
Dual Role of Thrombospondin-1 in Flow-Induced Remodeling

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
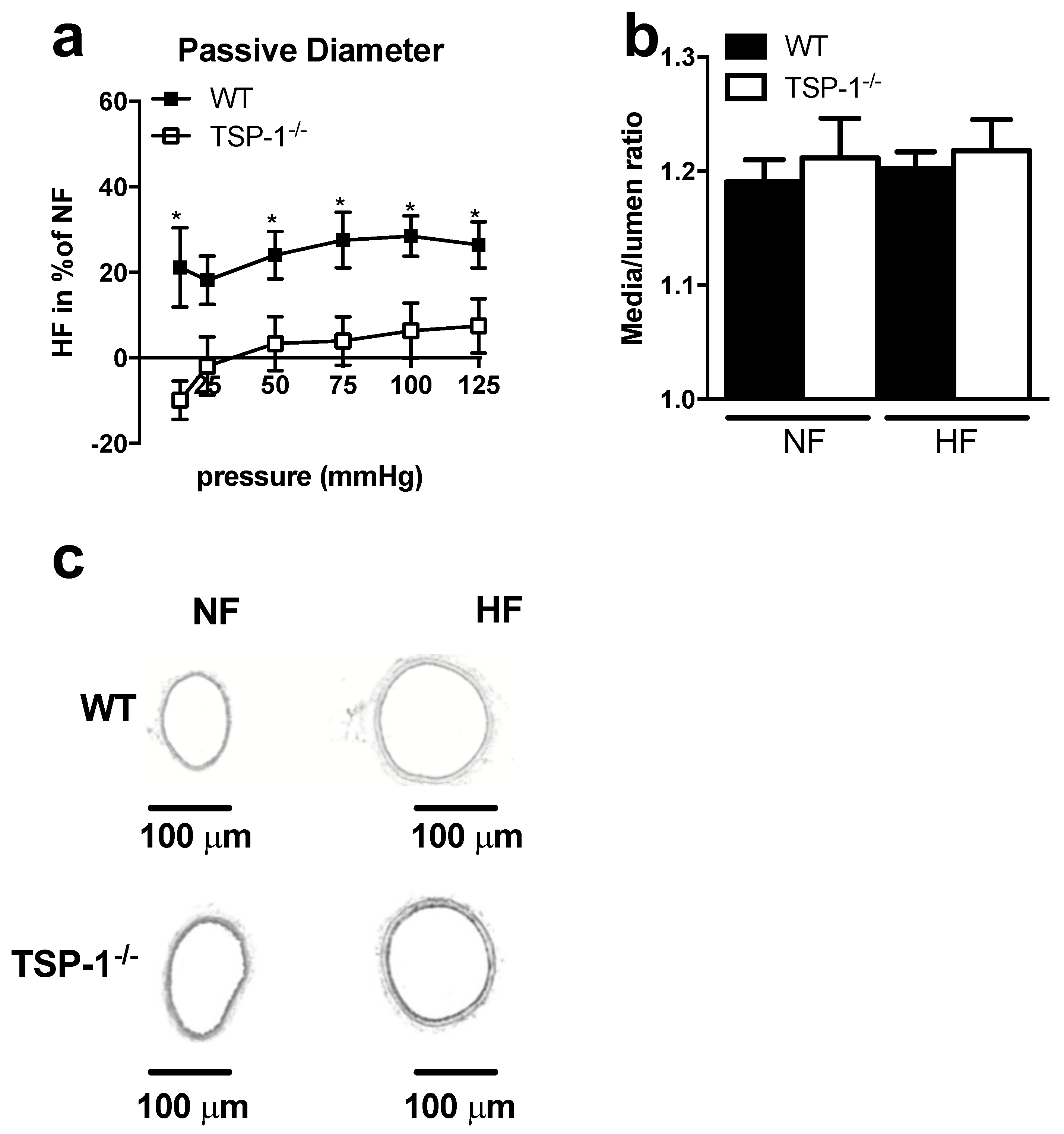
2.1. TSP-1 Modulates Flow-Mediated Remodeling
2.2. Circulating TSP-1 Induces Arterial Diameter Enlargement, but Tissue TSP-1 Participates in Arterial Parietal Thickening
- (1)
- We transferred WT bone marrow cells to TSP-1−/− mice to have WT circulating cells in TSP1−/− mice. The TSP-1 is only present in circulating cells but not in mice tissues. This group is named “WT BMC in TSP-1−/−“.
- (2)
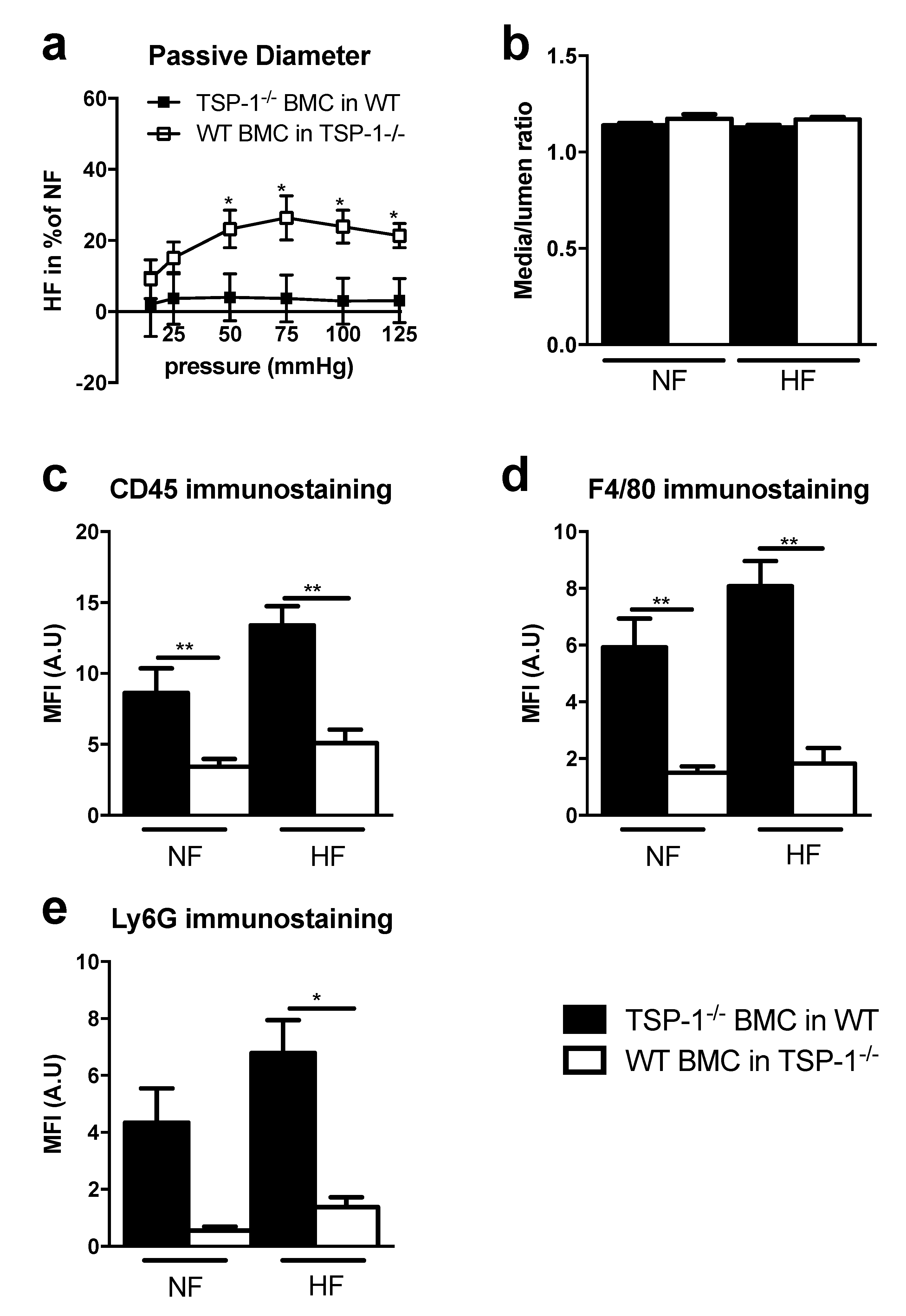
- We transferred TSP-1 bone marrow cells to WT mice to have TSP-1−/− circulating cells in WT mice. So, TSP-1 is only present in mice tissues but not in circulating cells. This group is named “TSP-1−/− BMC in WT“.Seven days after ligation, the HF/NF diameter ratio of mesenteric arteries in WT BMC in TSP-1−/− was increased compared to TSP-1−/− BMC in WT.Strikingly, there was no significant difference in TSP-1−/− BMC in the WT mice group (Figure 2a), showing a curve belonging to 0% of change. The two curves that were statistically different demonstrated that circulating WT cells, i.e., expressing TSP-1, were necessary for HF diameter expansion.Histomorphometry analysis revealed that media/lumen ratio (Figure 2b) and the cross-section area (Figure S3d) were not significantly different in NF and HF arteries of TSP-1−/− BMC in WT and WT BMC in TSP-1−/−. Media thickness was significantly increased in HF arteries in WT BMC in TSP-1−/− compared to TSP-1−/− BMC in WT. Furthermore, no differences were found between NF and HF arteries in TSP-1−/− BMC in WT or in WT BMC in TSP-1−/− (Figure S3c).
2.3. Hydralazine Does Not Improve Flow-Induced Remodeling in TSP-1−/− Mice
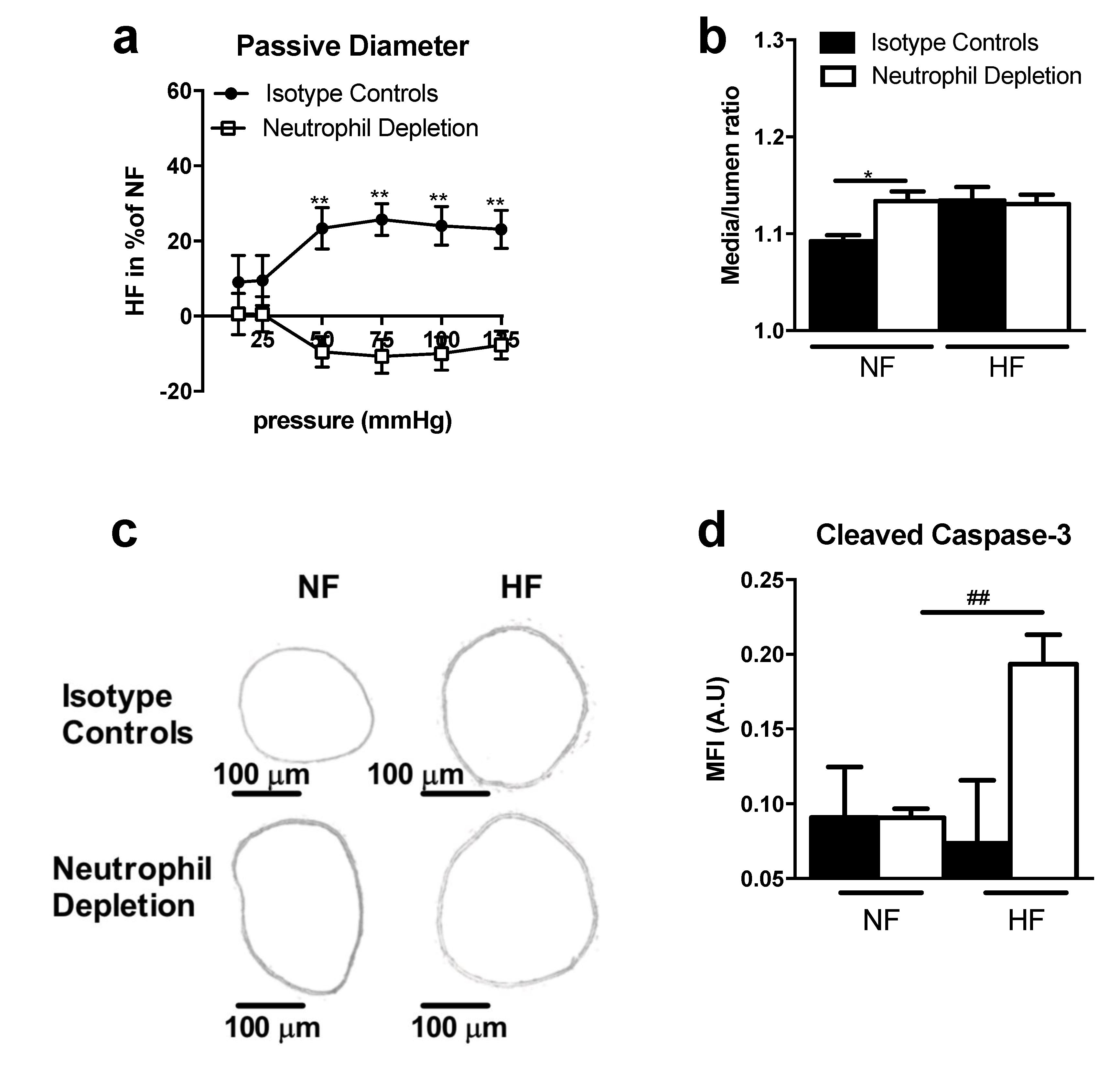
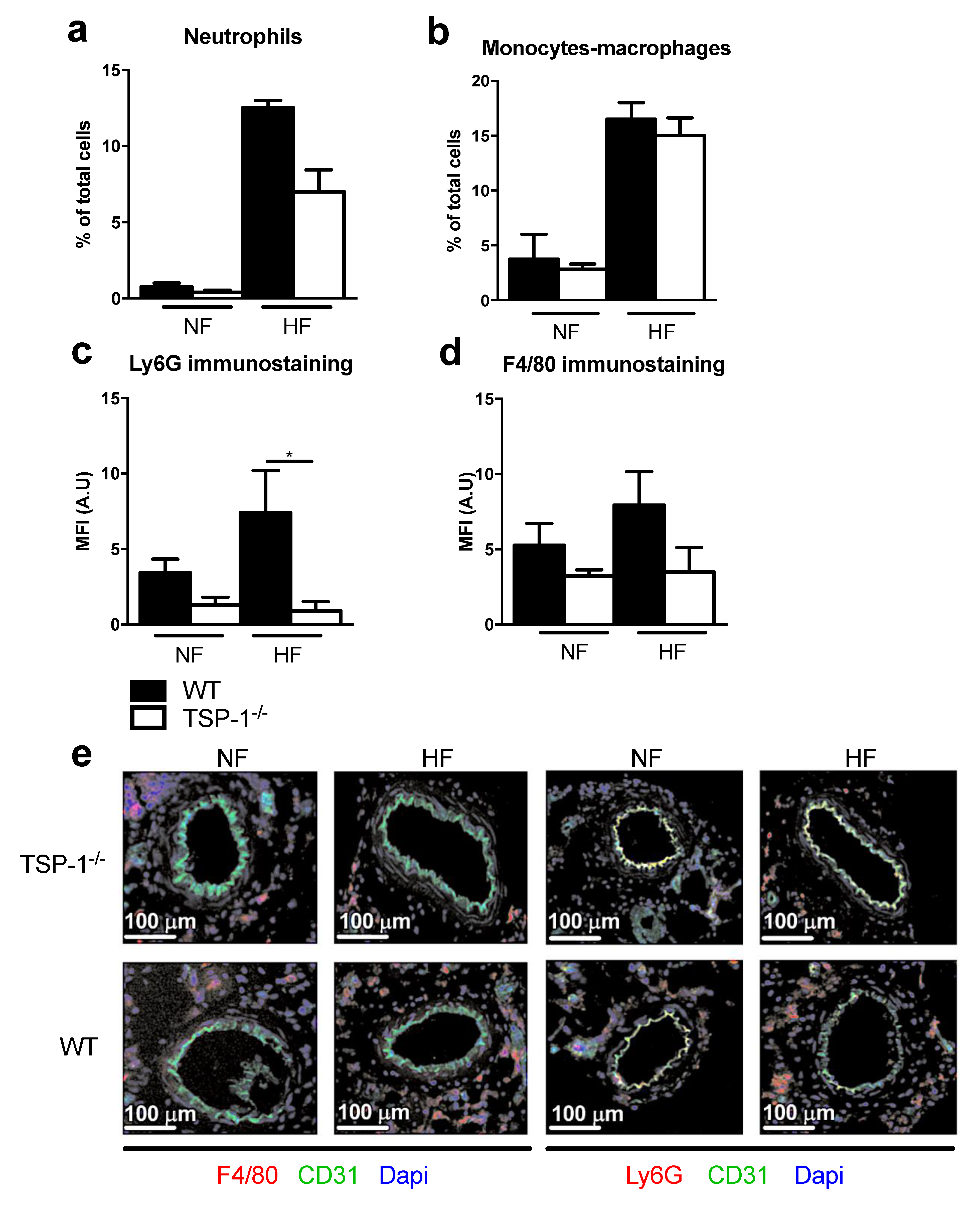
2.4. TSP-1 Mediates Flow-Induced Immune Cell Recruitment
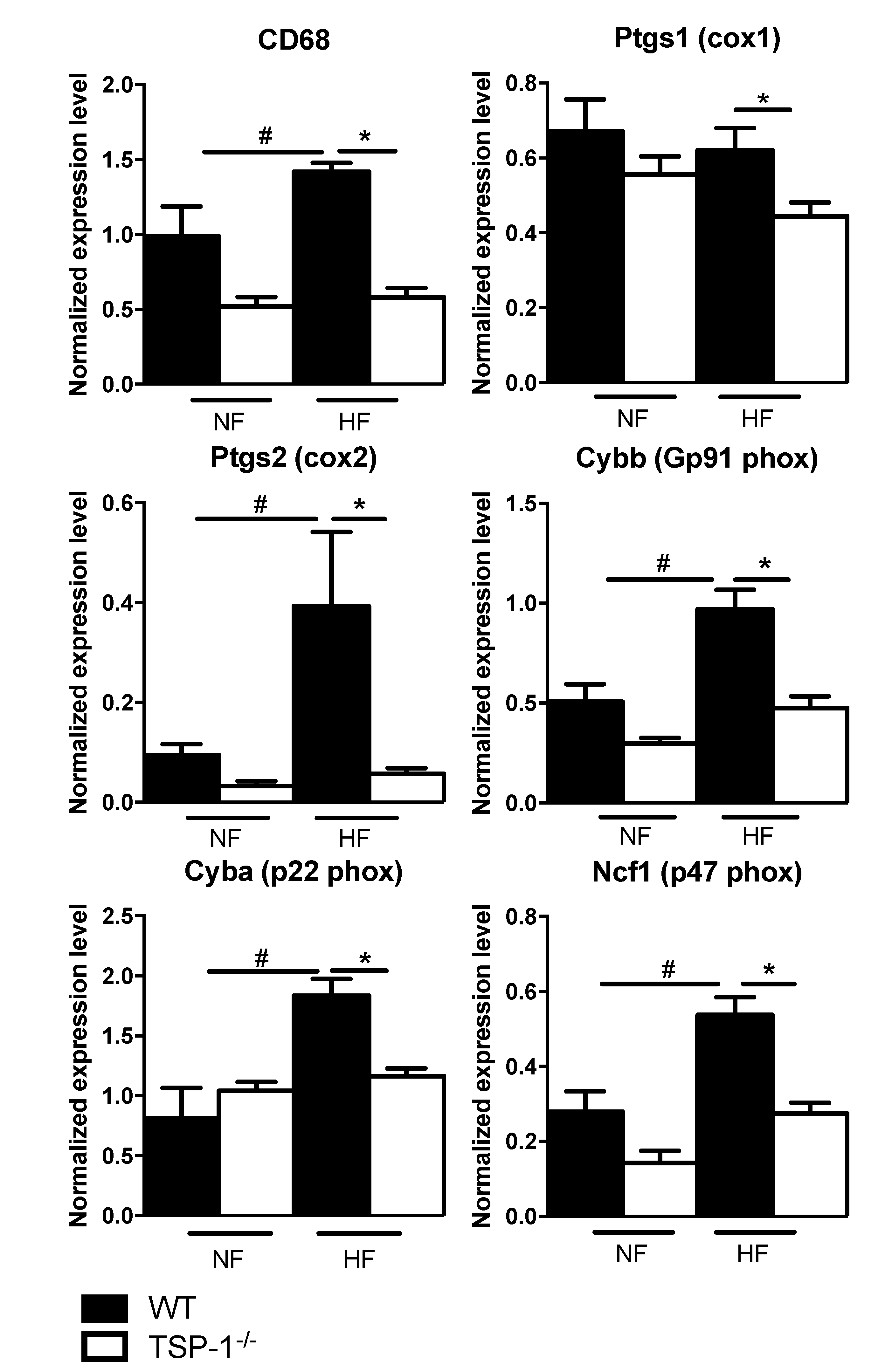
2.5. TSP-1 Deficiency Decreases Flow-Induced Inflammatory Gene Expression Levels
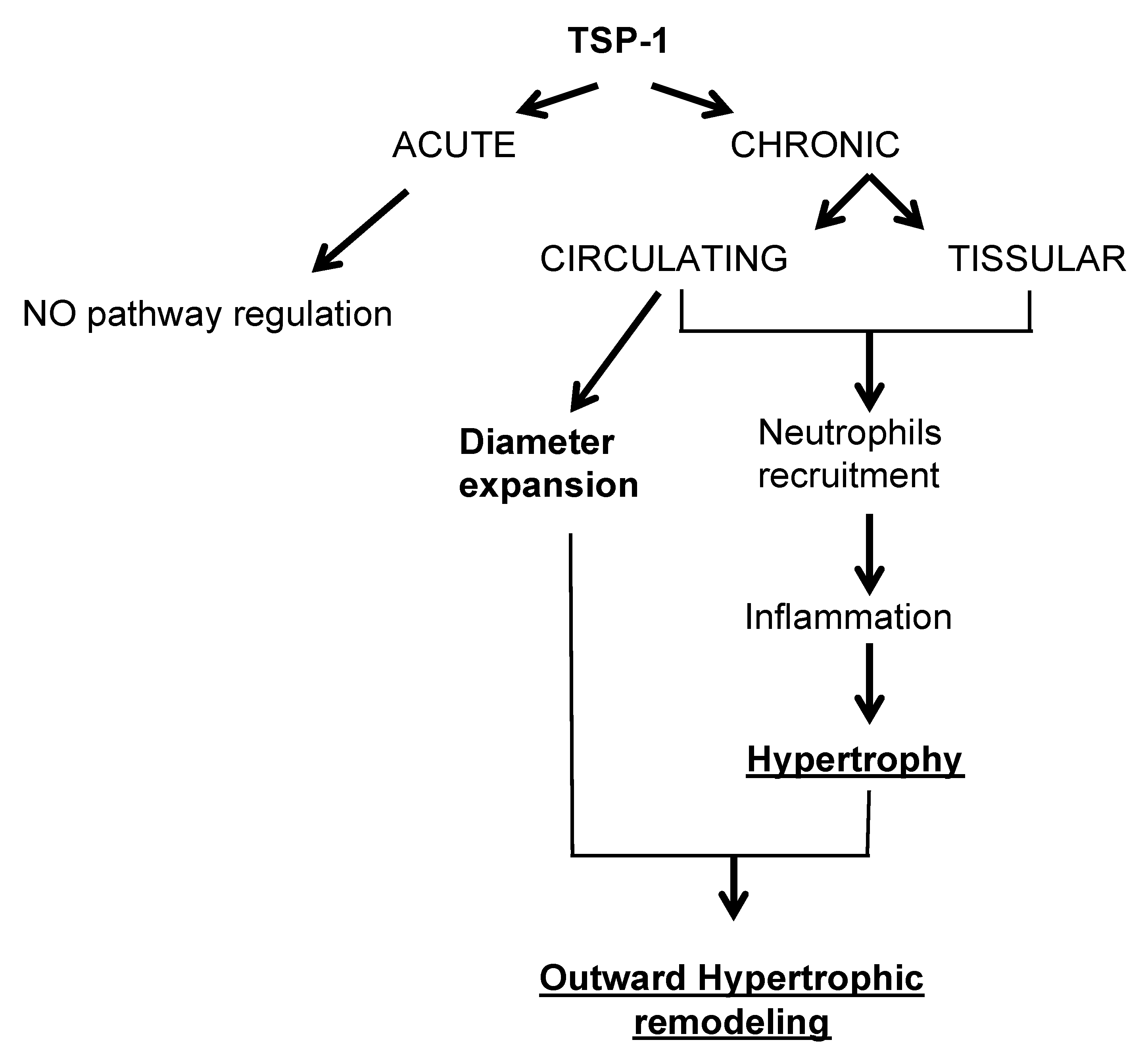
3. Discussion
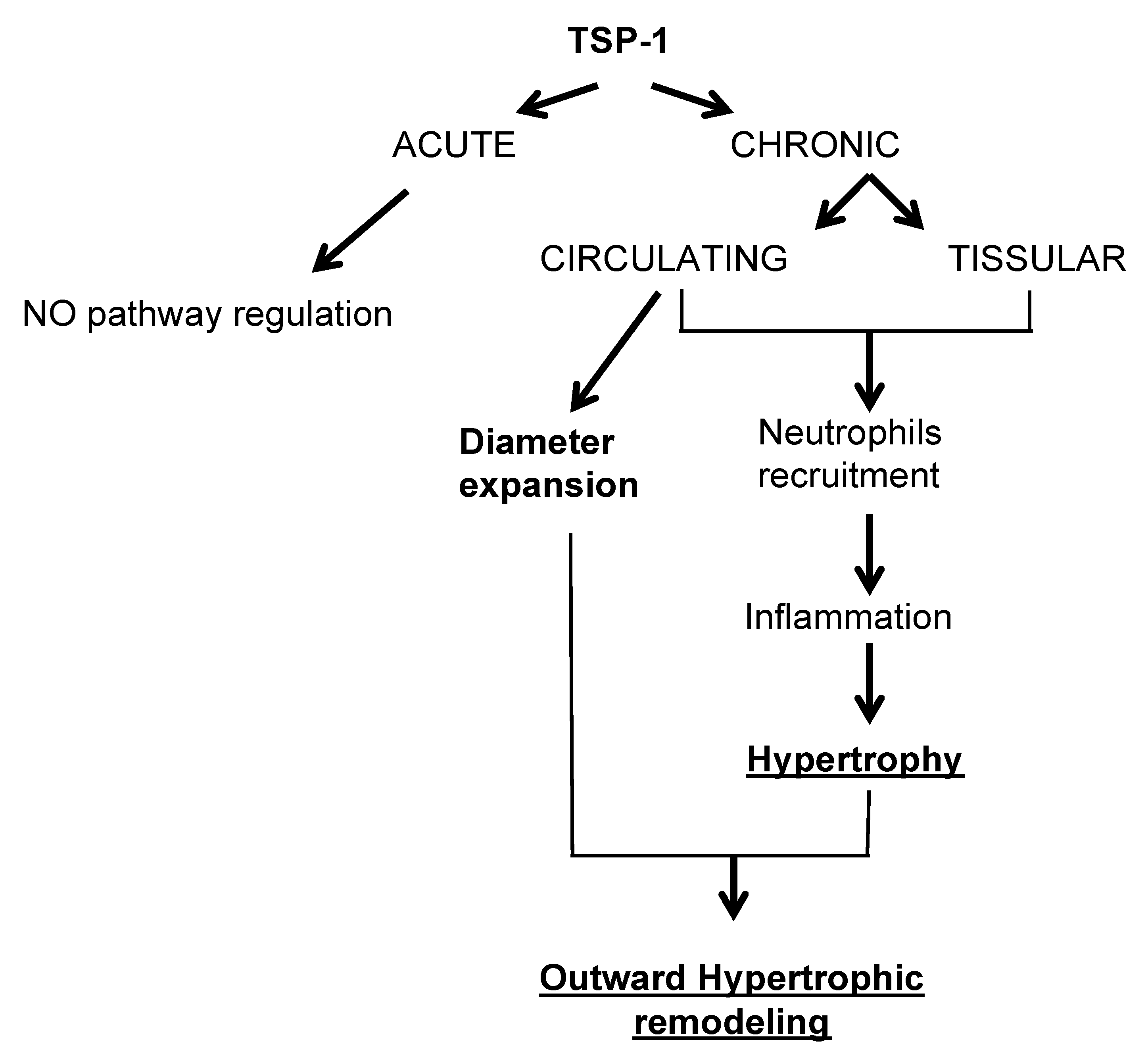
3.1. TSP-1 Is Essential for Small Vessel Remodeling during Adaptation to Blood Flow Increase
3.2. TSP-1 from Different Cellular Origins Impacts Small Vessel Remodeling in Different Ways
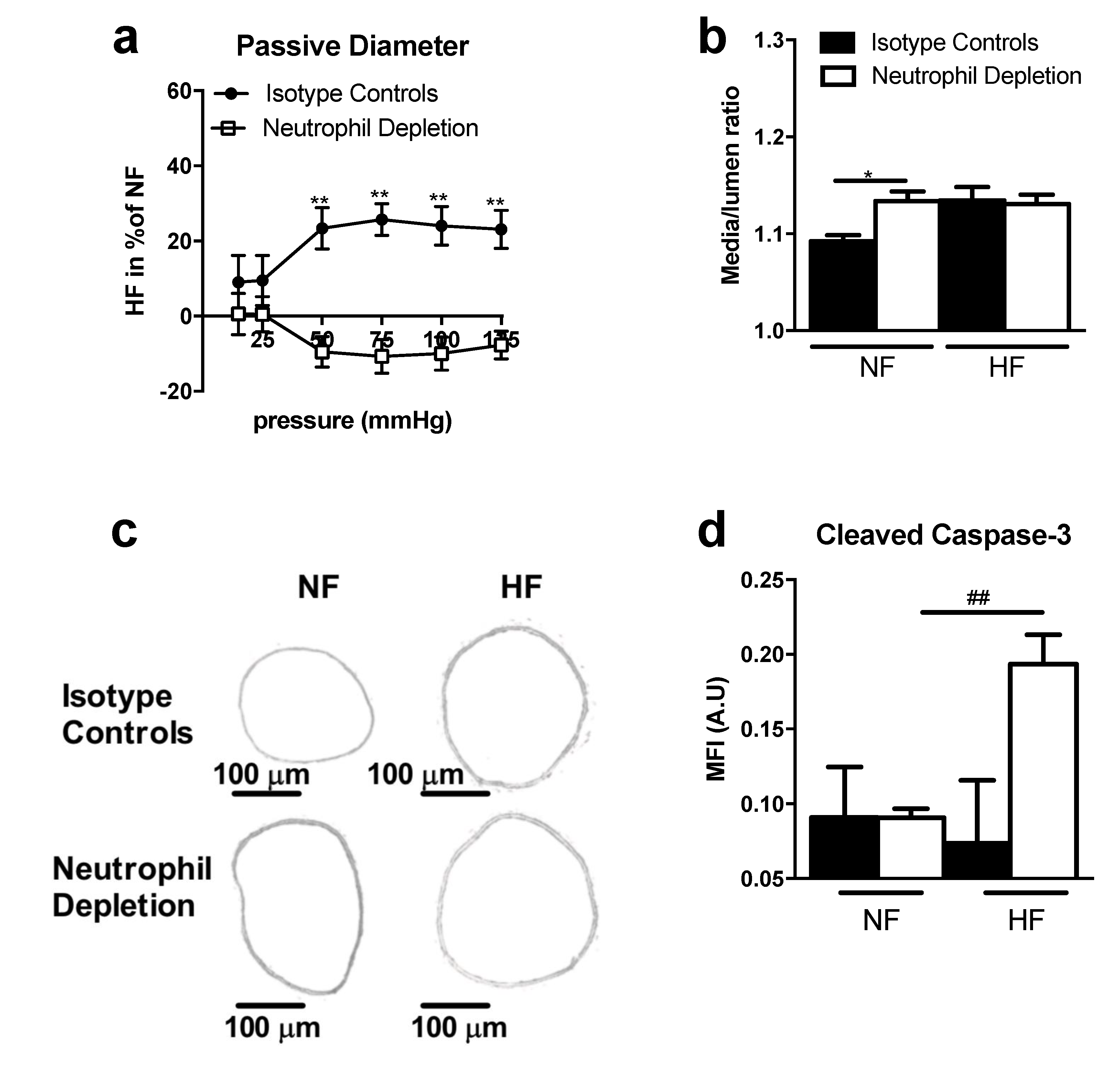
3.3. TSP-1 Participates in Blood Flow-Mediated Small Vessel Remodeling by Recruiting Inflammatory Cells
3.4. TSP-1 as a Therapeutic Target during Small Vessel Remodeling
4. Materials and Methods
4.1. Animal Models
4.2. Pressure–Diameter Relationship in Mesenteric Arteries
4.3. Histomorphometric Analyses
4.4. Quantitative Real-Time PCR
4.5. Neutrophil Depletion
4.6. Confocal Microscopy
4.7. Flow Cytometry Analysis
4.8. Mouse Irradiation and Adoptive Bone Marrow Transfer
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HF | High Flow |
| NF | Normal Flow |
| FMD | Flow-mediated dilation |
| CDR | Cumulative Dose Response |
| BM | Bone Marrow |
| MFI | Mean Fluorescence Intensity |
| TSP | Thrombospondin |
| WT | Wild Type |
| CD | Cluster of Differentiation |
| CSA | Cross Section Area |
References
- Whyte, J.J.; Laughlin, M.H. The effects of acute and chronic exercise on the vasculature. Acta Physiol. 2010, 199, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Osol, G.; Mandala, M. Maternal uterine vascular remodeling during pregnancy. Physiology 2009, 24, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Castorena-Gonzalez, J.A.; Staiculescu, M.C.; Foote, C.; Martinez-Lemus, L.A. Mechanisms of the inward remodeling process in resistance vessels: Is the actin cytoskeleton involved? Microcirculation 2014, 21, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Chantler, D.P.; Frisbee, J.C. Arterial function in cardio-metabolic diseases: From the micro-circulation to the large conduits. Prog. Cardiovasc. Dis. 2015, 57, 489–496. [Google Scholar] [CrossRef]
- Lehmann, M.V.; Schmieder, R.E. Remodeling of retinal small arteries in hypertension. Am. J. Hypertens. 2011, 24, 1267–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tronc, F.; Wassef, M.; Esposito, B.; Henrion, D.; Glagov, S.; Tedgui, A. Role of NO in flow-induced remodeling of the rabbit common carotid artery. Arter. Thromb. Vasc. Biol. 1996, 16, 1256–1262. [Google Scholar] [CrossRef]
- Martinez-Lemus, L.A.; Hill, M.; Meininger, G.A. The plastic nature of the vascular Wall: A continuum of remodeling events contributing to control of arteriolar diameter and structure. Physiology 2009, 24, 45–57. [Google Scholar] [CrossRef]
- Miyashiro, J.K.; Poppa, V.; Berk, B.C. Flow-induced vascular remodeling in the rat carotid artery diminishes with age. Circ. Res. 1997, 81, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Dumont, O.; Loufrani, L.; Henrion, D. Key role of the NO-pathway and matrix metalloprotease-9 in high blood flow-induced remodeling of rat resistance arteries. Arterioscler. Thrombos. Vasc. Biol. 2007, 27, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cousin, M.; Custaud, M.-A.; Baron-Menguy, C.; Toutain, B.; Dumont, O.; Guihot, A.-L.; Vessières, E.; Subra, J.-F.; Henrion, D.; Loufrani, L. Role of angiotensin II in the remodeling induced by a chronic increase in flow in rat mesenteric resistance arteries. Hypertension 2010, 55, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Caillon, A.; Grenier, C.; Grimaud, L.; Vessieres, E.; Guihot, A.-L.; Blanchard, S.; Lelievre, E.; Chabbert, M.; Foucher, E.D.; Jeannin, P.; et al. The angiotensin II type 2 receptor activates flow-mediated outward remodeling through T cells-dependent interleukin-17 production. Cardiovasc. Res. 2016, 112, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Abeles, D.; Kwei, S.; Stavrakis, G.; Zhang, Y.; Wang, E.T.; García-Cardeña, G. Gene expression changes evoked in a venous segment exposed to arterial flow. J. Vasc. Surg. 2006, 44, 863–870. [Google Scholar] [CrossRef] [Green Version]
- Isenberg, J.S.; Martin-Manso, G.; Maxhimer, J.B.; Roberts, D.D. Regulation of nitric oxide signalling by thrombospondin 1: Implications for anti-angiogenic therapies. Nat. Rev. Cancer 2009, 9, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Baenziger, N.L.; Brodie, G.; Majerus, P.W. Isolation and properties of a thrombin-sensitive protein of human platelets. J. Biol. Chem. 1972, 247, 2723–2731. [Google Scholar] [CrossRef]
- Lawler, J.; Slayter, H.; Coligan, J. Isolation and characterization of a high molecular weight glycoprotein from human blood platelets. J. Biol. Chem. 1978, 253, 8609–8616. [Google Scholar] [CrossRef]
- Raugi, G.J.; Mumby, S.M.; Abbott-Brown, D.; Bornstein, P. Thrombospondin: Synthesis and secretion by cells in culture. J. Cell Biol. 1982, 95, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Majack, A.R.; Cook, S.C.; Bornstein, P. Platelet-derived growth factor and heparin-like gly-cosaminoglycans regulate thrombospondin synthesis and deposition in the matrix by smooth muscle cells. J. Cell Biol. 1985, 101, 1059–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy-Ullrich, E.J.; Mosher, D.F. Localization of thrombospondin in clots formed in situ. Blood 1985, 66, 1098–1104. [Google Scholar] [CrossRef]
- Jaffe, A.E.; Ruggiero, J.T.; Falcone, D.J. Monocytes and macrophages synthesize and secrete thrombospondin. Blood 1985, 65, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Ticchioni, M.; Deckert, M.; Mary, F.; Bernard, G.; Brown, E.J.; Bernard, A. Integrin-associated protein (CD47) is a comitogenic molecule on CD3-activated human T cells. J. Immunol. 1997, 158, 677–684. [Google Scholar]
- Yoshida, H.; Tomiyama, Y.; Ishikawa, J.; Oritani, K.; Matsumura, I.; Shiraga, M.; Yokota, T.; Okajima, Y.; Ogawa, M.; Miyagawa, J.I.; et al. Integrin-associated protein/CD47 regulates motile activity in human B-cell lines through CDC42. Blood 2000, 96, 234–241. [Google Scholar] [CrossRef]
- Hermann, P.; Armant, M.; Brown, E.; Rubio, M.; Ishihara, H.; Ulrich, D.; Caspary, R.G.; Lindberg, F.P.; Armitage, R.; Maliszewski, C.; et al. The vitronectin receptor and its associated CD47 molecule mediates proin-flammatory cytokine synthesis in human monocytes by interaction with soluble CD23. J. Cell Biol. 1999, 144, 767–775. [Google Scholar] [CrossRef]
- Dorahy, D.J.; Thorne, R.F.; Fecondo, J.V.; Burns, G.F. Stimulation of platelet activation and aggregation by a carboxyl-terminal peptide from thrombospondin binding to the integrin-associated protein receptor. J. Biol. Chem. 1997, 272, 1323–1330. [Google Scholar] [CrossRef] [Green Version]
- Oldenborg, P.-A.; Zheleznyak, A.; Fang, Y.-F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Cooper, D.; Lindberg, F.P.; Gamble, J.R.; Brown, E.J.; Vadas, M.A. Transendothelial migration of neutrophils involves integrin-associated protein (CD47). Proc. Natl. Acad. Sci. USA 1995, 92, 3978–3982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.Q.; Frazier, W.A. The thrombospondin receptor CD47 (IAP) modulates and associ-ates with alpha2 beta1 integrin in vascular smooth muscle cells. Mol. Biol. Cell 1998, 9, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Phelan, M.W.; Forman, L.W.; Perrine, S.P.; Faller, D.V. Hypoxia increases thrombospondin-1 transcript and protein in cultured endothelial cells. J. Lab. Clin. Med. 1998, 132, 519–529. [Google Scholar] [CrossRef]
- Ridnour, L.A.; Isenberg, J.S.; Espey, M.G.; Thomas, D.D.; Roberts, D.; Wink, D.A. Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc. Natl. Acad. Sci. USA 2005, 102, 13147–13152. [Google Scholar] [CrossRef] [Green Version]
- Freyberg, M.A.; Kaiser, D.; Graf, R.; Vischer, P.; Friedl, P. Integrin-associated protein and thrombospondin-1 as endothelial mechano-sensitive death mediators. Biochem. Biophys. Res. Commun. 2000, 271, 584–588. [Google Scholar] [CrossRef]
- Freidja, M.L.; Vessieres, E.; Clere, N.; Desquiret, V.; Guihot, A.-L.; Toutain, B.; Loufrani, L.; Jardel, A.; Procaccio, V.; Faure, S.; et al. Heme oxygenase-1 induction restores high-blood-flow-dependent remodeling and endothelial function in mesenteric arteries of old rats. J. Hypertens. 2011, 29, 102–112. [Google Scholar] [CrossRef] [Green Version]
- Bakker, E.N.; Matlung, H.L.; Bonta, P.; De Vries, C.J.; Van Rooijen, N.; Van Bavel, E. Blood flow-dependent arterial remodelling is facilitated by inflammation but directed by vascular tone. Cardiovasc. Res. 2008, 78, 341–348. [Google Scholar] [CrossRef]
- Silvestre, J.-S.; Smadja, D.; Lévy, B.I. Postischemic revascularization: From cellular and molecular mechanisms to clinical applications. Physiol. Rev. 2013, 93, 1743–1802. [Google Scholar] [CrossRef] [PubMed]
- Idris-Khodja, N.; Mian, M.O.R.; Paradis, P.; Schiffrin, E.L. Dual opposing roles of adaptive immunity in hypertension. Eur. Heart J. 2014, 35, 1238–1244. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.; Nanan, R.K.H. Innate and adaptive immune interactions at the fetal-maternal inter-face in healthy human pregnancy and pre-eclampsia. Inflammation 2014, 5, 125. [Google Scholar]
- Osol, G.; Moore, L.G. Maternal uterine vascular remodeling during pregnancy. Microcirculation 2014, 21, 38–47. [Google Scholar] [CrossRef]
- Chiba, T.; Umegaki, K. Pivotal roles of monocytes/macrophages in stroke. Mediat. Inflamm. 2013, 2013, 759103. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Donegá, M.; Giusto, E.; Mallucci, G.; Marchetti, B.; Pluchino, S. The role of the immune system in central nervous system plasticity after acute injury. Neuroscience 2014, 283, 210–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorny, D.; Loufrani, L.; Kubis, N.; Lévy, B.I.; Henrion, D. Chronic hydralazine improves flow (shear stress)-induced endothelium-dependent dilation in mouse mesenteric resistance arteries in vitro. Microvasc. Res. 2002, 64, 127–134. [Google Scholar] [CrossRef]
- Van Royen, N.; Piek, J.J.; Buschmann, I.; Hoefer, I.; Voskuil, M.; Schaper, W. Stimulation of arteriogenesis; a new concept for the treatment of arterial occlusive disease. Cardiovasc. Res. 2001, 49, 543–553. [Google Scholar] [CrossRef]
- Unthank, J.L.; Fath, S.W.; Burkhart, H.M.; Miller, S.C.; Dalsing, M.C. Wall remodeling during luminal expansion of mesenteric arterial collaterals in the rat. Circ. Res. 1996, 79, 1015–1023. [Google Scholar] [CrossRef]
- Dolan, J.M.; Sim, F.J.; Meng, H.; Kolega, J. Endothelial cells express a unique transcriptional profile under very high wall shear stress known to induce expansive arterial remodeling. Am. J. Physiol. Physiol. 2012, 302, C1109–C1118. [Google Scholar] [CrossRef] [Green Version]
- Félétou, M.; Huang, Y.; Vanhoutte, P.M. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br. J. Pharmacol. 2011, 164, 894–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caughey, G.; Cleland, L.G.; Penglis, P.S.; Gamble, J.R.; James, M.J. Roles of cyclooxygenase (COX)-1 and COX-2 in prostanoid production by human endothelial cells: Selective up-regulation of prostacyclin synthesis by COX-2. J. Immunol. 2001, 167, 2831–2838. [Google Scholar] [CrossRef]
- Busse, R.; Edwards, G.; Félétou, M.; Fleming, I.; Vanhoutte, P.M.; Weston, A.H. EDHF: Bringing the concepts together. Trends Pharmacol. Sci. 2002, 23, 374–380. [Google Scholar] [CrossRef]
- Loufrani, L.; Henrion, D. Vasodilator treatment with hydralazine increases blood flow in mdx mice resistance arteries without vascular wall remodelling or endothelium function improvement. J. Hypertens. 2005, 23, 1855–1860. [Google Scholar] [CrossRef] [PubMed]
- Dumont, O.; Pinaud, F.; Guihot, A.-L.; Baufreton, C.; Loufrani, L.; Henrion, D. Alteration in flow (shear stress)-induced remodelling in rat resistance arteries with aging: Improvement by a treatment with hydralazine. Cardiovasc. Res. 2007, 77, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Pomares, L.; Platt, N.; Mcknight, A.; da Silva, R.; Gordon, S. CHAPTER 2: Macrophage membrane molecules: Markers of tissue Differentiation and heterogeneity. Immunobiology 1996, 195, 407–416. [Google Scholar] [CrossRef]
- Saito, N.; Pulford, K.A.; Breton-Gorius, J.; Masse, J.M.; Mason, D.Y.; Cramer, E.M. Ultrastructural localization of the CD68 macrophage-associated antigen in human blood neutrophils and monocytes. Am. J. Pathol. 1991, 139, 1053–1059. [Google Scholar]
- Groemping, Y.; Rittinger, K. Activation and assembly of the NADPH oxidase: A structural perspective. Biochem. J. 2005, 386, 401–416. [Google Scholar] [CrossRef] [Green Version]
- Hattori, H.; Subramanian, K.K.; Sakai, J.; Jia, Y.; Li, Y.; Porter, T.F.; Loison, F.; Sarraj, B.; Kasorn, A.; Jo, H.; et al. Small-molecule screen identifies reactive oxygen species as key regulators of neutrophil chemotaxis. Proc. Natl. Acad. Sci. USA 2010, 107, 3546–3551. [Google Scholar] [CrossRef] [Green Version]
- Buda, V.; Andor, M.; Cristescu, C.; Tomescu, M.C.; Muntean, D.M.; Bâibâță, D.E.; Bordejevic, D.A.; Danciu, C.; Dalleur, O.; Coricovac, D.; et al. Thrombospondin-1 serum levels in hypertensive patients with endothelial dysfunction after one year of treatment with perindopril. Drug Des. Dev. Ther. 2019, 13, 3515–3526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, Y.; Tanaka, M.; Yamakage, H.; Sasaki, Y.; Muranaka, K.; Hata, H.; Ikai, I.; Shimatsu, A.; Inoue, M.; Chun, T.H.; et al. Thrombospondin 1 as a novel biological marker of obesity and metabolic syndrome. Metabolism 2015, 64, 1490–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghimire, K.; Li, Y.; Chiba, T.; Julovi, S.M.; Li, J.; Ross, M.A.; Straub, A.C.; O’Connell, P.J.; Rüegg, C.; Pagano, P.J.; et al. CD47 promotes age-associated deterioration in angiogenesis, blood flow and glucose homeostasis. Cells 2020, 9, 1695. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; He, J.; Zhang, X.; Li, J.; Zhao, P.; Fei, P. TSP1 ameliorates age-related macular degeneration by regulating the STAT3-iNOS signaling pathway. Exp. Cell Res. 2020, 388, 111811. [Google Scholar] [CrossRef]
- Meijles, D.N.; Sahoo, S.; Al Ghouleh, I.; Amaral, J.H.; Bienes-Martinez, R.; Knupp, H.E.; Attaran, S.; Sembrat, J.C.; Nouraie, S.M.; Rojas, M.M.; et al. The matricellular protein TSP1 promotes human and mouse endothelial cell senescence through CD47 and Nox1. Sci. Signal. 2017, 10, eaaj1784. [Google Scholar] [CrossRef] [Green Version]
- Kale, A.; Rogers, N.M.; Ghimire, K. Thrombospondin-1 CD47 signalling: From mechanisms to medicine. Int. J. Mol. Sci. 2021, 22, 4062. [Google Scholar] [CrossRef]
- Lawler, J.; Sunday, M.E.; Thibert, V.; Duquette, M.; George, E.L.; Rayburn, H.; Hynes, R.O. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J. Clin. Investig. 1998, 101, 982–992. [Google Scholar] [CrossRef]
- Bonnefoy, A.; Daenens, K.; Feys, H.B.; De Vos, R.; Vandervoort, P.; Vermylen, J.; Lawler, J.; Hoylaerts, M.F. Thrombospondin-1 controls vascular platelet recruitment and thrombus adherence in mice by protecting (sub)endothelial VWF from cleavage by ADAMTS13. Blood 2005, 107, 955–964. [Google Scholar] [CrossRef]
- Loufrani, L.; Li, Z.; Lévy, B.I.; Paulin, D.; Henrion, D. Excessive microvascular adaptation to changes in blood flow in mice lacking gene encoding for desmin. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1579–1584. [Google Scholar] [CrossRef] [Green Version]
- Ceiler, D.L.; De Mey, J.G.R. Chronic N G -nitro- l -arginine methyl ester treatment does not prevent flow-induced remodeling in mesenteric feed arteries and arcading arterioles. Arter. Thromb. Vasc. Biol. 2000, 20, 2057–2063. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.M.; Thomay, A.A.; Connolly, M.D.; Reichner, J.S.; Albina, J.E. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 2008, 83, 64–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grenier, C.; Caillon, A.; Munier, M.; Grimaud, L.; Champin, T.; Toutain, B.; Fassot, C.; Blanc-Brude, O.; Loufrani, L. Dual Role of Thrombospondin-1 in Flow-Induced Remodeling. Int. J. Mol. Sci. 2021, 22, 12086. https://doi.org/10.3390/ijms222112086
Grenier C, Caillon A, Munier M, Grimaud L, Champin T, Toutain B, Fassot C, Blanc-Brude O, Loufrani L. Dual Role of Thrombospondin-1 in Flow-Induced Remodeling. International Journal of Molecular Sciences. 2021; 22(21):12086. https://doi.org/10.3390/ijms222112086
Chicago/Turabian StyleGrenier, Céline, Antoine Caillon, Mathilde Munier, Linda Grimaud, Tristan Champin, Bertrand Toutain, Céline Fassot, Olivier Blanc-Brude, and Laurent Loufrani. 2021. "Dual Role of Thrombospondin-1 in Flow-Induced Remodeling" International Journal of Molecular Sciences 22, no. 21: 12086. https://doi.org/10.3390/ijms222112086
APA StyleGrenier, C., Caillon, A., Munier, M., Grimaud, L., Champin, T., Toutain, B., Fassot, C., Blanc-Brude, O., & Loufrani, L. (2021). Dual Role of Thrombospondin-1 in Flow-Induced Remodeling. International Journal of Molecular Sciences, 22(21), 12086. https://doi.org/10.3390/ijms222112086