
ELF3 Is a Target That Promotes Therapeutic Efficiency in EGFR Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer Cells via Inhibiting PKCί


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
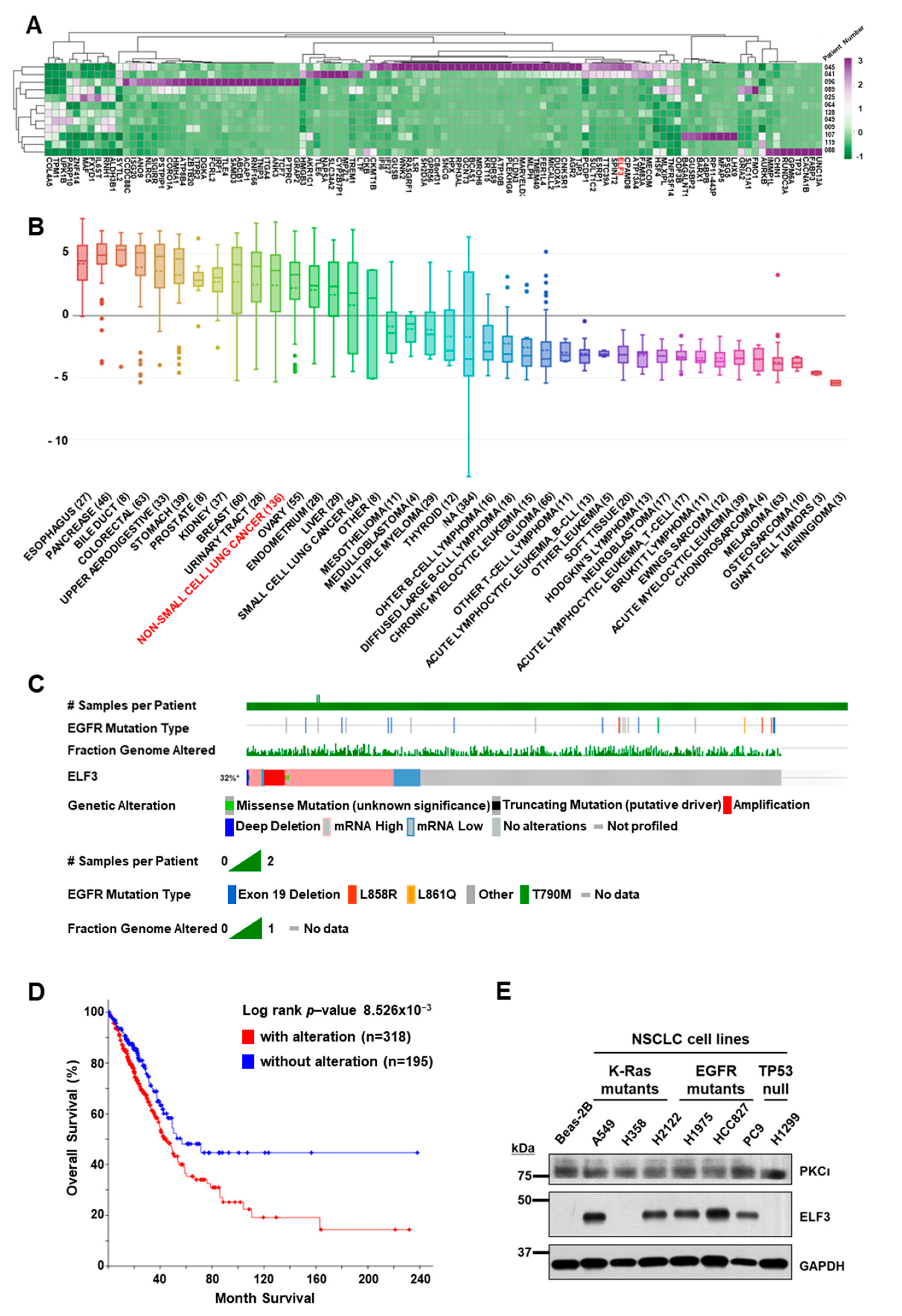
2.1. Transcriptome Analyses of PDCs from Lung Cancer Patients
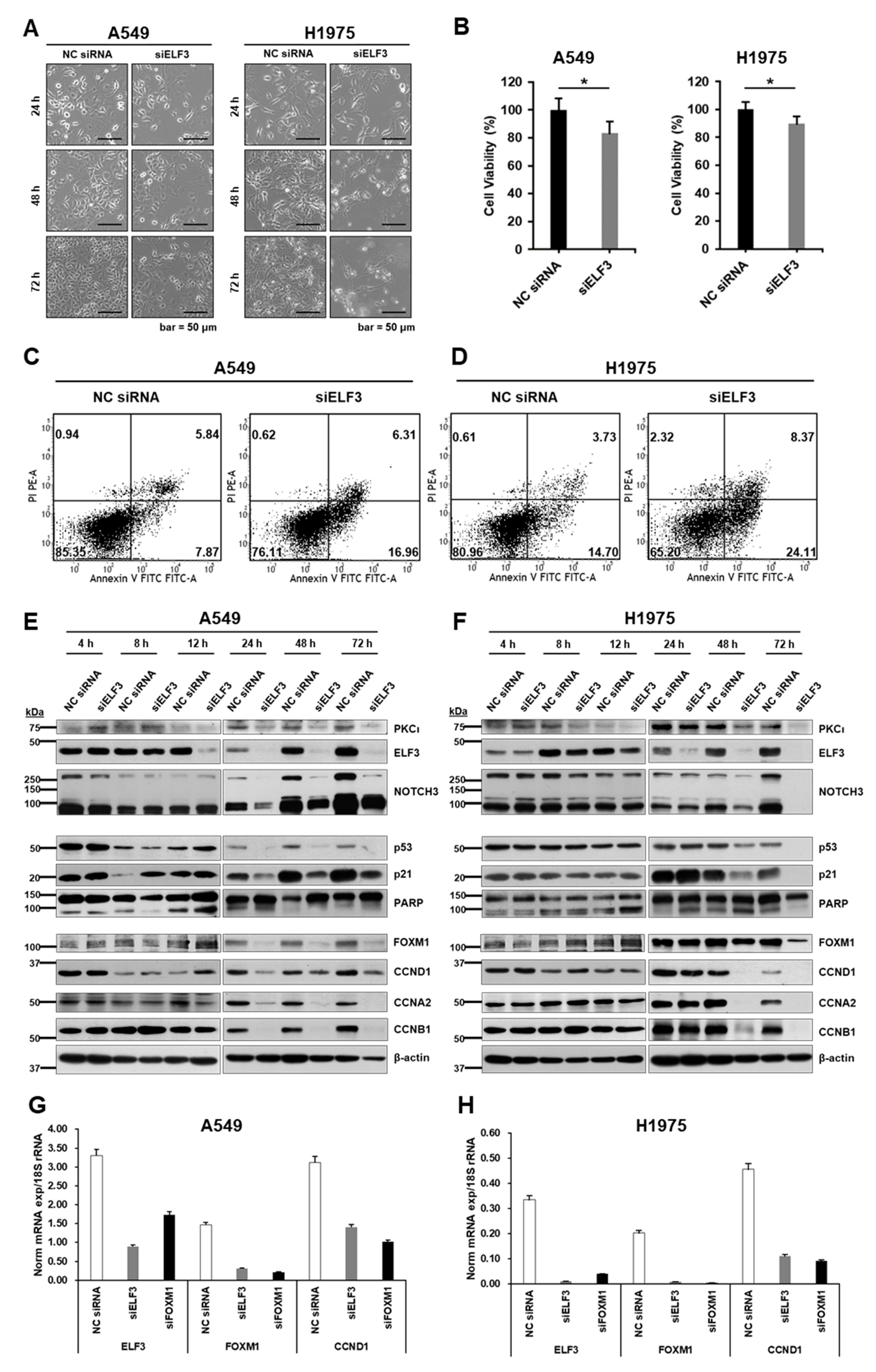
2.2. ELF3 Knockdown Induced Cell Death and Reduced Cell Cycle Progression in Lung Cancer Cells
2.3. ANF Represses ELF3 via Inhibition of PKCί
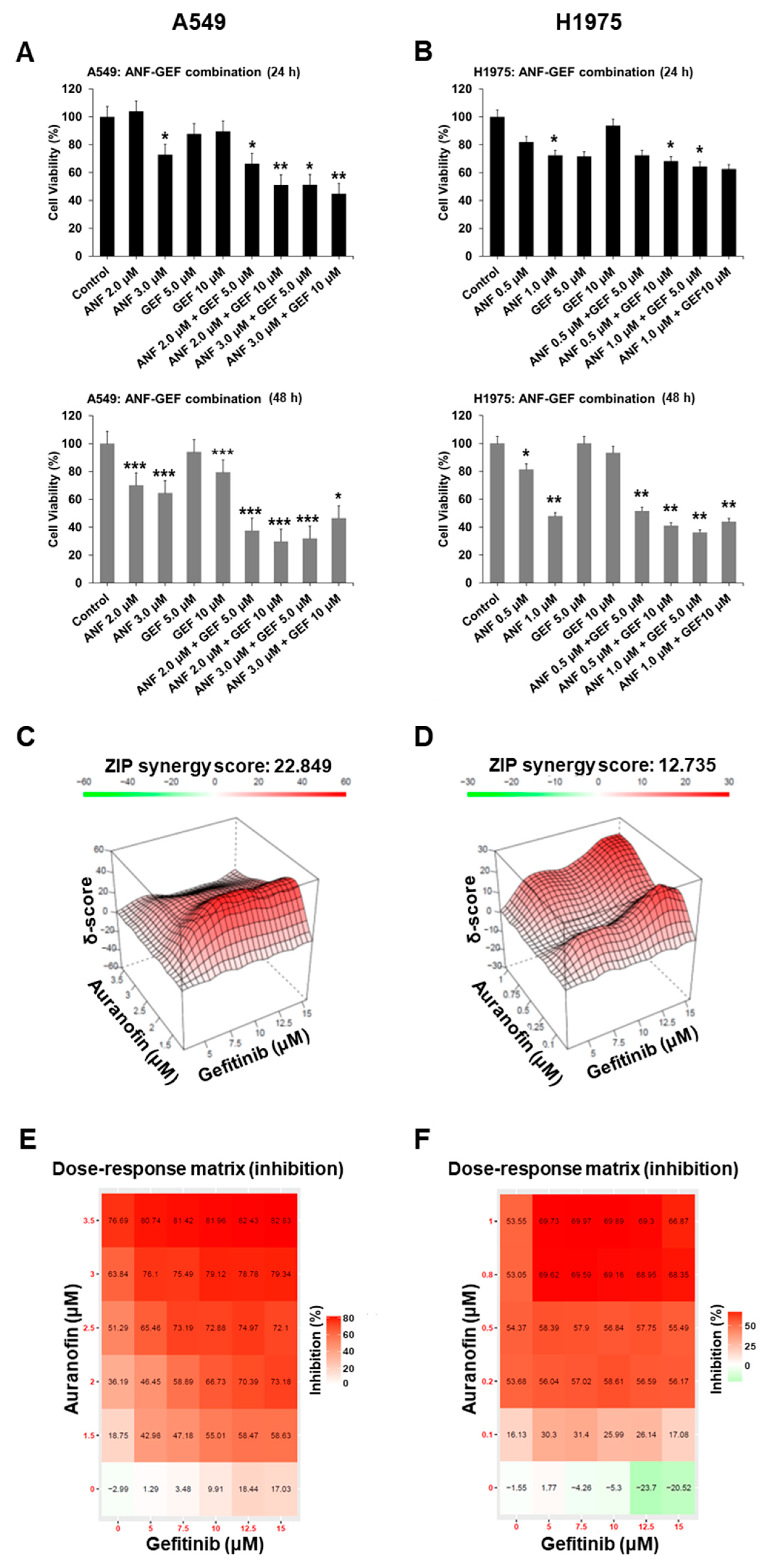
2.4. ANF and GEF Synergistically Inhibit TKI Resistance in Cells
2.5. ANF and GEF Combination Induces Apoptosis and Repression of Cell Cycle
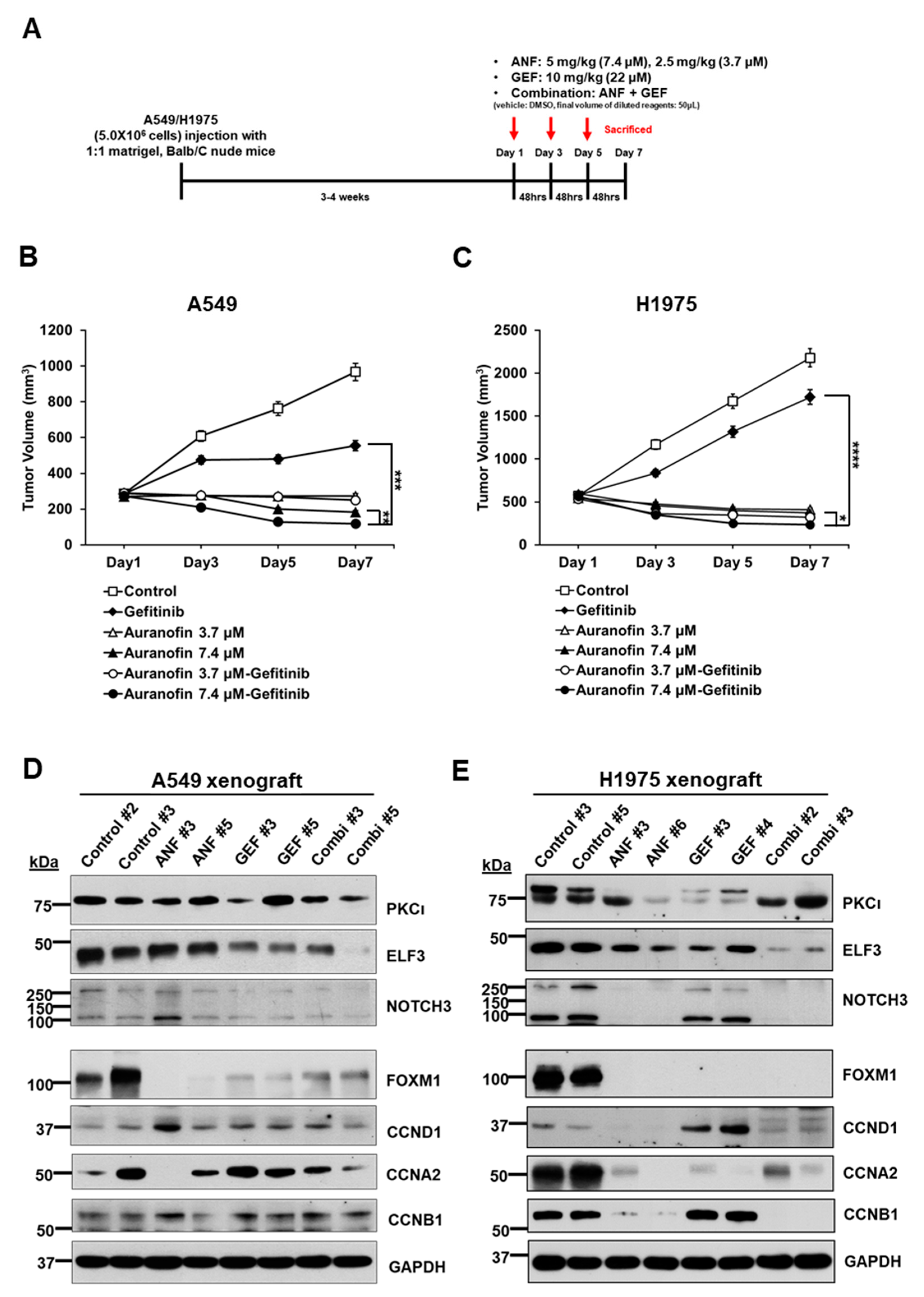
2.6. ANF and GEF Combination Reduces Cell Growth in an In Vivo Mouse Model
3. Discussion
4. Materials and Methods
4.1. Transcriptome Analyses from Patient-Derived Cells
4.2. Cell Lines and Maintenance
4.3. Antibodies for Immunoblot and Immunocytochemistry
4.4. ELF3 and FOXM1 Expression Knockdown
4.5. Treatment of Auranofin and Gefitinib
4.6. Quantitative Reverse Transcription PCR
4.7. Analyses of Cell Cycle and Apoptosis
4.8. In Vivo Analysis of Auranofin-Gefitinib Combination
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Li, K.; Yang, M.; Liang, N.; Li, S. Determining EGFR-TKI sensitivity of G719X and other uncommon EGFR mutations in non-small cell lung cancer: Perplexity and solution. Oncol. Rep. 2017, 37, 1347–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Yang, N.; Ou, Q.; Xiang, Y.; Jiang, T.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Shao, Y.W.; et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018, 24, 3097–3107. [Google Scholar] [CrossRef] [Green Version]
- Tymms, M.J.; Ng, A.Y.; Thomas, R.S.; Schutte, B.C.; Zhou, J.; Eyre, H.J.; Sutherland, G.R.; Seth, A.; Rosenberg, M.; Papas, T.; et al. A novel epithelial-expressed ETS gene, ELF3: Human and murine cDNA sequences, murine genomic organization, human mapping to 1q32.3 and expression in tissue and cancer. Oncogene 1997, 15, 2449–2462. [Google Scholar] [CrossRef] [Green Version]
- Yachida, S.; Wood, L.D.; Suzuki, M.; Takai, E.; Totoki, Y.; Kato, M.; Luchini, C.; Arai, Y.; Nakamura, H.; Hama, N. Genomic sequencing identifies ELF3 as a driver of ampullary carcinoma. Cancer Cell 2016, 29, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, S.; Amekawa, S.; Yomogida, K.; Sumi, T.; Nakazawa, M.; Yura, Y.; Nishimune, Y.; Nozaki, M. ESE-1 inhibits the invasion of oral squamous cell carcinoma in conjugation with MMP-9 suppression. Oral Dis. 2008, 14, 144–149. [Google Scholar] [CrossRef]
- Liu, J.; Cho, S.N.; Akkanti, B.; Jin, N.; Mao, J.; Long, W.; Chen, T.; Zhang, Y.; Tang, X.; Wistub, I.I.; et al. ErbB2 pathway activation upon Smad4 loss promotes lung tumor growth and metastasis. Cell Rep. 2015, 10, 1599–1913. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.L.; Chen, Z.F.; Chen, H.M.; Wang, M.Y.; Kong, X.; Wang, Y.C.; Sun, T.T.; Hong, J.; You, W.; Xu, J.; et al. Elf3 drives β-catenin transactivation and associates with poor prognosis in colorectal cancer. Cell Death Dis. 2014, 5, e1263. [Google Scholar] [CrossRef] [Green Version]
- Mesquita, B.; Lopes, P.; Rodrigues, A.; Pereira, D.; Afonso, M.; Leal, C.; Henrique, R.; Lind, G.E.; Jerónimo, C.; Lothe, R.A.; et al. Frequent copy number gains at 1q21 and 1q32 are associated with overexpression of the ETS transcription factors ETV3 and ELF3 in breast cancer irrespective of molecular subtypes. Breast Cancer Res. Treat. 2013, 138, 3–45. [Google Scholar] [CrossRef]
- Wang, H.; Yu, Z.; Huo, S.; Chen, Z.; Ou, Z.; Mai, J.; Ding, S.; Zhang, J. Overexpression of ELF3 facilitates cell growth and metastasis through PI3K/Akt and ERK signaling pathways in non-small cell lung cancer. Int. J. Biochem. Cell Biol. 2018, 94, 98–106. [Google Scholar] [CrossRef]
- Yu, X.M.; Wu, Y.C.; Liu, X.; Huang, X.C.; Hou, X.X.; Wang, J.L.; Cheng, X.L.; Mao, W.M.; Ling, Z.Q. Cell-free RNA content in peripheral blood as potential biomarkers for detecting circulating tumor cells in non-small cell lung carcinoma. Int. J. Mol. Sci. 2016, 17, 1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enfield, K.; Marshall, E.A.; Anderson, C.; Ng, K.W.; Rahmati, S.; Xu, Z.; Fuller, M.; Milne, K.; Lu, D.; Shi, R.; et al. Epithelial tumor suppressor ELF3 is a lineage-specific amplified oncogene in lung adenocarcinoma. Nat. Commun. 2019, 10, 5438. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Justilien, V.; Jamieson, L.; Murray, N.R.; Fields, A.P. Protein kinase Cι drives a NOTCH3-dependent stem-like phenotype in mutant KRAS lung adenocarcinoma. Cancer Cell 2016, 29, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Chen, P.; Zhang, Y.; Lu, W.; Ding, W.; Luo, Y.; Wen, S.; Xu, R.; Liu, P.; Huang, P. Synergy between auranofin and celecoxib against colon cancer in vitro and in vivo through a novel redox-mediated mechanism. Cancers 2019, 11, 931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signature database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Justilien, V.; Walsh, M.P.; Ali, S.A.; Thompson, E.A.; Murray, N.R.; Fields, A.P. The PRKCI and SOX2 oncogenes are coamplifed and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell 2014, 25, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Ono, M.; Hirata, A.; Kometani, T.; Miyagawa, M.; Ueda, S.; Kinoshita, H.; Fujii, T.; Kuwano, M.; Michihiko, K. Sensitivity to gefitinib (Iressa, AZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGR receptor/Akt pathway for proliferation. Mol. Cancer Ther. 2004, 3, 465–472. [Google Scholar]
- Liao, G.B.; Li, X.Z.; Zeng, S.; Liu, C.; Yang, S.M.; Yang, L.; Hu, C.J.; Bai, J.Y. Regulation of the master regulator of FOXM1 in cancer. Cell Commun. Signal. 2018, 16, 57. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, H.; Shimozato, O.; Ishii, T.; Kamoda, H.; Hagiwara, Y.; Tsukanishi, T.; Ohtori, S.; Yonemoto, T. The thioredoxin reductase inhibitor auranofin suppresses pulmonary metastasis of osteosarcoma, but no local progression. Anticancer Res. 2021, 41, 4947–4955. [Google Scholar] [CrossRef]
- Regala, R.P.; Weems, C.; Jamieson, L.; Copland, J.A.; Thompson, E.A.; Fields, A.P. Atypical protein kinase Ci plays a critical role in human lung cancer cell growth and tumorigenicity. J. Biol. Chem. 2005, 280, 31109–31115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regala, R.P.; Davis, R.K.; Kunz, A.; Khoor, A.; Leitges, M.; Fields, A.P. Atypical protein kinase Cι is required for bronchioalveolar stem cell expansion and lung tumorigenesis. Cancer Res. 2009, 69, 7603–7611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sizemore, G.M.; Pitarresi, J.R.; Balakrishnan, S.; Ostrowski, M.C. The ETS family of oncogenic transcription factors in solid tumours. Nat. Rev. Cancer 2017, 17, 337–351. [Google Scholar] [CrossRef]
- Oh, S.; Shin, S.; Janknecht, R. ETV1, 4, and 5: An oncogenic subfamily of ETS transcription factors. Biochim. Biophys. Acta 2012, 1826, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamura-Saito, M.; Yamazaki, Y.; Kaneko, K.; Kawaguchi, N.; Kanda, H.; Mukai, H.; Gotoh, T.; Motoi, T.; Fukayama, M.; Aburatani, H.; et al. Fusion between CIC and DUX4 upregulates PEA3 family genes in Ewing-like sarcoma with t(4;19)(q35;q13) translocation. Hum. Mol. Genet. 2006, 15, 2125–2137. [Google Scholar] [CrossRef]
- Shaikhibrahim, Z.; Lindstrot, A.; Langer, B.; Buettner, R.; Wernert, N. Differential expression of ETS family members in prostate cancer tissues and androgen-sensitive and insensitive prostate cancer cell lines. Int. J. Mol. Med. 2011, 28, 89–93. [Google Scholar]
- Li, Z.; Zhang, L.; Ma, Z.; Yang, M.; Tang, J.; Fu, Y.; Mao, Y.; Hong, X.; Zhang, Y. ETV1 induces epithelial to mesenchymal transition in human gastric cancer cells through the upregulation of Snail expression. Oncol. Rep. 2013, 30, 2859–2863. [Google Scholar] [CrossRef]
- Kim, E.; Kim, D.; Lee, J.S.; Yoe, J.; Park, J.; Kim, C.J.; Jeong, D.; Kim, S.; Lee, Y. Capicua suppresses hepatocellular carcinoma progression by controlling the ETV4-MMP1 axis. Hepatology 2018, 67, 2287–2301. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Kim, E.; Lee, J.; Kim, D.; Kim, H.; Kim, C.J.; Kim, S.; Jeong, D.; Lee, Y. Capicua suppresses colorectal cancer progression via repression of ETV4 expression. Cancer Cell Int. 2020, 20, 42. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.E.; Madhi, H.; Kim, H.; Lee, J.S.; Kim, M.H.; Kim, Y.N.; Goh, S.H. FAM188B expression is critical for cell growth via FOXM1 regulation in lung cancer. Biomedicines 2020, 8, 465. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, H.; Cao, M.; Wang, L.; Wu, S.; Fang, B. Auranofin enhances ibrutinib’s anticancer activity in EGFR-mutant lung adenocarcinoma. Mol. Cancer Ther. 2018, 17, 2156–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Monica, S.; Madeddu, D.; Tiseo, M.; Vivo, V.; Galetti, M.; Cretella, D.; Bonelli, M.; Fumarola, C.; Cavazzoni, A.; Falco, A.; et al. Combination of gefitinib and pemetrexed prevents the acquisition of TKI resistance in NSCLC cell lines carrying EGFR-activating mutation. J. Thorac. Oncol. 2016, 11, 1051–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-S.; Choi, Y.E.; Kim, S.; Han, J.-Y.; Goh, S.-H. ELF3 Is a Target That Promotes Therapeutic Efficiency in EGFR Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer Cells via Inhibiting PKCί. Int. J. Mol. Sci. 2021, 22, 12287. https://doi.org/10.3390/ijms222212287
Lee J-S, Choi YE, Kim S, Han J-Y, Goh S-H. ELF3 Is a Target That Promotes Therapeutic Efficiency in EGFR Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer Cells via Inhibiting PKCί. International Journal of Molecular Sciences. 2021; 22(22):12287. https://doi.org/10.3390/ijms222212287
Chicago/Turabian StyleLee, Jeon-Soo, Young Eun Choi, Sunshin Kim, Ji-Youn Han, and Sung-Ho Goh. 2021. "ELF3 Is a Target That Promotes Therapeutic Efficiency in EGFR Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer Cells via Inhibiting PKCί" International Journal of Molecular Sciences 22, no. 22: 12287. https://doi.org/10.3390/ijms222212287