
Leaf Apoplast of Field-Grown Potato Analyzed by Quantitative Proteomics and Activity-Based Protein Profiling

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Analysis of Apoplastic Peptides
2.2. Effect of Fungicide Application on Potato Apoplastic Proteome
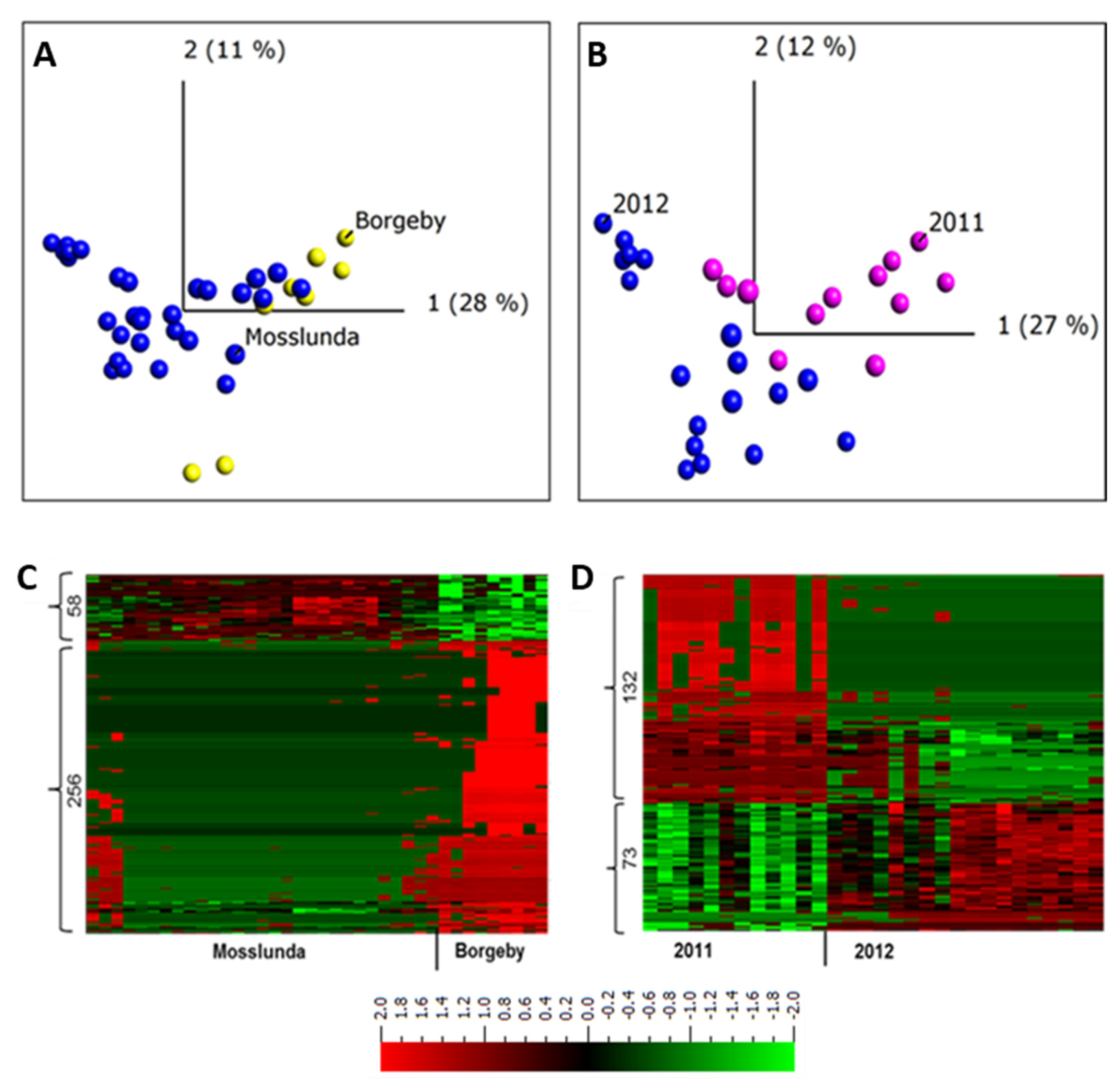
2.3. Apoplastic Proteome Differences between Growing Sites and between Years
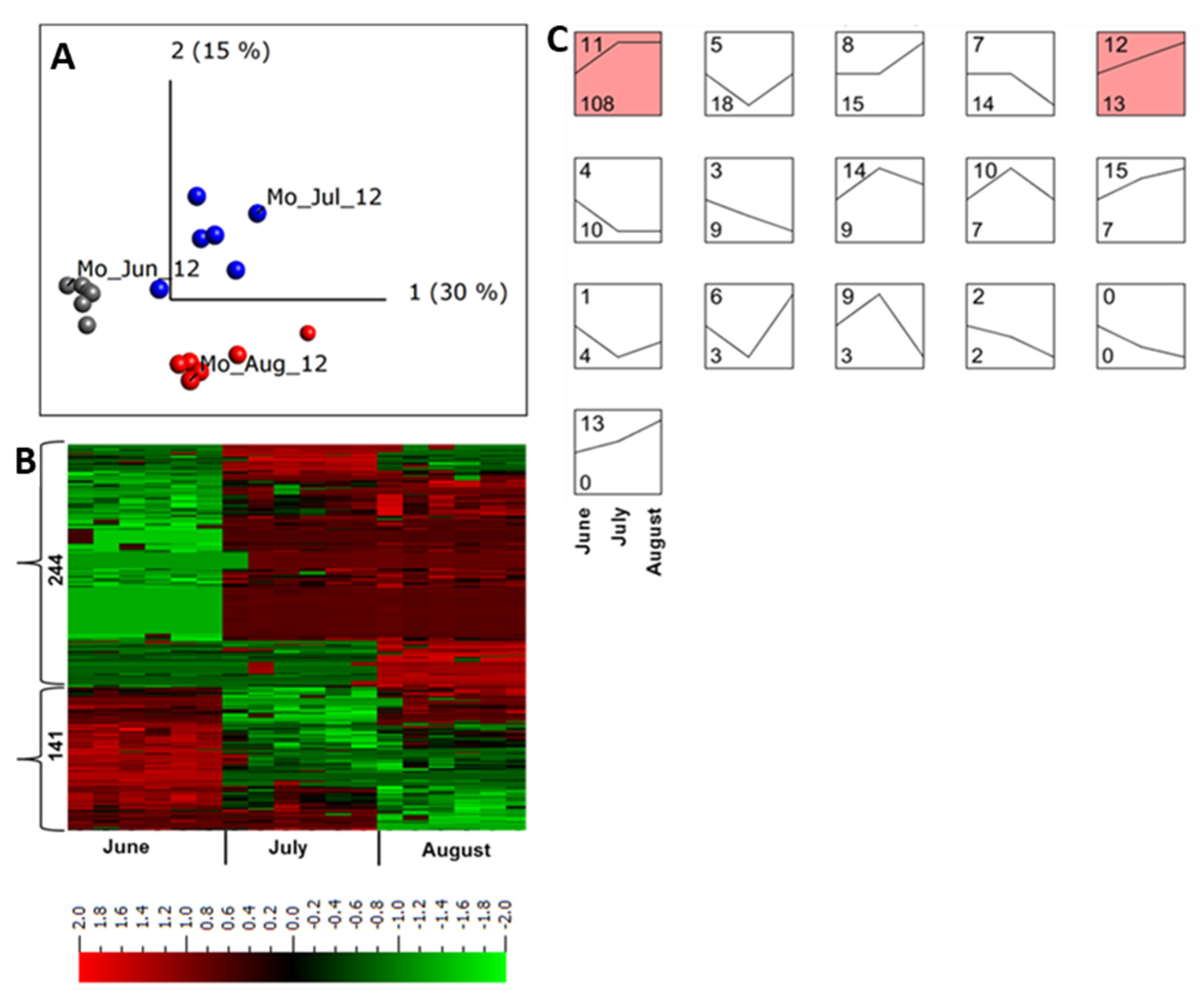
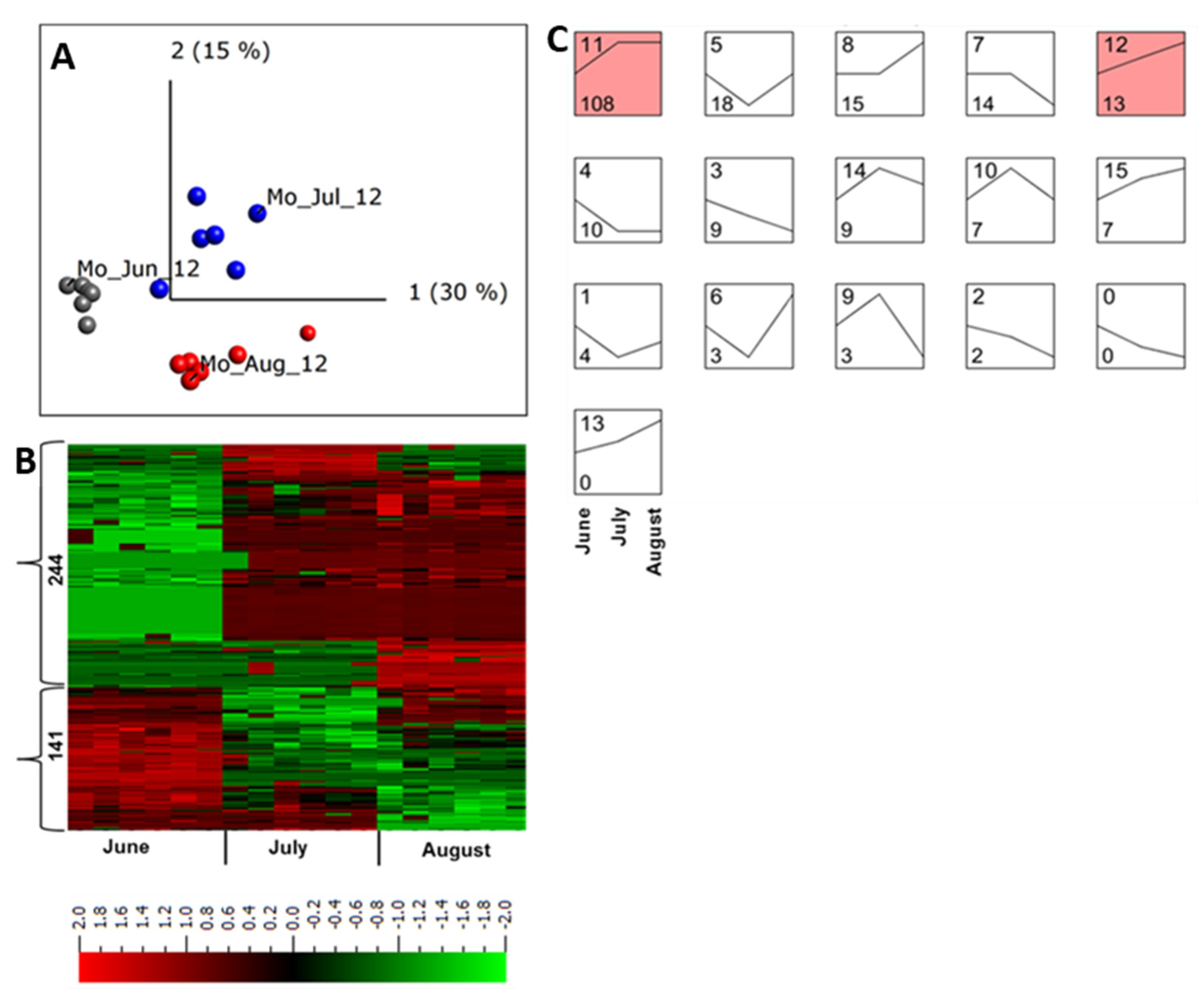
2.4. Abundance of Apoplastic Proteins across a Growing Season
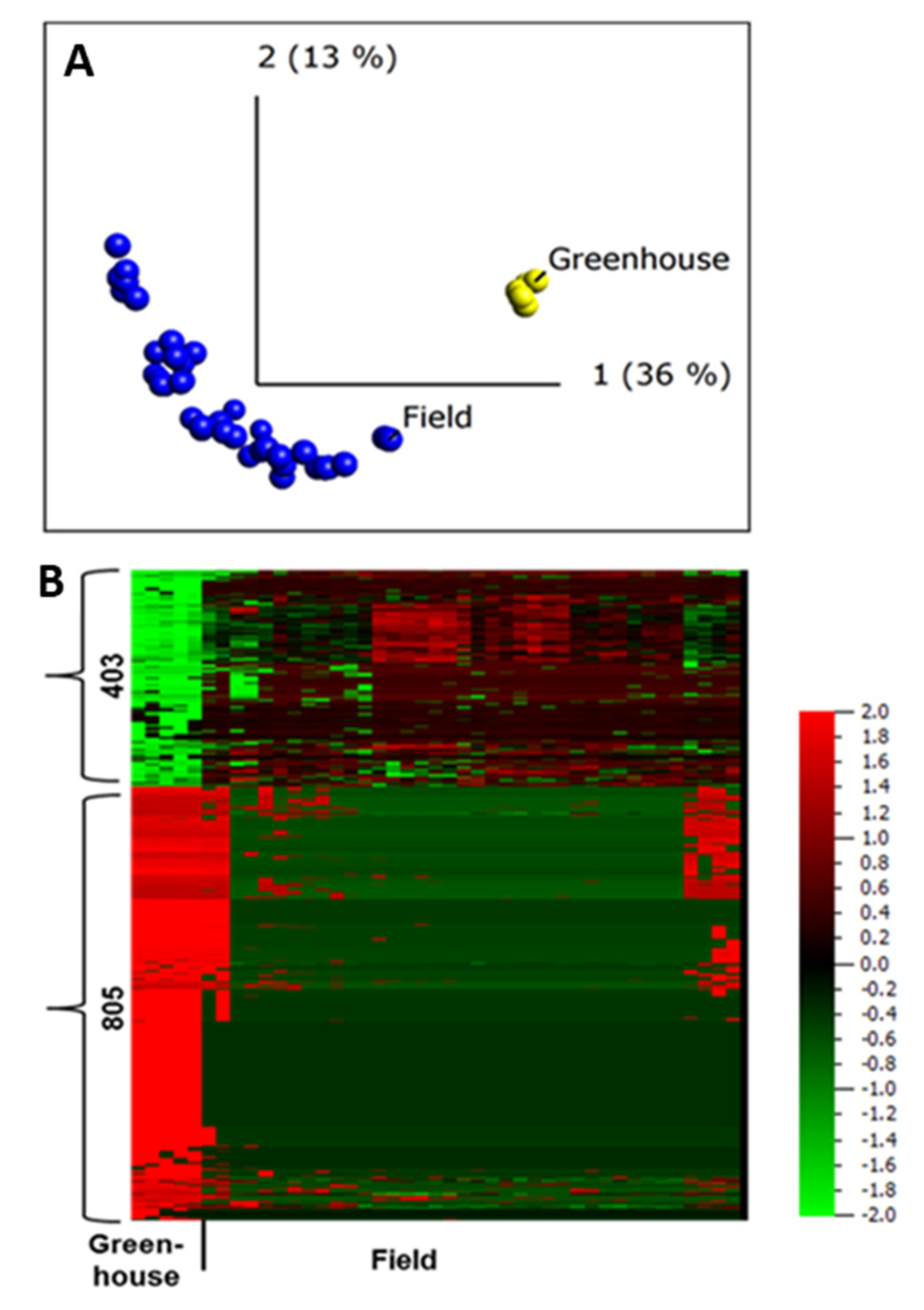
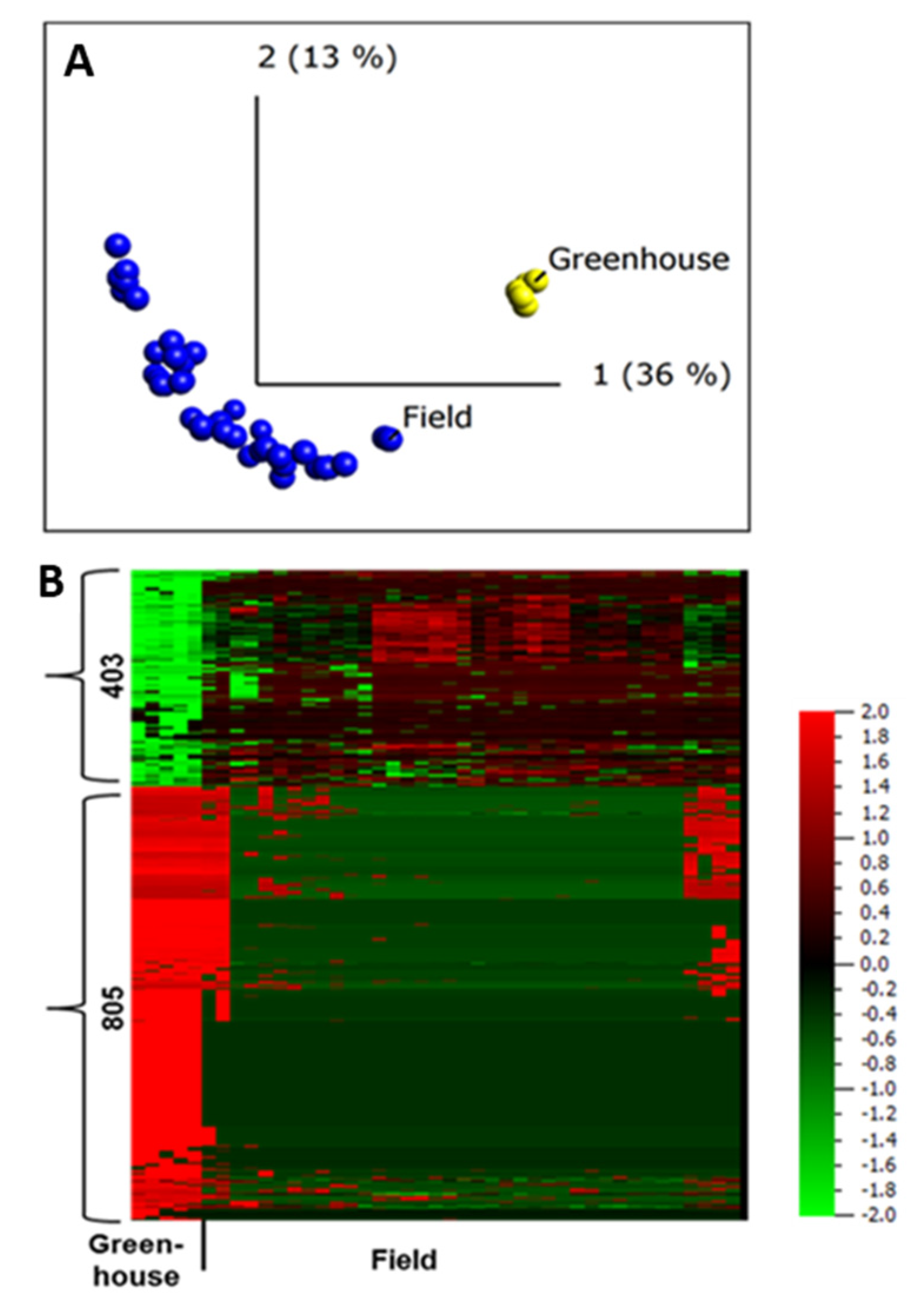
2.5. Difference in Protein Abundance under Field and Greenhouse Conditions
2.6. ABPP Reveals Seasonal Effects
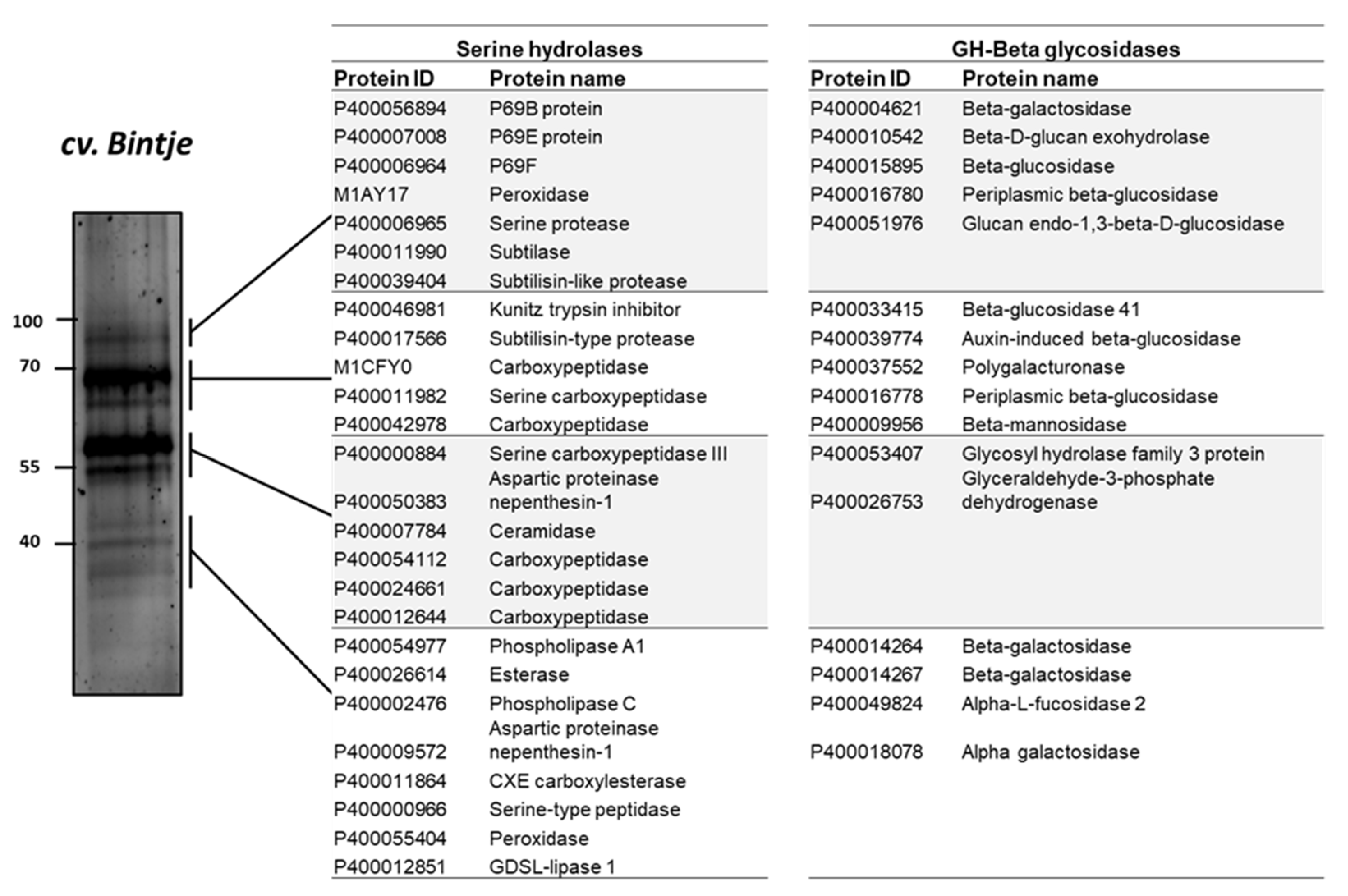
2.7. Serine Hydrolases and β-Glycosidases Identified by ABPP and MS
3. Materials and Methods
3.1. Plant Material and Field Sites
3.2. Apoplast Isolation and Protein Digestion
3.3. Mass Spectrometry
3.4. Quantitative Analysis of Peptides
3.5. Labelling of Apoplastic Proteome Activity
3.6. Affinity Purification and Identification of Serine Hydrolases and β-glycosidases
3.7. Bioinformatics Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mishra, Y.; Jankanpaa, H.J.; Kiss, A.Z.; Funk, C.; Schroder, W.P.; Jansson, S. Arabidopsis plants grown in the field and climate chambers significantly differ in leaf morphology and photosystem components. BMC Plant Biol. 2012, 12, 6. [Google Scholar] [CrossRef] [Green Version]
- Ruhe, J.; Agler, M.T.; Placzek, A.; Kramer, K.; Finkemeier, I.; Kemen, E.M. Obligate Biotroph Pathogens of the Genus Albugo Are Better Adapted to Active Host Defense Compared to Niche Competitors. Front. Plant Sci. 2016, 7, 820. [Google Scholar] [CrossRef] [Green Version]
- Poorter, H.; Fiorani, F.; Pieruschka, R.; Wojciechowski, T.; van der Putten, W.H.; Kleyer, M.; Schurr, U.; Postma, J. Pampered inside, pestered outside? Differences and similarities between plants growing in controlled conditions and in the field. New Phytol. 2016, 212, 838–855. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.W.; Antle, J.M.; Basso, B.; Boote, K.J.; Conant, R.T.; Foster, I.; Godfray, H.C.J.; Herrero, M.; Howitt, R.E.; Janssen, S.; et al. Brief history of agricultural systems modeling. Agric. Syst. 2016, 155, 240–254. [Google Scholar] [CrossRef]
- Oerke, E.C. Crop losses to pests. J. Agric. Sci. 2005, 144, 31. [Google Scholar] [CrossRef]
- Alexandersson, E.; Ali, A.; Resjo, S.; Andreasson, E. Plant secretome proteomics. Front. Plant Sci. 2013, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- De Wit, P.J. Apoplastic fungal effectors in historic perspective; a personal view. New Phytol. 2016, 212, 805–813. [Google Scholar] [CrossRef] [Green Version]
- Jashni, M.K.; Mehrabi, R.; Collemare, J.; Mesarich, C.H.; de Wit, P.J.G.M. The battle in the apoplast: Further insights into the roles of proteases and their inhibitors in plant-pathogen interactions. Front. Plant Sci. 2015, 6, 584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Zhang, C.J.; Ge, W.N.; Zhang, Y.F.; Burlingame, A.L.; Guo, Y. Identification of NaCl stress-responsive apoplastic proteins in rice shoot stems by 2D-DIGE. J. Proteom. 2011, 74, 1045–1067. [Google Scholar] [CrossRef] [Green Version]
- Delaunois, B.; Jeandet, P.; Clement, C.; Baillieul, F.; Dorey, S.; Cordelier, S. Uncovering plant-pathogen crosstalk through apoplastic proteomic studies. Front. Plant Sci. 2014, 5, 249. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, Y.C.; Wang, Y.M. Apoplastic Proteases: Powerful Weapons against Pathogen Infection in Plants. Plant Commun. 2020, 1, 100085. [Google Scholar] [CrossRef]
- Bengtsson, T.; Weighill, D.; Proux-Wera, E.; Levander, F.; Resjo, S.; Burra, D.D.; Moushib, L.I.; Hedley, P.E.; Liljeroth, E.; Jacobson, D.; et al. Proteomics and transcriptomics of the BABA-induced resistance response in potato using a novel functional annotation approach. BMC Genom. 2014, 15, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burra, D.D.; Berkowitz, O.; Hedley, P.E.; Morris, J.; Resjo, S.; Levander, F.; Liljeroth, E.; Andreasson, E.; Alexandersson, E. Phosphite-induced changes of the transcriptome and secretome in Solanum tuberosum leading to resistance against Phytophthora infestans. BMC Plant Biol. 2014, 14, 254. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Alexandersson, E.; Sandin, M.; Resjo, S.; Lenman, M.; Hedley, P.; Levander, F.; Andreasson, E. Quantitative proteomics and transcriptomics of potato in response to Phytophthora infestans in compatible and incompatible interactions. BMC Genom. 2014, 15, 497. [Google Scholar] [CrossRef] [Green Version]
- Lankinen, A.; Abreha, K.B.; Masini, L.; Ali, A.; Resjo, S.; Andreasson, E. Plant immunity in natural populations and agricultural fields: Low presence of pathogenesis-related proteins in Solanum leaves. PLoS ONE 2018, 13, e0207253. [Google Scholar] [CrossRef]
- Frades, I.; Abreha, K.B.; Proux-Wéra, E.; Lankinen, Å.; Andreasson, E.; Alexandersson, E. A novel workflow correlating RNA-seq data to resistance levels of wild Solanum species and potato clones to Phytophthora infestans. Front. Plant Sci. 2015, 6, 718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yogendra, K.N.; Kushalappa, A.C. Integrated transcriptomics and metabolomics reveal induction of hierarchies of resistance genes in potato against late blight. Funct Plant Biol. 2016, 43, 766–782. [Google Scholar] [CrossRef]
- Evers, D.; Legay, S.; Lamoureux, D.; Hausman, J.F.; Hoffmann, L.; Renaut, J. Towards a synthetic view of potato cold and salt stress response by transcriptomic and proteomic analyses. Plant Mol. Biol. 2012, 78, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Zhang, H.X.; Gan, X.Y.; Zhang, L.; Chen, Y.C.; Nie, F.J.; Shi, L.; Li, M.; Guo, Z.Q.; Zhang, G.H.; et al. Transcriptome Profiling of the Potato (Solanum tuberosum L.) Plant under Drought Stress and Water-Stimulus Conditions. PLoS ONE 2015, 10, e0128041. [Google Scholar]
- Boguszewska-Mankowska, D.; Gietler, M.; Nykiel, M. Comparative proteomic analysis of drought and high temperature response in roots of two potato cultivars. Plant Growth Regul. 2020, 92, 345–363. [Google Scholar] [CrossRef]
- Alexandersson, E.; Jacobson, D.; Vivier, M.A.; Weckwerth, W.; Andreasson, E. Field-omics-understanding large-scale molecular data from field crops. Front. Plant Sci. 2014, 5, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.H.; Smith, J.W.; Huang, C.M. Mass Spectrometry-Based Label-Free Quantitative Proteomics. J. Biomed. Biotechnol. 2010, 840518. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Lu, S.; Liu, K.F.; Wang, S.; Huang, L.Q.; Guo, L.P. Proteomics: A powerful tool to study plant responses to biotic stress. Plant Methods 2019, 15, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoorn, R.A.; Colby, T.; Nickel, S.; Richau, K.H.; Schmidt, J.; Kaiser, M. Mining the Active Proteome of Arabidopsis thaliana. Front. Plant Sci. 2011, 2, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozkurt, T.O.; Schornack, S.; Win, J.; Shindo, T.; Ilyas, M.; Oliva, R.; Cano, L.M.; Jones, A.M.E.; Huitema, E.; van der Hoorn, R.A.L.; et al. Phytophthora infestans effector AVRblb2 prevents secretion of a plant immune protease at the haustorial interface. Proc. Natl. Acad. Sci. USA 2011, 108, 20832–20837. [Google Scholar] [CrossRef] [Green Version]
- Kaschani, F.; Gu, C.; Niessen, S.; Hoover, H.; Cravatt, B.F.; van der Hoorn, R.A. Diversity of serine hydrolase activities of unchallenged and botrytis-infected Arabidopsis thaliana. Mol. Cell. Proteom. MCP 2009, 8, 1082–1093. [Google Scholar] [CrossRef] [Green Version]
- Sueldo, D.; Ahmed, A.; Misas-Villamil, J.; Colby, T.; Tameling, W.; Joosten, M.H.A.J.; van der Hoorn, R.A.L. Dynamic hydrolase activities precede hypersensitive tissue collapse in tomato seedlings. New Phytol. 2014, 203, 913–925. [Google Scholar] [CrossRef]
- Chandrasekar, B.; Colby, T.; Emran Khan Emon, A.; Jiang, J.; Hong, T.N.; Villamor, J.G.; Harzen, A.; Overkleeft, H.S.; van der Hoorn, R.A. Broad-range glycosidase activity profiling. Mol. Cell. Proteom. MCP 2014, 13, 2787–2800. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Moushib, L.I.; Lenman, M.; Levander, F.; Olsson, K.; Carlson-Nilson, U.; Zoteyeva, N.; Liljeroth, E.; Andreasson, E. Paranoid potato: Phytophthora-resistant genotype shows constitutively activated defense. Plant Signal. Behav. 2012, 7, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Andreasson, E.; Abreha, K.B.; Resjö, S. Isolation of Apoplast. In Isolation of Plant Organelles and Structures: Methods and Protocols; Taylor, N.L., Millar, A.H., Eds.; Springer: New York, NY, USA, 2017; pp. 233–240. [Google Scholar]
- Tyers, M.; Mann, M. From genomics to proteomics. Nature 2003, 422, 193–197. [Google Scholar] [CrossRef]
- Chawade, A.; Alexandersson, E.; Bengtsson, T.; Andreasson, E.; Levander, F. Targeted Proteomics Approach for Precision Plant Breeding. J. Proteome Res. 2016, 15, 638–646. [Google Scholar] [CrossRef] [Green Version]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Webb-Robertson, B.J.M.; Wiberg, H.K.; Matzke, M.M.; Brown, J.N.; Wang, J.; McDermott, J.E.; Smith, R.D.; Rodland, K.D.; Metz, T.O.; Pounds, J.G.; et al. Review, Evaluation, and Discussion of the Challenges of Missing Value Imputation for Mass Spectrometry-Based Label-Free Global Proteomics. J. Proteome Res. 2015, 14, 1993–2001. [Google Scholar] [CrossRef] [Green Version]
- Doherty, J.R.; Botti-Marino, M.; Kerns, J.P.; Ritchie, D.F.; Roberts, J.A. Response of Microbial Populations on the Creeping Bentgrass Phyllosphere to Periodic Fungicide Applications. Plant Health Prog. 2017, 18, 44–49. [Google Scholar] [CrossRef]
- Byrnes, C. Public release summary of the evaluation by the NRA of the new active constituent: Fluazinam in the product: SHIRLAN FUNGICIDE. Natl. Regist. Auth. Agric. Vet. Chem. 2011, 1–28. Available online: https://apvma.gov.au/node/13751 (accessed on 1 November 2021).
- Hoehenwarter, W.; Larhlimi, A.; Hummel, J.; Egelhofer, V.; Selbig, J.; van Dongen, J.T.; Wienkoop, S.; Weckwerth, W. MAPA Distinguishes Genotype-Specific Variability of Highly Similar Regulatory Protein Isoforms in Potato Tuber. J. Proteome Res. 2011, 10, 2979–2991. [Google Scholar] [CrossRef] [PubMed]
- Dal Santo, S.; Tornielli, G.B.; Zenoni, S.; Fasoli, M.; Farina, L.; Anesi, A.; Guzzo, F.; Delledonne, M.; Pezzotti, M. The plasticity of the grapevine berry transcriptome. Genome Biol. 2013, 14, r54. [Google Scholar] [CrossRef] [Green Version]
- Wiik, L. Potato Late Blight and Tuber Yield: Results from 30 Years of Field Trials. Potato Res. 2014, 57, 77–98. [Google Scholar] [CrossRef]
- Zhang, B.; He, H.B.; Ding, X.L.; Zhang, X.D.; Zhang, X.P.; Yang, X.M.; Filley, T.R. Soil microbial community dynamics over a maize (Zea mays L.) growing season under conventional- and no-tillage practices in a rainfed agroecosystem. Soil Till Res. 2012, 124, 153–160. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [Green Version]
- Thimm, O.; Blasing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Kruger, P.; Selbig, J.; Muller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Kwon, S.J.; Jin, H.C.; Lee, S.; Nam, M.H.; Chung, J.H.; Kwon, S.I.; Ryu, C.M.; Park, O.K. GDSL lipase-like 1 regulates systemic resistance associated with ethylene signaling in Arabidopsis. Plant J. 2009, 58, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Liljeroth, E.; Bengtsson, T.; Wiik, L.; Andreasson, E. Induced resistance in potato to Phytphthora infestans-effects of BABA in greenhouse and field tests with different potato varieties. Eur. J. Plant Pathol. 2010, 127, 171–183. [Google Scholar] [CrossRef]
- Resjo, S.; Ali, A.; Meijer, H.J.G.; Seidl, M.F.; Snel, B.; Sandin, M.; Levander, F.; Govers, F.; Andreasson, E. Quantitative Label-Free Phosphoproteomics of Six Different Life Stages of the Late Blight Pathogen Phytophthora infestans Reveals Abundant Phosphorylation of Members of the CRN Effector Family. J. Proteome Res. 2014, 13, 1848–1859. [Google Scholar] [CrossRef]
- Martens, L.; Chambers, M.; Sturm, M.; Kessner, D.; Levander, F.; Shofstahl, J.; Tang, W.H.; Ropp, A.; Neumann, S.; Pizarro, A.D.; et al. mzML-a Community Standard for Mass Spectrometry Data. Mol. Cell. Proteom. 2011, 10, R110.000133. [Google Scholar] [CrossRef] [Green Version]
- French, W.R.; Zimmerman, L.J.; Schilling, B.; Gibson, B.W.; Miller, C.A.; Townsend, R.R.; Sherrod, S.D.; Goodwin, C.R.; McLean, J.A.; Tabb, D.L. Wavelet-Based Peak Detection and a New Charge Inference Procedure for MS/MS Implemented in ProteoWizard’s msConvert. J. Proteome Res. 2015, 14, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Hakkinen, J.; Vincic, G.; Mansson, O.; Warell, K.; Levander, F. The Proteios Software Environment: An Extensible Multiuser Platform for Management and Analysis of Proteomics Data. J. Proteome Res. 2009, 8, 3037–3043. [Google Scholar] [CrossRef]
- Teleman, J.; Chawade, A.; Sandin, M.; Levander, F.; Malmstrom, J. Dinosaur: A Refined Open-Source Peptide MS Feature Detector. J. Proteome Res. 2016, 15, 2143–2151. [Google Scholar] [CrossRef]
- Sandin, M.; Ali, A.; Hansson, K.; Mansson, O.; Andreasson, E.; Resjo, S.; Levander, F. An Adaptive Alignment Algorithm for Quality-controlled Label-free LC-MS. Mol. Cell. Proteom. 2013, 12, 1407–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawade, A.; Alexandersson, E.; Levander, F. Normalyzer: A Tool for Rapid Evaluation of Normalization Methods for Omics Data Sets. J. Proteome Res. 2014, 13, 3114–3120. [Google Scholar] [CrossRef]
- Smyth, G.K.; Ritchie, M.; Thorne, N.; Wettenhall, J. LIMMA: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Stat. Biol. Health 2005, 397–420. [Google Scholar]
- Kaschani, F.; Nickel, S.; Pandey, B.; Cravatt, B.F.; Kaiser, M.; van der Hoorn, R.A.L. Selective inhibition of plant serine hydrolases by agrochemicals revealed by competitive ABPP. Bioorganic Med. Chem. 2012, 20, 597–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalski, A.; Damoc, E.; Lange, O.; Denisov, E.; Nolting, D.; Muller, M.; Viner, R.; Schwartz, J.; Remes, P.; Belford, M.; et al. Ultra High Resolution Linear Ion Trap Orbitrap Mass Spectrometer (Orbitrap Elite) Facilitates Top Down LC MS/MS and Versatile Peptide Fragmentation Modes. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.V.; de Godoy, L.M.F.; Li, G.Q.; Macek, B.; Mortensen, P.; Pesch, R.; Makarov, A.; Lange, O.; Horning, S.; Mann, M. Parts per million mass accuracy on an orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteom. 2005, 4, 2010–2021. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [Green Version]
- Ramsak, Z.; Baebler, S.; Rotter, A.; Korbar, M.; Mozetic, I.; Usadel, B.; Gruden, K. GoMapMan: Integration, consolidation and visualization of plant gene annotations within the MapMan ontology. Nucleic Acids Res. 2014, 42, D1167–D1175. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Sequence | Protein IDs | Protein Name | Genome | Signal P | Log2 Fold Change | |
|---|---|---|---|---|---|---|
| Location | July | August | ||||
| TDPNQNTGIVIQK | DMP400016183 | Pectinesterase | chr03 | Yes | 4.72 | 4.73 |
| DGQPSEQHFGLFYPDQR | Q70BW9 | 1,3-beta-glucan glucanohydrolase | 4.71 | 4.81 | ||
| GQTWVIDAPR | DMP400005465 | Osmotin | chr08 | Yes | 4.68 | 4.83 |
| GLTWSVPTGR | DMP400022299 | Peroxidase | chr01 | Yes | 4.68 | 4.63 |
| RLDPGQTWVIDAPR | Q5XUH0 | Osmotin-like protein | 4.65 | 4.84 | ||
| MLNEGFVPDDVSLK | Q9FHR3 | Putative pentatricopeptide repeat-containing protein At5g37570 | 4.65 | 4.56 | ||
| NIQNAISGAGLGNQIK | DMP400051976 | Glucan endo-1,3-beta-D-glucosidase | chr10 | Yes | 4.64 | 4.64 |
| TSNLYAIGEMEIEENKK | DMP400023312 | DUF26 domain-containing protein 2 | chr12 | Yes | 4.62 | 4.67 |
| LLALSDTPYK | DMP400046980 | Kunitz trypsin inhibitor | chr06 | Yes | 4.62 | 4.57 |
| VCWPVPNK | DMP400033260 | Xylem serine proteinase 1 | chr10 | No | 4.61 | 4.65 |
| SPSAYLNNPAGER | DMP400007784 | Ceramidase | chr03 | Yes | 4.61 | 4.24 |
| RYCGMLNVPTGEN- LDCNNQR | DMP400002757 | Class II chitinase | chr02 | Yes | 4.6 | 4.72 |
| QRCPDAYSYPQDD- PTSTFTCPSDSTNYR | DMP400005463 | Osmotin OSML13 | chr08 | Yes | 4.59 | 4.34 |
| GVIFFGDSPYVFLPGMDVSK | DMP400015799 | Xyloglucan-specific endoglucanase inhibitor 4 | chr01 | Yes | 4.58 | 4.49 |
| IFESCSTDTFQIR | DMP400041178 | Embryo-specific 3 | chr01 | Yes | 4.57 | 4.51 |
| YCGICCEECK | DMP400037307 | Snakin-1 | chr04 | Yes | 4.57 | 4.45 |
| ALPTYTPESPADATR | DMP400038185 | Transketolase, chloroplastic | chr10 | No | 4.56 | 4.62 |
| VITSSTEAQAYTPGR | Q43143 | Pectinesterase/pectinesterase inhibitor U1 | 4.53 | 4.54 | ||
| GFEAAPSVSFTVDGEEK | DMP400000884 | Serine carboxypeptidase III | chr11 | No | 4.52 | 4.62 |
| FVVVVDDSK | M1BPR5 | Uncharacterized protein (Solanum tuberosum) | 4.52 | 4.58 | ||
| AETWVQEETRALISLR | Q43326 | Box II Factor | 4.52 | 4.56 | ||
| KFGLTVDNVLDAR | DMP400031346 | Reticuline oxidase | chr02 | Yes | 4.52 | 4.52 |
| LCPQGGDGGTFANLDK | DMP400055305 | Peroxidase | chr01 | Yes | 4.51 | 4.61 |
| CLCGSPLPDCK | DMP400038422 | Polygalacturonase inhibitor protein | chr07 | Yes | 4.51 | 4.49 |
| TVTNLGDGQSTYTAK | DMP400027005 | Subtilisin-like protease preproenzyme | chr12 | Yes | 4.48 | 4.51 |
| LCGEIPKGEYMK | DMP400014905 | Polygalacturonase inhibiting protein | chr09 | Yes | 4.45 | 4.17 |
| ADNLDTCYR | DMP400025990 | 41 kD chloroplast nucleoid DNA binding protein (CND41) | chr08 | Yes | 4.43 | 4.23 |
| GTGDFTGR | SW_g323.t1 | Pathogenesis-related protein 1b (Solanum tuberosum) | 4.41 | 4.49 | ||
| RIVDIPAGAFSFNSNT- GAGTIIDSGTVFTR | DMP400009572 | Aspartic proteinase nepenthesin-1 | chr01 | Yes | 4.38 | 4.55 |
| VIIADIQNDLGNSLVK | DMP400032777 | Short chain alcohol dehydrogenase | chr12 | No | 4.37 | 4.56 |
| TLPESTTNENK | K7WVA0 | Acyl-CoA-binding protein (Solanum tuberosum) | 4.37 | 4.42 | ||
| CHAVQCTANINGECPGQLK | DMP400023388 | Osmotin | Yes | 4.35 | 4.68 | |
| TNCNFDGDGR | Q01591 | Osmotin-like protein TPM-1 | 4.35 | 4.41 | ||
| LSEDGQVLEVLEDVEGK | DMP400030201 | Strictosidine synthase | chr07 | Yes | 4.31 | 4.59 |
| SMVGTPLMPGISVDTYIF- ALYDEDLKPGPGSER | DMP400001406 | Glucan endo-1,3-beta-glucosidase | chr01 | Yes | 4.3 | 4.65 |
| GNLDIFSGR | DMP400035839 | Wound/stress protein | chr04 | Yes | 4.27 | 4.6 |
| ITGNDYSSGVR | DMP400007118 | Citrate binding protein | chr11 | Yes | 4.26 | 4.54 |
| AVGEAGLGNDIK | DMP400062364 | Glucan endo-1,3-beta-glucosidase, basic isoform 2 | chr01 | No | 4.24 | 4.58 |
| HAGPQFDYLEK | DMP400019521 | Glutathione S-transferase omega | chr10 | No | 4.23 | 4.58 |
| SSSTDVFGR | DMP400043338 | Subtilisin-like protease | chr02 | Yes | 4.21 | 4.61 |
| YLVTIGGVEGNPGR | DMP400017956 | Miraculin | chr03 | Yes | 4.21 | 4.52 |
| MYQLSFK | DMP400050666 | Unidentified | chr08 | Yes | 4.21 | 4.48 |
| ADAGHVLVEK | DMP400022826 | MRNA binding protein | chr09 | No | 4.15 | 4.49 |
| GQGTVGTEINR | DMP400023006 | Threonine dehydratase biosynthetic, chloroplastic | chr09 | No | 4.14 | 4.52 |
| WQPSGADQAANR | P52405 | Endochitinase 3 | 4.1 | 4.45 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abreha, K.B.; Alexandersson, E.; Resjö, S.; Lankinen, Å.; Sueldo, D.; Kaschani, F.; Kaiser, M.; van der Hoorn, R.A.L.; Levander, F.; Andreasson, E. Leaf Apoplast of Field-Grown Potato Analyzed by Quantitative Proteomics and Activity-Based Protein Profiling. Int. J. Mol. Sci. 2021, 22, 12033. https://doi.org/10.3390/ijms222112033
Abreha KB, Alexandersson E, Resjö S, Lankinen Å, Sueldo D, Kaschani F, Kaiser M, van der Hoorn RAL, Levander F, Andreasson E. Leaf Apoplast of Field-Grown Potato Analyzed by Quantitative Proteomics and Activity-Based Protein Profiling. International Journal of Molecular Sciences. 2021; 22(21):12033. https://doi.org/10.3390/ijms222112033
Chicago/Turabian StyleAbreha, Kibrom B., Erik Alexandersson, Svante Resjö, Åsa Lankinen, Daniela Sueldo, Farnusch Kaschani, Markus Kaiser, Renier A. L. van der Hoorn, Fredrik Levander, and Erik Andreasson. 2021. "Leaf Apoplast of Field-Grown Potato Analyzed by Quantitative Proteomics and Activity-Based Protein Profiling" International Journal of Molecular Sciences 22, no. 21: 12033. https://doi.org/10.3390/ijms222112033
APA StyleAbreha, K. B., Alexandersson, E., Resjö, S., Lankinen, Å., Sueldo, D., Kaschani, F., Kaiser, M., van der Hoorn, R. A. L., Levander, F., & Andreasson, E. (2021). Leaf Apoplast of Field-Grown Potato Analyzed by Quantitative Proteomics and Activity-Based Protein Profiling. International Journal of Molecular Sciences, 22(21), 12033. https://doi.org/10.3390/ijms222112033