018, 6, 116–133. [Google Scholar] [CrossRef]
- Donn, S.; Kirkegaard, J.A.; Perera, G.; Richardson, A.E.; Watt, M. Evolution of bacterial communities in the wheat crop rhizosphere. Environ. Microbiol. 2015, 17, 610–621. [Google Scholar] [CrossRef]
- Mahoney, A.K.; Yin, C.; Hulbert, S.H. Community structure, species variation, and potential functions of rhizosphere-associated bacteria of different winter wheat (Triticum aestivum) cultivars. Front. Plant. Sci. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Waghmode, T.R.; Sun, R.; Kuramae, E.E.; Hu, C.; Liu, B. Root-associated microbiomes of wheat under the combined effect of plant development and nitrogen fertilization. Microbiome 2019, 7, 136. [Google Scholar] [CrossRef] [Green Version]
- Kirby, R. Actinomycetes and Lignin Degradation. Adv. Appl. Microbiol. 2005, 58, 125–168. [Google Scholar]
- Watt, M.; Hugenholtz, P.; White, R.; Vinall, K. Numbers and locations of native bacteria on field-grown wheat roots quantified by fluorescence in situ hybridization (FISH). Environ. Microbiol. 2006, 8, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Karnaouri, A.; Matsakas, L.; Krikigianni, E.; Rova, U.; Christakopoulos, P. Valorization of waste forest biomass toward the production of cello-oligosaccharides with potential prebiotic activity by utilizing customized enzyme cocktails. Biotechnol. Biofuels 2019, 12, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Usyskin-Tonne, A.; Hadar, Y.; Yermiyahu, U.; Minz, D. Elevated CO2 has a significant impact on denitrifying bacterial community in wheat roots. Soil Biol. Biochem. 2020, 142, 107697. [Google Scholar] [CrossRef]
- Rocha, J.; Cacace, D.; Kampouris, I.; Guilloteau, H.; Jäger, T.; Marano, R.; Karaolia, P.; Manaia, C.M.; Merlin, C.; Fatta-Kassinos, D.; et al. Inter-laboratory calibration of quantitative analyses of antibiotic resistance genes. J. Environ. Chem. Eng. 2020, 8, 102214. [Google Scholar] [CrossRef]
- Tovi, N.; Frenk, S.; Hadar, Y.; Minz, D. Host Specificity and Spatial Distribution Preference of Three Pseudomonas Isolates. Front. Microbiol. 2019, 9, 3263. [Google Scholar] [CrossRef]
- Usyskin-Tonne, A.; Hadar, Y.; Yermiyahu, U.; Minz, D. Elevated CO2 and nitrate levels increase wheat root-associated bacterial abundance and impact rhizosphere microbial community composition and function. ISME J. 2020, 15, 1073–1084. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 15 September 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN community edition—Interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [Green Version]
- Clarke, K.; Ainsworth, M. A method of linking multivariate community structure to environmental variables. Mar. Ecol. Prog Ser. 1993, 92, 205–219. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 2001, 26, 32–46. [Google Scholar]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. CoNet app: Inference of biological association networks using Cytoscape. F1000Research 2016, 5, 1519. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usyskin-Tonne, A.; Hadar, Y.; Minz, D. Spike Formation Is a Turning Point Determining Wheat Root Microbiome Abundance, Structures and Functions. Int. J. Mol. Sci. 2021, 22, 11948. https://doi.org/10.3390/ijms222111948
Usyskin-Tonne A, Hadar Y, Minz D. Spike Formation Is a Turning Point Determining Wheat Root Microbiome Abundance, Structures and Functions. International Journal of Molecular Sciences. 2021; 22(21):11948. https://doi.org/10.3390/ijms222111948
Chicago/Turabian StyleUsyskin-Tonne, Alla, Yitzhak Hadar, and Dror Minz. 2021. "Spike Formation Is a Turning Point Determining Wheat Root Microbiome Abundance, Structures and Functions" International Journal of Molecular Sciences 22, no. 21: 11948. https://doi.org/10.3390/ijms222111948