
Photopharmacology of Ion Channels through the Light of the Computational Microscope

 , , , and
, , , and
Abstract
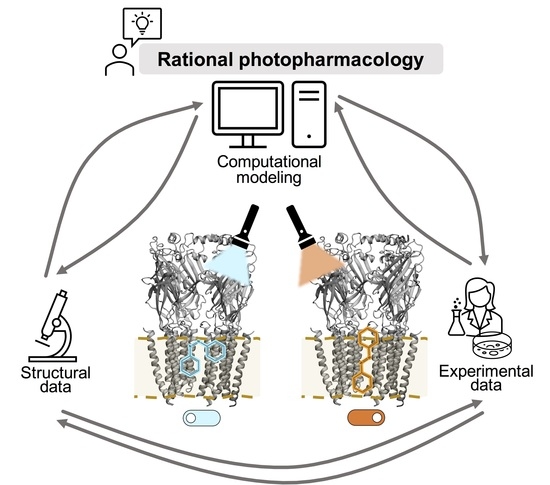
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Modeling
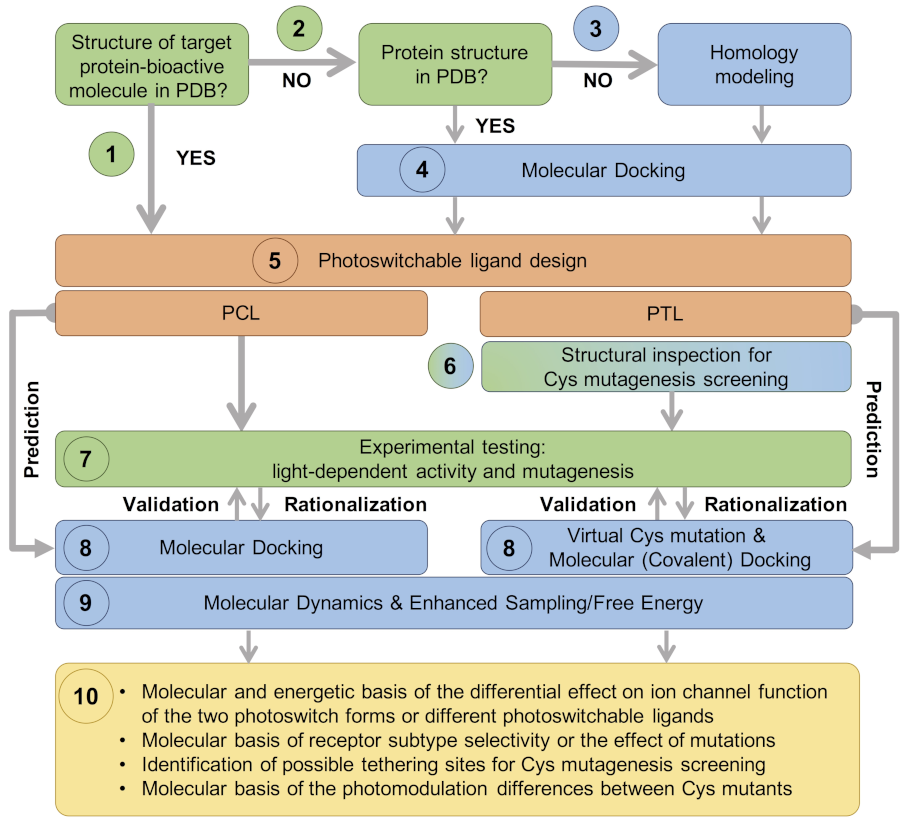
- (1)
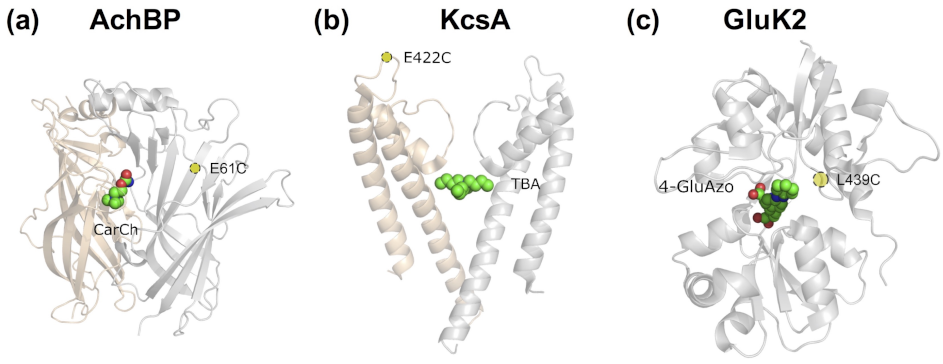
- In the most straightforward case, a search in the Protein Data Bank (PDB) yields an experimental structure of the target protein in complex with the bioactive molecule to be used as a basic module of the photoswitchable ligand. A structure of the target protein bound to a similar molecule (in terms of chemical structure and activity) or a structure of a homologous protein–ligand complex can also be used as a surrogate, as demonstrated by the examples mentioned in the Introduction.
- (2)
- In the absence of an experimental structure of the target protein–bioactive molecule complex, an experimental structure of the apo protein can alternatively be used; ideally, this structure contains the appropriate subunit composition and was captured in the relevant functional state.
- (3)
- When no experimental structure is available, homology modeling can generate a structural model of the target protein based on the experimental structure of a homologous template protein. When selecting the template structure, one should consider the sequence identity between the target and template and, additionally, other features of the template structure, such as the functional state and the bound ligand(s).
- (4)
- Although the (experimental or computational) structure of the protein alone is already informative, carrying out a computational molecular docking of the bioactive molecule can help to further optimize the photoswitchable ligand design. In particular, the predicted binding mode can be used to identify the optimal position to introduce the photochromic group and/or estimate the length of the linker between the different modules of the PCL or PTL, as well as pinpoint potential residues for Cys screening.
- (5)
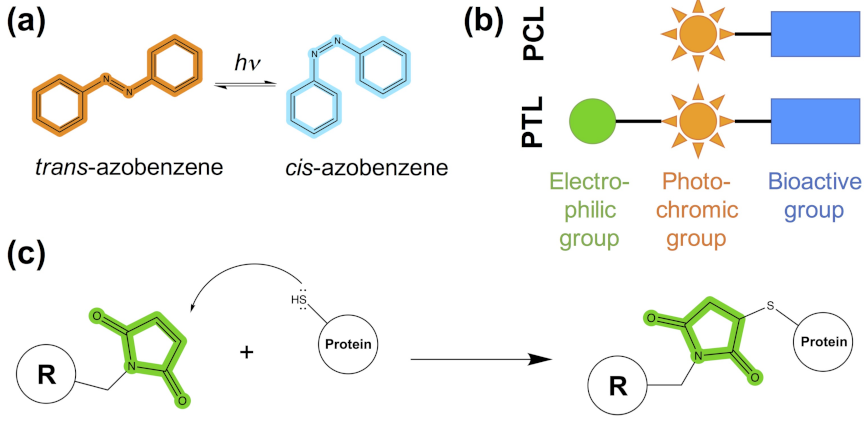
- The photoswitchable ligand (PCL or PTL) design follows the modular approach depicted in Figure 1b. As mentioned in steps (1)–(4), structural information on the binding mode of the bioactive module to the target protein can be used to guide such a design.
- (6)
- In the case of PTLs, their design additionally includes an inspection of the structure of the target protein, either experimental or computational, in order to identify putative tethering positions, i.e., residues near the ligand binding site amenable for cysteine mutagenesis screening.
- (7)
- Upon design of the photoswitchable ligand, synthesis and experimental testing can already be performed; the latter includes measuring the modulatory effect of the ligand under different light conditions, as well as site-directed mutagenesis (either Cys mutations for PTL covalent attachment or other mutations to confirm the binding site location and PCL/PTL ligand binding mode).
- (8)
- The observed light-dependent activity (or lack thereof), as well as the effect of mutations, can be rationalized a posteriori by performing a molecular docking of the PCL or virtual Cys mutation combined with covalent docking for the PTL. The resulting model of the target protein–photoswitch complex can be inspected to design additional site-directed mutations to validate the predicted PCL/PTL predicted binding mode. Alternatively, molecular docking can be used a priori (i.e., before experimental testing) to select the best candidate among several possible photoswitchable ligand designs (for subsequent experimental testing), as well as to explore alternative Cys tethering sites.
- (9)
- Though the (static) computational models described so far are already useful to understand the molecular basis of light-controlled ion channel modulation, they can additionally be refined by molecular dynamics. Such simulations, alone or in combination with enhanced sampling and free energy techniques, can provide further dynamical and energetic insights into the photoswitch effect, as explained earlier in this section.
- (10)
- This integrative computational-experimental approach offers a comprehensive understanding of the PCL/PTL effect on ion channel function, including but not limited to the information listed in the last step of the proposed workflow (see Figure 3).
3. Computational Modeling of Photoswitchable Ligands Targeting Voltage-Gated Ion Channels
3.1. Photoswitchable Pore Blockers
3.2. Photoswitchable Modulators
4. Computational Modeling of Photoswitchable Ligands Targeting Ligand-Gated Ion Channels
4.1. Nicotinic Acetylcholine Receptors
4.2. 5-HT3 Receptors
4.3. GABAA Receptors
4.4. Glycine Receptors
4.5. Ionotropic Glutamate Receptors
4.6. P2X Receptors
5. Conclusions and Perspectives
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Velema, W.A.; Szymanski, W.; Feringa, B.L. Photopharmacology: Beyond Proof of Principle. J. Am. Chem. Soc. 2014, 136, 2178–2191. [Google Scholar] [CrossRef] [Green Version]
- Broichhagen, J.; Frank, J.A.; Trauner, D. A Roadmap to Success in Photopharmacology. Acc. Chem. Res. 2015, 48, 1947–1960. [Google Scholar] [CrossRef] [PubMed]
- Hüll, K.; Morstein, J.; Trauner, D. In Vivo Photopharmacology. Chem. Rev. 2018, 118, 10710–10747. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Ellis-Davies, G.C.R.; Mourot, A. Optical control of neuronal ion channels and receptors. Nat. Rev. Neurosci. 2019, 20, 514–532. [Google Scholar] [CrossRef]
- Beharry, A.A.; Woolley, G.A. Azobenzene photoswitches for biomolecules. Chem. Soc. Rev. 2011, 40, 4422. [Google Scholar] [CrossRef]
- Fehrentz, T.; Schönberger, M.; Trauner, D. Optochemical Genetics. Angew. Chem. Int. Ed. 2011, 50, 12156–12182. [Google Scholar] [CrossRef] [PubMed]
- Ellis-Davies, G.C. Caged compounds: Photorelease technology for control of cellular chemistry and physiology. Nat. Methods 2007, 4, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Young, D.D.; Deiters, A. Photochemical control of biological processes. Org. Biomol. Chem. 2007, 5, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Klán, P.; Solomek, T.; Bochet, C.G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Photoremovable protecting groups in chemistry and biology: Reaction mechanisms and efficacy. Chem. Rev. 2013, 113, 119–191. [Google Scholar] [CrossRef]
- Welleman, I.M.; Hoorens, M.W.H.; Feringa, B.L.; Boersma, H.H.; Szymański, W. Photoresponsive molecular tools for emerging applications of light in medicine. Chem. Sci. 2020, 11, 11672–11691. [Google Scholar] [CrossRef] [PubMed]
- Deal, W.J.; Erlanger, B.F.; Nachmansohn, D. Photoregulation of biological activity by photochromic reagents, III. Photoregulation of bioelectricity by acetylcholine receptor inhibitors. Proc. Natl. Acad. Sci. USA 1969, 64, 1230–1234. [Google Scholar] [CrossRef] [Green Version]
- Bartels, E.; Wassermann, N.H.; Erlanger, B.F. Photochromic Activators of the Acetylcholine Receptor. Proc. Natl. Acad. Sci. USA 1971, 68, 1820–1823. [Google Scholar] [CrossRef] [Green Version]
- Silman, I.; Karlin, A. Acetylcholine Receptor: Covalent Attachment of Depolarizing Groups at the Active Site. Science 1969, 164, 1420–1421. [Google Scholar] [CrossRef] [PubMed]
- Brejc, K.; van Dijk, W.J.; Klaassen, R.V.; Schuurmans, M.; van der Oost, J.; Smit, A.B.; Sixma, T.K. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001, 411, 269–276. [Google Scholar] [CrossRef]
- Celie, P.H.; van Rossum-Fikkert, S.E.; van Dijk, W.J.; Brejc, K.; Smit, A.B.; Sixma, T.K. Nicotine and Carbamylcholine Binding to Nicotinic Acetylcholine Receptors as Studied in AChBP Crystal Structures. Neuron 2004, 41, 907–914. [Google Scholar] [CrossRef] [Green Version]
- Bourne, Y.; Talley, T.T.; Hansen, S.B.; Taylor, P.; Marchot, P. Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake α-neurotoxins and nicotinic receptors. EMBO J. 2005, 24, 1512–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, S.B.; Sulzenbacher, G.; Huxford, T.; Marchot, P.; Taylor, P.; Bourne, Y. Structures of Aplysia AChBP Complexes Nicotinic Agon. Antagon. Reveal Distinctive Bind. Interfaces Conform. EMBO J. 2005, 24, 3635–3646. [Google Scholar] [CrossRef]
- Lenaeus, M.J.; Vamvouka, M.; Focia, P.J.; Gross, A. Structural basis of TEA blockade in a model potassium channel. Nat. Struct. Mol. Biol. 2005, 12, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Reiter, A.; Skerra, A.; Trauner, D.; Schiefner, A. A Photoswitchable Neurotransmitter Analogue Bound to Its Receptor. Biochemistry 2013, 52, 8972–8974. [Google Scholar] [CrossRef] [PubMed]
- Doyle, D.A. The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Banghart, M.; Borges, K.; Isacoff, E.; Trauner, D.; Kramer, R.H. Light-activated ion channels for remote control of neuronal firing. Nat. Neurosci. 2004, 7, 1381–1386. [Google Scholar] [CrossRef] [Green Version]
- Chambers, J.J.; Banghart, M.R.; Trauner, D.; Kramer, R.H. Light-Induced Depolarization of Neurons Using a Modified Shaker K+ Channel and a Molecular Photoswitch. J. Neurophysiol. 2006, 96, 2792–2796. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.L. Crystal Structures of the GluR5 and GluR6 Ligand Binding Cores: Molecular Mechanisms Underlying Kainate Receptor Selectivity. Neuron 2005, 45, 539–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volgraf, M.; Gorostiza, P.; Szobota, S.; Helix, M.R.; Isacoff, E.Y.; Trauner, D. Reversibly Caged Glutamate: A Photochromic Agonist of Ionotropic Glutamate Receptors. J. Am. Chem. Soc. 2006, 129, 260–261. [Google Scholar] [CrossRef]
- Volgraf, M.; Gorostiza, P.; Numano, R.; Kramer, R.H.; Isacoff, E.Y.; Trauner, D. Allosteric control of an ionotropic glutamate receptor with an optical switch. Nat. Chem. Biol. 2005, 2, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Morstein, J.; Awale, M.; Reymond, J.L.; Trauner, D. Mapping the Azolog Space Enables the Optical Control of New Biological Targets. ACS Cent. Sci. 2019, 5, 607–618. [Google Scholar] [CrossRef]
- Lee, E.H.; Hsin, J.; Sotomayor, M.; Comellas, G.; Schulten, K. Discovery through the computational microscope. Structure 2009, 17, 1295–1306. [Google Scholar] [CrossRef] [Green Version]
- Gorostiza, P.; Isacoff, E. Optical switches and triggers for the manipulation of ion channels and pores. Mol. Biosyst. 2007, 3, 686. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Barrufet, A.; Izquierdo-Serra, M.; Gorostiza, P. Photoswitchable ion channels and receptors. In Novel Approaches for Single Molecule Activation and Detection; Benfenati, F., Di Fabrizio, E., Torre, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 169–188. [Google Scholar]
- Lerch, M.M.; Hansen, M.J.; van Dam, G.M.; Szymanski, W.; Feringa, B.L. Emerging Targets in Photopharmacology. Angew. Chem. Int. Ed. 2016, 55, 10978–10999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kienzler, M.A.; Isacoff, E.Y. Precise modulation of neuronal activity with synthetic photoswitchable ligands. Curr. Opin. Neurobiol. 2017, 45, 202–209. [Google Scholar] [CrossRef]
- Bregestovski, P.; Maleeva, G.; Gorostiza, P. Light-induced regulation of ligand-gated channel activity. Br. J. Pharmacol. 2017, 175, 1892–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bregestovski, P.D.; Maleeva, G.V. Photopharmacology: A Brief Review Using the Control of Potassium Channels as an Example. Neurosci. Behav. Physiol. 2019, 49, 184–191. [Google Scholar] [CrossRef]
- Bregestovski, P.D.; Ponomareva, D.N. Photochromic Modulation of Cys-loop Ligand-gated Ion Channels. J. Evol. Biochem. Physiol. 2021, 57, 354–371. [Google Scholar] [CrossRef]
- Olivella, M.; Gonzalez, A.; Pardo, L.; Deupi, X. Relation between sequence and structure in membrane proteins. Bioinformatics 2013, 29, 1589–1592. [Google Scholar] [CrossRef] [Green Version]
- Piccoli, S.; Suku, E.; Garonzi, M.; Giorgetti, A. Genome-wide Membrane Protein Structure Prediction. Curr. Genom. 2013, 14, 324–329. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.; Ribeiro, A.; Coimbra, J.; Neves, R.; Martins, S.; Moorthy, N.; Fernandes, P.; Ramos, M. Protein-Ligand Docking in the New Millennium—A Retrospective of 10 Years in the Field. Curr. Med. Chem. 2013, 20, 2296–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucher, D.; Guidoni, L.; Carloni, P.; Rothlisberger, U. Coordination Numbers of K+ and Na+ Ions Inside the Selectivity Filter of the KcsA Potassium Channel: Insights from First Principles Molecular Dynamics. Biophys. J. 2010, 98, L47–L49. [Google Scholar] [CrossRef] [Green Version]
- Salari, R.; Murlidaran, S.; Brannigan, G. Pentameric ligand-gated ion channels: Insights from computation. Mol. Simul. 2014, 40, 821–829. [Google Scholar] [CrossRef]
- Cournia, Z.; Allen, T.W.; Andricioaei, I.; Antonny, B.; Baum, D.; Brannigan, G.; Buchete, N.V.; Deckman, J.T.; Delemotte, L.; del Val, C.; et al. Membrane Protein Structure, Function, and Dynamics: A Perspective from Experiments and Theory. J. Membr. Biol. 2015, 248, 611–640. [Google Scholar] [CrossRef] [Green Version]
- Comitani, F.; Melis, C.; Molteni, C. Elucidating ligand binding and channel gating mechanisms in pentameric ligand-gated ion channels by atomistic simulations. Biochem. Soc. Trans. 2015, 43, 151–156. [Google Scholar] [CrossRef]
- Crnjar, A.; Comitani, F.; Melis, C.; Molteni, C. Mutagenesis computer experiments in pentameric ligand-gated ion channels: The role of simulation tools with different resolution. Interface Focus 2019, 9, 20180067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, R.J.; Carnevale, V.; Delemotte, L.; Hellmich, U.A.; Rothberg, B.S. Permeating disciplines: Overcoming barriers between molecular simulations and classical structure-function approaches in biological ion transport. Biochim. Biophys. Acta Biomembr. 2018, 1860, 927–942. [Google Scholar] [CrossRef]
- Carnevale, V.; Delemotte, L.; Howard, R.J. Molecular Dynamics Simulations of Ion Channels. Trends Biochem. Sci. 2021, 46, 621–622. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; Catterall, W.A. The VGL-Chanome: A Protein Superfamily Specialized for Electrical Signaling and Ionic Homeostasis. Sci. Signal. 2004, 2004, re15. [Google Scholar] [CrossRef] [PubMed]
- Moreau, A.; Gosselin-Badaroudine, P.; Chahine, M. Biophysics, pathophysiology, and pharmacology of ion channel gating pores. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Alexander, S.P.H.; Mathie, A.; Peters, J.A.; Veale, E.L.; Striessnig, J.; Kelly, E.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; et al. The Concise Guide to Pharmacology 2019/20: Ion channels. Br. J. Pharmacol. 2019, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Liu, D.; Wu, K.; Lei, J.; Yan, N. Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science 2019, 363, 1303–1308. [Google Scholar] [CrossRef]
- Mourot, A.; Herold, C.; Kienzler, M.A.; Kramer, R.H. Understanding and improving photo-control of ion channels in nociceptors with azobenzene photo-switches. Br. J. Pharmacol. 2017, 175, 2296–2311. [Google Scholar] [CrossRef]
- Fehrentz, T.; Huber, F.M.E.; Hartrampf, N.; Bruegmann, T.; Frank, J.A.; Fine, N.H.F.; Malan, D.; Danzl, J.G.; Tikhonov, D.B.; Sumser, M.; et al. Optical control of L-type Ca2+ channels using a diltiazem photoswitch. Nat. Chem. Biol. 2018, 14, 764–767. [Google Scholar] [CrossRef] [Green Version]
- Palmisano, V.F.; Gómez-Rodellar, C.; Pollak, H.; Cárdenas, G.; Corry, B.; Faraji, S.; Nogueira, J.J. Binding of azobenzene and P-Diaminoazobenzene Hum. Volt.-Gated Sodium Channel Nav1.4. Phys. Chem. Chem. Phys. 2021, 23, 3552–3564. [Google Scholar] [CrossRef]
- Trads, J.B.; Hüll, K.; Matsuura, B.S.; Laprell, L.; Fehrentz, T.; Görldt, N.; Kozek, K.A.; Weaver, C.D.; Klöcker, N.; Barber, D.M.; et al. Sign Inversion in Photopharmacology: Incorporation of Cyclic Azobenzenes in Photoswitchable Potassium Channel Blockers and Openers. Angew. Chem. Int. Ed. 2019, 58, 15421–15428. [Google Scholar] [CrossRef]
- Stein, M.; Breit, A.; Fehrentz, T.; Gudermann, T.; Trauner, D. Optical Control of TRPV1 Channels. Angew. Chem. Int. Ed. 2013, 52, 9845–9848. [Google Scholar] [CrossRef]
- Kokel, D.; Cheung, C.Y.J.; Mills, R.; Coutinho-Budd, J.; Huang, L.; Setola, V.; Sprague, J.; Jin, S.; Jin, Y.N.; Huang, X.P.; et al. Photochemical activation of TRPA1 channels in neurons and animals. Nat. Chem. Biol. 2013, 9, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, M.R.; Bhardwaj, R.; Lindinger, S.; Butorac, C.; Romanin, C.; Hediger, M.A.; Reymond, J.L. Photoswitchable Inhibitor of the Calcium Channel TRPV6. ACS Med. Chem. Lett. 2019, 10, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- Curcic, S.; Tiapko, O.; Groschner, K. Photopharmacology and opto-chemogenetics of TRPC channels-some therapeutic visions. Pharmacol. R. 2019, 200, 13–26. [Google Scholar] [CrossRef]
- Lichtenegger, M.; Tiapko, O.; Svobodova, B.; Stockner, T.; Glasnov, T.N.; Schreibmayer, W.; Platzer, D.; de la Cruz, G.G.; Krenn, S.; Schober, R.; et al. An optically controlled probe identifies lipid-gating fenestrations within the TRPC3 channel. Nat. Chem. Biol. 2018, 14, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, C.; Domene, C. Location and Character of Volatile General Anesthetics Binding Sites in the Transmembrane Domain of TRPV1. Mol. Pharm. 2018, 15, 3920–3930. [Google Scholar] [CrossRef] [Green Version]
- Oakes, V.; Domene, C. Combining Structural Data with Computational Methodologies to Investigate Structure–Function Relationships in TRP Channels. In TRP Channels; Springer: New York, NY, USA, 2019; Volume 1987, pp. 65–82. [Google Scholar] [CrossRef]
- Koleva, M.N.; Fernandez-Ballester, G. In Silico Approaches for TRP Channel Modulation. In TRP Channels; Springer: New York, NY, USA, 2019; Volume 1987, pp. 187–206. [Google Scholar] [CrossRef]
- Chernov-Rogan, T.; Gianti, E.; Liu, C.; Villemure, E.; Cridland, A.P.; Hu, X.; Ballini, E.; Lange, W.; Deisemann, H.; Li, T.; et al. TRPA1 modulation by piperidine carboxamides suggests an evolutionarily conserved binding site and gating mechanism. Proc. Natl. Acad. Sci. USA 2019, 116, 26008–26019. [Google Scholar] [CrossRef] [Green Version]
- Yazici, A.T.; Gianti, E.; Kasimova, M.A.; Lee, B.H.; Carnevale, V.; Rohacs, T. Dual regulation of TRPV1 channels by phosphatidylinositol via functionally distinct binding sites. J. Biol. Chem. 2021, 296, 100573. [Google Scholar] [CrossRef]
- Collingridge, G.L.; Olsen, R.W.; Peters, J.; Spedding, M. A nomenclature for ligand-gated ion channels. Neuropharmacology 2009, 56, 2–5. [Google Scholar] [CrossRef] [Green Version]
- Jaiteh, M.; Taly, A.; Hénin, J. Evolution of Pentameric Ligand-Gated Ion Channels: Pro-Loop Receptors. PLoS ONE 2016, 11, e0151934. [Google Scholar] [CrossRef] [Green Version]
- Although eukaryotic pLGICs have been traditionally denoted as Cys-loop receptors, the lack of such disulfide bond-containing loop in the prokaryotic members of this family makes the term pLGIC preferable, according to the latest guidelines of the NC-IUPHAR; see reference [47]. Alternatively, the name Pro-loop receptors has been proposed in reference [64].
- Zhao, Y.; Liu, S.; Zhou, Y.; Zhang, M.; Chen, H.; Xu, H.E.; Sun, D.; Liu, L.; Tian, C. Structural basis of human α7 nicotinic acetylcholine receptor activation. Cell Res. 2021, 31, 713–716. [Google Scholar] [CrossRef]
- Sobolevsky, A.I.; Rosconi, M.P.; Gouaux, E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 2009, 462, 745–756. [Google Scholar] [CrossRef] [Green Version]
- Mansoor, S.E.; Lü, W.; Oosterheert, W.; Shekhar, M.; Tajkhorshid, E.; Gouaux, E. X-ray structures define human P2X3 receptor gating cycle and antagonist action. Nature 2016, 538, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Tochitsky, I.; Banghart, M.R.; Mourot, A.; Yao, J.Z.; Gaub, B.; Kramer, R.H.; Trauner, D. Optochemical control of genetically engineered neuronal nicotinic acetylcholine receptors. Nat. Chem. 2012, 4, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Law, R.J.; Henchman, R.H.; McCammon, J.A. A gating mechanism proposed from a simulation of a human α7 nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 6813–6818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Shi, L.; Jiang, D.; Cheng, J.; Shao, X.; Li, Z. Azobenzene Modified Imidacloprid Derivatives as Photoswitchable Insecticides: Steering Molecular Activity in a Controllable Manner. Sci. Rep. 2015, 5, 13962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Xu, Q.; Xu, Z.; Wang, L.; Liu, Z.; Li, Z.; Shao, X. Optical Control of Invertebrate nAChR and Behaviors with Dithienylethene-Imidacloprid. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kesters, D.; Thompson, A.J.; Brams, M.; van Elk, R.; Spurny, R.; Geitmann, M.; Villalgordo, J.M.; Guskov, A.; Danielson, U.H.; Lummis, S.C.R.; et al. Structural basis of ligand recognition in 5-HT3 receptors. EMBO Rep. 2013, 14, 49–56. [Google Scholar] [CrossRef]
- Rustler, K.; Maleeva, G.; Bregestovski, P.; König, B. Azologization of serotonin 5-HT3 receptor antagonists. Beilstein J. Org. Chem. 2019, 15, 780–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassaine, G.; Deluz, C.; Grasso, L.; Wyss, R.; Tol, M.B.; Hovius, R.; Graff, A.; Stahlberg, H.; Tomizaki, T.; Desmyter, A.; et al. X-ray structure of the mouse serotonin 5-HT3 receptor. Nature 2014, 512, 276–281. [Google Scholar] [CrossRef]
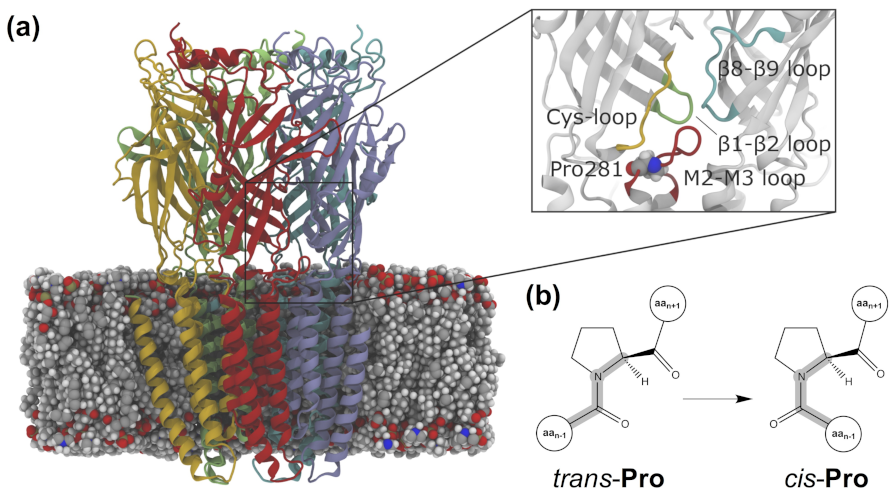
- Lummis, S.C.R.; Beene, D.L.; Lee, L.W.; Lester, H.A.; Broadhurst, R.W.; Dougherty, D.A. Cis-Trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature 2005, 438, 248–252. [Google Scholar] [CrossRef] [Green Version]
- Melis, C.; Bussi, G.; Lummis, S.C.R.; Molteni, C. Trans-Cis Switching Mechanisms in Proline Analogs and Their Relevance for the Gating of the 5-HT3 Receptor. J. Phys. Chem. B 2009, 113, 12148–12153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crnjar, A.; Comitani, F.; Hester, W.; Molteni, C. Trans-Cis Proline Switch. A Pentameric Ligand-Gated Ion Channel: How They Are Affect. How They Affect Biomol. Environ. J. Phys. Chem. Lett. 2019, 10, 694–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidpeter, P.A.M.; Rheinberger, J.; Nimigean, C.M. Prolyl isomerization controls activation kinetics of a cyclic nucleotide-gated ion channel. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Leone, V.; Lattanzi, G.; Molteni, C.; Carloni, P. Mechanism of Action of Cyclophilin A Explored by Metadynamics Simulations. PLoS Comput. Biol. 2009, 5, e1000309. [Google Scholar] [CrossRef] [Green Version]
- Maschio, M.C.; Fregoni, J.; Molteni, C.; Corni, S. Proline isomerization effects in the amyloidogenic protein β2-microglobulin. Phys. Chem. Chem. Phys. 2021, 23, 356–367. [Google Scholar] [CrossRef]
- Klippenstein, V.; Mony, L.; Paoletti, P. Probing Ion Channel Structure and Function Using Light-Sensitive Amino Acids. Trends Biochem. Sci. 2018, 43, 436–451. [Google Scholar] [CrossRef] [Green Version]
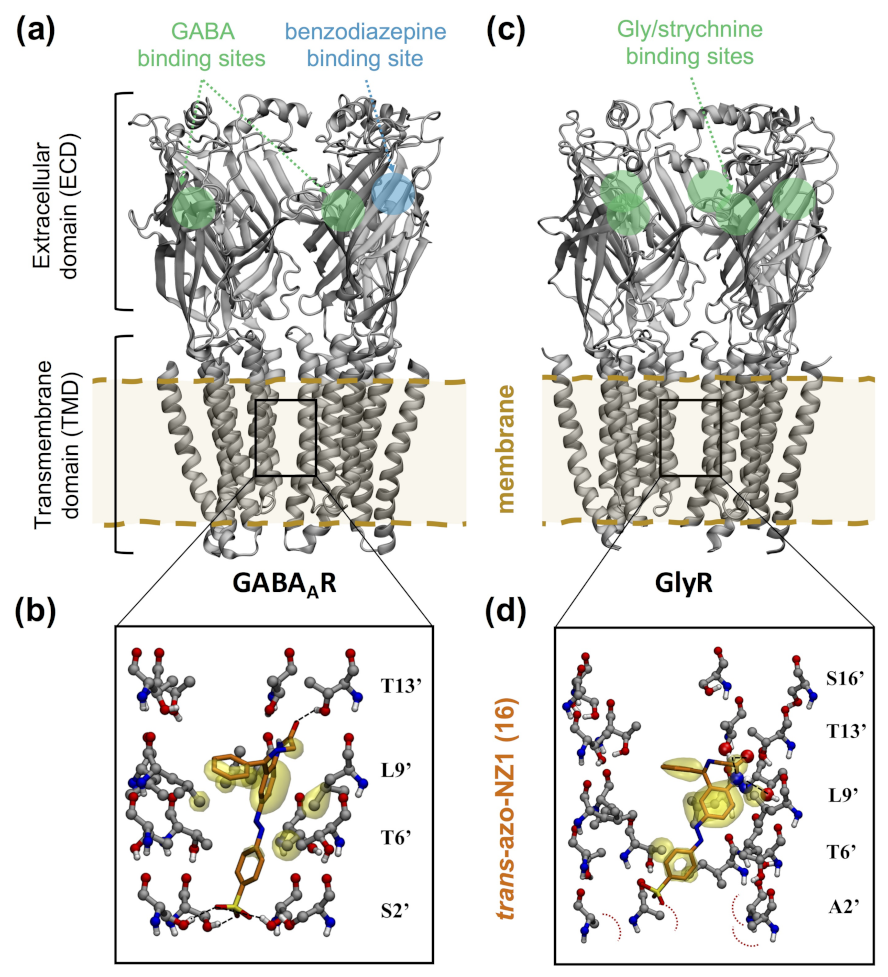
- Sieghart, W.; Savić, M.M. International Union of Basic and Clinical Pharmacology. CVI: GABAA Receptor Subtype- and Function-selective Ligands: Key Issues in Translation to Humans. Pharmacol. Rev. 2018, 70, 836–878. [Google Scholar] [CrossRef] [Green Version]
- Olsen, R.W.; Sieghart, W. GABAA receptors: Subtypes provide diversity of function and pharmacology. Neuropharmacology 2009, 56, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Belelli, D.; Hales, T.G.; Lambert, J.J.; Luscher, B.; Olsen, R.; Peters, J.A.; Rudolph, U.; Sieghart, W. GABAA receptors (version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS Guide Pharmacol. CITE 2019, 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Puthenkalam, R.; Hieckel, M.; Simeone, X.; Suwattanasophon, C.; Feldbauer, R.V.; Ecker, G.F.; Ernst, M. Structural Studies of GABAA Receptor Binding Sites: Which Experimental Structure Tells us What? Front. Mol. Neurosci. 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Scott, S.; Aricescu, A.R. A structural perspective on GABAA receptor pharmacology. Curr. Opin. Struct. Biol. 2019, 54, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Hibbs, R.E. Direct Structural Insights into GABAA Receptor Pharmacology. Trends Biochem. Sci. 2021, 46, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Davenport, C.M.; Mourot, A.; Vytla, D.; Smith, C.M.; Medeiros, K.A.; Chambers, J.J.; Kramer, R.H. Engineering a Light-Regulated GABAA Receptor for Optical Control of Neural Inhibition. ACS Chem. Biol. 2014, 9, 1414–1419. [Google Scholar] [CrossRef]
- Lin, W.C.; Tsai, M.C.; Davenport, C.M.; Smith, C.M.; Veit, J.; Wilson, N.M.; Adesnik, H.; Kramer, R.H. A Comprehensive Optogenetic Pharmacology Toolkit for In Vivo Control of GABAA Receptors and Synaptic Inhibition. Neuron 2015, 88, 879–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.C.; Tsai, M.C.; Rajappa, R.; Kramer, R.H. Design of a Highly Bistable Photoswitchable Tethered Ligand for Rapid and Sustained Manipulation of Neurotransmission. J. Am. Chem. Soc 2018, 140, 7445–7448. [Google Scholar] [CrossRef]
- Yue, L.; Pawlowski, M.; Dellal, S.S.; Xie, A.; Feng, F.; Otis, T.S.; Bruzik, K.S.; Qian, H.; Pepperberg, D.R. Robust photoregulation of GABAA receptors by allosteric modulation with a propofol analogue. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Stein, M.; Middendorp, S.J.; Carta, V.; Pejo, E.; Raines, D.E.; Forman, S.A.; Sigel, E.; Trauner, D. Azo-Propofols: Photochromic Potentiators of GABAA Receptors. Angew. Chem. Int. Ed. 2012, 51, 10500–10504. [Google Scholar] [CrossRef] [Green Version]
- Rustler, K.; Maleeva, G.; Gomila, A.M.J.; Gorostiza, P.; Bregestovski, P.; König, B. Optical Control of GABAA Receptors with a Fulgimide-Based Potentiator. Chem. Eur. J. 2020, 26, 12722–12727. [Google Scholar] [CrossRef]
- O’Mara, M.; Cromer, B.; Parker, M.; Chung, S.H. Homology Model of the GABAA Receptor Examined Using Brownian Dynamics. Biophys. J. 2005, 88, 3286–3299. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, M.; Huckvale, R.; Pandurangan, A.P.; Baker, J.R.; Smart, T.G. Optopharmacology reveals a differential contribution of native GABAA receptors to dendritic and somatic inhibition using azogabazine. Neuropharmacology 2020, 176, 108135. [Google Scholar] [CrossRef]
- Laverty, D.; Desai, R.; Uchański, T.; Masiulis, S.; Stec, W.J.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Steyaert, J.; Miller, K.W.; et al. Cryo-EM structure of the human α1β3γ2 GABAA receptor in a lipid bilayer. Nature 2019, 565, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Borghese, C.M.; Wang, H.Y.L.; McHardy, S.F.; Messing, R.O.; Trudell, J.R.; Harris, R.A.; Bertaccini, E.J. Modulation of α1β3γ2 GABAA receptors expressed in X. Laevis Oocytes Using A Propofol Photoswitch Tethered Transmembrane Helix. Proc. Natl. Acad. Sci. USA 2021, 118, e2008178118. [Google Scholar] [CrossRef]
- Bertaccini, E.J.; Yoluk, O.; Lindahl, E.R.; Trudell, J.R. Assessment of Homology Templates and an Anesthetic Binding Site within the γ-Aminobutyric Acid Receptor. Anesthesiology 2013, 119, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- Cayla, N.S.; Dagne, B.A.; Wu, Y.; Lu, Y.; Rodriguez, L.; Davies, D.L.; Gross, E.R.; Heifets, B.D.; Davies, M.F.; MacIver, M.B.; et al. A newly developed anesthetic based on a unique chemical core. Proc. Natl. Acad. Sci. USA 2019, 116, 15706–15715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GABA receptors composed by α, β and γ subunits are assembled in a β-α-γ-β-α heteropentameric arrangement. In order to distinguish the different subunit interfaces, a principal (+) and a complementary (-) sides are defined, so that the heteropentamer contains the following interfaces: β+α-, α+γ-, γ+β- and α+β-. The two GABA binding sites are then located at the two β+α- interfaces in the extracellular domain.
- Kim, J.J.; Gharpure, A.; Teng, J.; Zhuang, Y.; Howard, R.J.; Zhu, S.; Noviello, C.M.; Walsh, R.M.; Lindahl, E.; Hibbs, R.E. Shared structural mechanisms of general anaesthetics and benzodiazepines. Nature 2020, 585, 303–308. [Google Scholar] [CrossRef]
- Maleeva, G.; Wutz, D.; Rustler, K.; Nin-Hill, A.; Rovira, C.; Petukhova, E.; Bautista-Barrufet, A.; Gomila-Juaneda, A.; Scholze, P.; Peiretti, F.; et al. A photoswitchable GABA receptor channel blocker. Br. J. Pharmacol. 2019, 176, 2661–2677. [Google Scholar] [CrossRef] [Green Version]
- Although GABA receptors formed by ρ-subunits have been traditionally refer to as GABAC receptors, nowadays the NC-IUPHAR recommends their classification as GABAA receptors, based on structural and functional criteria [85].
- Bergmann, R.; Kongsbak, K.; Sørensen, P.L.; Sander, T.; Balle, T. A Unified Model of the GABAA Receptor Comprising Agonist and Benzodiazepine Binding Sites. PLoS ONE 2013, 8, e52323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naffaa, M.M.; Chebib, M.; Hibbs, D.E.; Hanrahan, J.R. Comparison of templates for homology model of ρ1 GABAC receptors: More insights to the orthosteric binding site’s structure and functionality. J. Mol. Graph. Model. 2015, 62, 43–55. [Google Scholar] [CrossRef]
- Hibbs, R.E.; Eric, G. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 2011, 474, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Laverty, D.; Thomas, P.; Field, M.; Andersen, O.J.; Gold, M.G.; Biggin, P.C.; Gielen, M.; Smart, T.G. Crystal structures of a GABAA-receptor chimera reveal new endogenous neurosteroid-binding sites. Nat. Struct. Mol. Biol. 2017, 24, 977–985. [Google Scholar] [CrossRef]
- Du, J.; Lü, W.; Wu, S.; Cheng, Y.; Gouaux, E. Glycine receptor mechanism elucidated by electron cryo-microscopy. Nature 2015, 526, 224–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maleeva, G.; Nin-Hill, A.; Rustler, K.; Petukhova, E.; Ponomareva, D.; Mukhametova, E.; Gomila, A.M.; Wutz, D.; Alfonso-Prieto, M.; König, B.; et al. Subunit-Specific Photocontrol of Glycine Receptors by Azobenzene-Nitrazepam Photoswitcher. eNeuro 2020, 8. [Google Scholar] [CrossRef]
- Lynch, J.W.; Zhang, Y.; Talwar, S.; Estrada-Mondragon, A. Glycine Receptor Drug Discovery. In Advances in Pharmacology; Geraghty, D.P., Rash, L.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 79, pp. 225–253. [Google Scholar] [CrossRef]
- Zeilhofer, H.U.; Acuña, M.A.; Gingras, J.; Yévenes, G.E. Glycine receptors and glycine transporters: Targets for novel analgesics? Cell. Mol. Life Sci. 2017, 75, 447–465. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Shaffer, P.L.; Ayube, S.; Bregman, H.; Chen, H.; Lehto, S.G.; Luther, J.A.; Matson, D.J.; McDonough, S.I.; Michelsen, K.; et al. Crystal structures of human glycine receptor α3 bound to a novel class of analgesic potentiators. Nat. Struct. Mol. Biol. 2016, 24, 108–113. [Google Scholar] [CrossRef]
- Gomila, A.M.; Rustler, K.; Maleeva, G.; Nin-Hill, A.; Wutz, D.; Bautista-Barrufet, A.; Rovira, X.; Bosch, M.; Mukhametova, E.; Petukhova, E.; et al. Photocontrol of Endogenous Glycine Receptors In Vivo. Cell Chem. Biol. 2020, 27, 1425–1433.e7. [Google Scholar] [CrossRef]
- Zhorov, B.S.; Bregestovski, P.D. Chloride Channels of Glycine and GABA Receptors with Blockers: Monte Carlo Minimization and Structure-Activity Relationships. Biophys. J. 2000, 78, 1786–1803. [Google Scholar] [CrossRef] [Green Version]
- Gielen, M.; Corringer, P.J. The dual-gate model for pentameric ligand-gated ion channels activation and desensitization. J. Physiol. 2018, 596, 1873–1902. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Wolter, T.; Kubař, T.; Sumser, M.; Trauner, D.; Elstner, M. Molecular Dynamics Investigation of gluazo, a Photo-Switchable Ligand for the Glutamate Receptor GluK2. PLoS ONE 2015, 10, e0135399. [Google Scholar] [CrossRef] [Green Version]
- Numano, R.; Szobota, S.; Lau, A.Y.; Gorostiza, P.; Volgraf, M.; Roux, B.; Trauner, D.; Isacoff, E.Y. Nanosculpting reversed wavelength sensitivity into a photoswitchable iGluR. Proc. Natl. Acad. Sci. USA 2009, 106, 6814–6819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlin, S.; Szobota, S.; Reiner, A.; Carroll, E.C.; Kienzler, M.A.; Guyon, A.; Xiao, T.; Trauner, D.; Isacoff, E.Y. A family of photoswitchable NMDA receptors. eLife 2016, 5, e12040. [Google Scholar] [CrossRef]
- Stawski, P.; Sumser, M.; Trauner, D. A Photochromic Agonist of AMPA Receptors. Angew. Chem. Int. Ed. 2012, 51, 5748–5751. [Google Scholar] [CrossRef] [PubMed]
- Wolter, T.; Steinbrecher, T.; Trauner, D.; Elstner, M. Ligand Photo-Isomerization Triggers Conformational Changes in iGluR2 Ligand Binding Domain. PLoS ONE 2014, 9, e92716. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, D.; Mondoloni, S.; Tange, J.; Lambolez, B.; Faure, P.; Taly, A.; Tricoire, L.; Mourot, A. Probing the ionotropic activity of glutamate GluD2 receptor in HEK cells with genetically-engineered photopharmacology. eLife 2020, 9, e59026. [Google Scholar] [CrossRef]
- Wang, T.; Ulrich, H.; Semyanov, A.; Illes, P.; Tang, Y. Optical control of purinergic signaling. Purinergic Signal. 2021, 17, 385–392. [Google Scholar] [CrossRef]
- Habermacher, C.; Martz, A.; Calimet, N.; Lemoine, D.; Peverini, L.; Specht, A.; Cecchini, M.; Grutter, T. Photo-switchable tweezers illuminate pore-opening motions of an ATP-gated P2X ion channel. eLife 2016, 5, e11050. [Google Scholar] [CrossRef]
- Lemoine, D.; Habermacher, C.; Martz, A.; Mery, P.F.; Bouquier, N.; Diverchy, F.; Taly, A.; Rassendren, F.; Specht, A.; Grutter, T. Optical control of an ion channel gate. Proc. Natl. Acad. Sci. USA 2013, 110, 20813–20818. [Google Scholar] [CrossRef] [Green Version]
- Kawate, T.; Carlisle Michel, J.; Birdsong, W.T.; Eric, G. Crystal structure of the ATP-gated P2X4 ion channel in the closed state. Nature 2009, 460, 592–598. [Google Scholar] [CrossRef]
- Hattori, M.; Eric, G. Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 2012, 485, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Egea, P.F.; Vecchio, A.J.; Asial, I.; Gupta, M.; Paulino, J.; Bajaj, R.; Dickinson, M.S.; Ferguson-Miller, S.; Monk, B.C.; et al. Highlighting membrane protein structure and function: A celebration of the Protein Data Bank. J. Biol. Chem. 2021, 296, 100557. [Google Scholar] [CrossRef]
- Arkhipova, V.; Fu, H.; Hoorens, M.W.H.; Trinco, G.; Lameijer, L.N.; Marin, E.; Feringa, B.L.; Poelarends, G.J.; Szymanski, W.; Slotboom, D.J.; et al. Structural Aspects of Photopharmacology: Insight into the Binding of Photoswitchable and Photocaged Inhibitors to the Glutamate Transporter Homologue. J. Am. Chem. Soc. 2021, 143, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Bianco, G.; Goodsell, D.S.; Forli, S. Selective and Effective: Current Progress in Computational Structure-Based Drug Discovery of Targeted Covalent Inhibitors. Trends Pharmacol. Sci. 2020, 41, 1038–1049. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, A.; Ferenczy, G.G.; Keseru, G.M. Binding Mode Prediction and Virtual Screening Applications by Covalent Docking. In Protein-Ligand Interactions and Drug Design; Ballante, F., Ed.; Springer: New York, NY, USA, 2021; Volume 2266, pp. 73–88. [Google Scholar] [CrossRef]
- Bryce, R.A. What Next for Quantum Mechanics in Structure-Based Drug Discovery. In Quantum Mechanics in Drug Discovery; Heifetz, A., Ed.; Springer: New York, NY, USA, 2020; Volume 2114, pp. 339–353. [Google Scholar] [CrossRef]
- Aucar, M.G.; Cavasotto, C.N. Molecular Docking Using Quantum Mechanical-Based Methods. In Quantum Mechanics in Drug Discovery; Heifetz, A., Ed.; Springer: New York, NY, USA, 2020; Volume 2114, pp. 269–284. [Google Scholar] [CrossRef]
- Vivo, M.D.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- Gioia, D.; Bertazzo, M.; Recanatini, M.; Masetti, M.; Cavalli, A. Dynamic Docking: A Paradigm Shift in Computational Drug Discovery. Molecules 2017, 22, 2029. [Google Scholar] [CrossRef] [Green Version]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef]
- Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int. J. Mol. Sci. 2020, 21, 6339. [Google Scholar] [CrossRef]
- Klaja, O.; Frank, J.A.; Trauner, D.; Bondar, A.N. Potential energy function for a photo-switchable lipid molecule. J. Comput. Chem. 2020, 41, 2336–2351. [Google Scholar] [CrossRef] [PubMed]
- Kotev, M.; Sarrat, L.; Gonzalez, C.D. User-Friendly Quantum Mechanics: Applications for Drug Discovery. In Quantum Mechanics in Drug Discovery; Heifetz, A., Ed.; Springer: New York, NY, USA, 2020; Volume 2114, pp. 231–255. [Google Scholar] [CrossRef]
- Cembran, A.; Bernardi, F.; Garavelli, M.; Gagliardi, L.; Orlandi, G. On the Mechanism of the Cis-Trans Isomerization Lowest Electron. States Azobenzene: S0, S1, T1. J. Am. Chem. Soc 2004, 126, 3234–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quick, M.; Dobryakov, A.L.; Gerecke, M.; Richter, C.; Berndt, F.; Ioffe, I.N.; Granovsky, A.A.; Mahrwald, R.; Ernsting, N.P.; Kovalenko, S.A. Photoisomerization Dynamics and Pathways of Trans-cis-Azobenzene in Solution from Broadband Femtosecond Spectroscopies and Calculations. J. Phys. Chem. B 2014, 118, 8756–8771. [Google Scholar] [CrossRef] [PubMed]
- Morstein, J.; Impastato, A.C.; Trauner, D. Photoswitchable Lipids. ChemBioChem 2020, 22, 73–83. [Google Scholar] [CrossRef]
- Dämgen, M.A.; Biggin, P.C. Computational methods to examine conformational changes and ligand-binding properties: Examples in neurobiology. Neurosci. Lett. 2019, 700, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.L.; Song, W.; Sansom, M.S. Lipid-Dependent Regulation of Ion Channels and G Protein–Coupled Receptors: Insights from Structures and Simulations. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 31–50. [Google Scholar] [CrossRef]
- Lachmann, D.; Lahmy, R.; König, B. Fulgimides as Light-Activated Tools in Biological Investigations. Eur. J. Org. Chem. 2019, 2019, 5018–5024. [Google Scholar] [CrossRef]
- Komarov, I.V.; Afonin, S.; Babii, O.; Schober, T.; Ulrich, A.S. Efficiently Photocontrollable or Not? Biological Activity of Photoisomerizable Diarylethenes. Chem. Eur. J. 2018, 24, 11245–11254. [Google Scholar] [CrossRef]
- Villarón, D.; Wezenberg, S.J. Stiff-Stilbene Photoswitches: From Fundamental Studies to Emergent Applications. Angew. Chem. Int. Ed. 2020, 59, 13192–13202. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nin-Hill, A.; Mueller, N.P.F.; Molteni, C.; Rovira, C.; Alfonso-Prieto, M. Photopharmacology of Ion Channels through the Light of the Computational Microscope. Int. J. Mol. Sci. 2021, 22, 12072. https://doi.org/10.3390/ijms222112072
Nin-Hill A, Mueller NPF, Molteni C, Rovira C, Alfonso-Prieto M. Photopharmacology of Ion Channels through the Light of the Computational Microscope. International Journal of Molecular Sciences. 2021; 22(21):12072. https://doi.org/10.3390/ijms222112072
Chicago/Turabian StyleNin-Hill, Alba, Nicolas Pierre Friedrich Mueller, Carla Molteni, Carme Rovira, and Mercedes Alfonso-Prieto. 2021. "Photopharmacology of Ion Channels through the Light of the Computational Microscope" International Journal of Molecular Sciences 22, no. 21: 12072. https://doi.org/10.3390/ijms222112072