
The Role of Cannabinoids in Bone Metabolism: A New Perspective for Bone Disorders

 ,
,  , , , ,
, , , , 
Abstract
1. Introduction
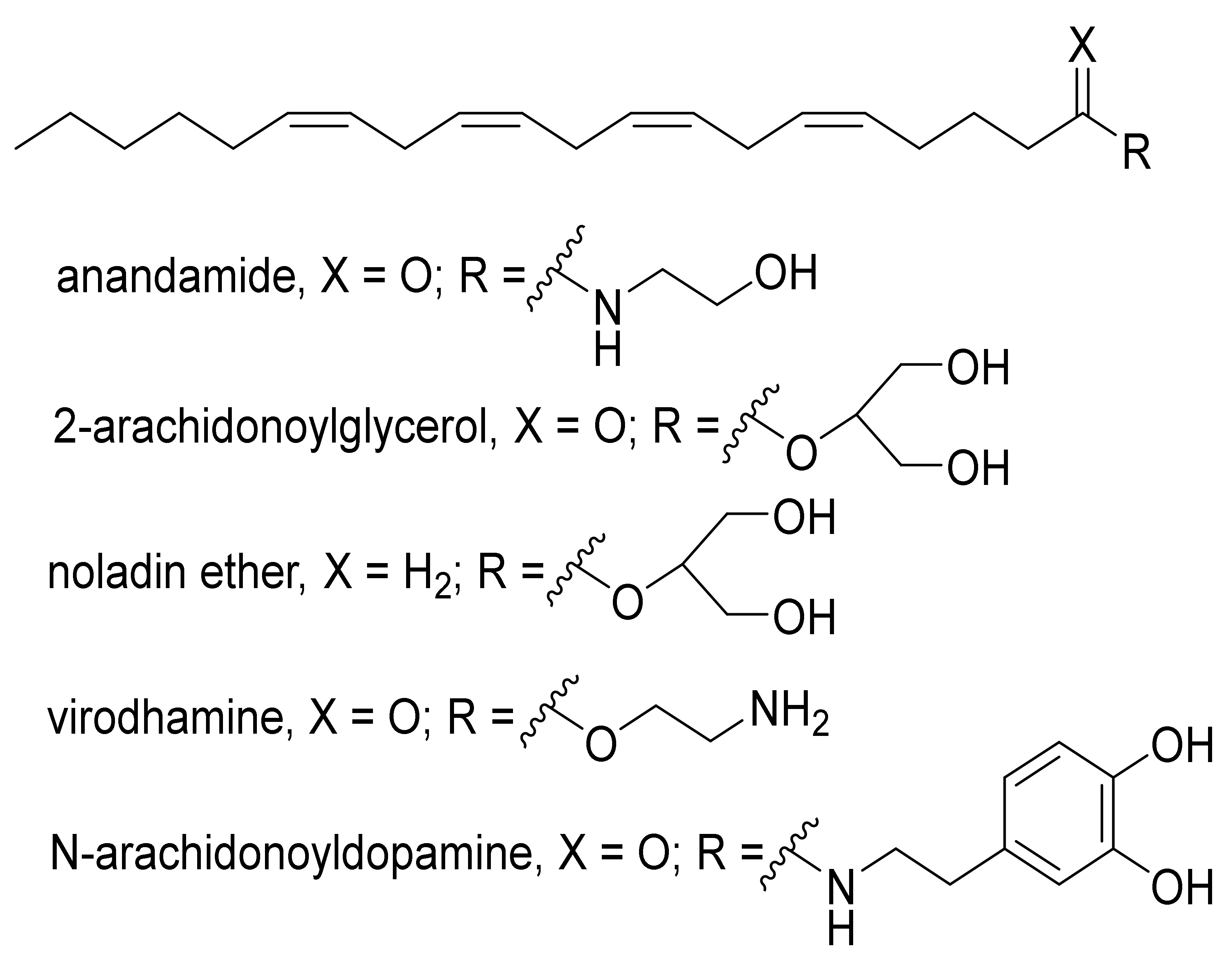
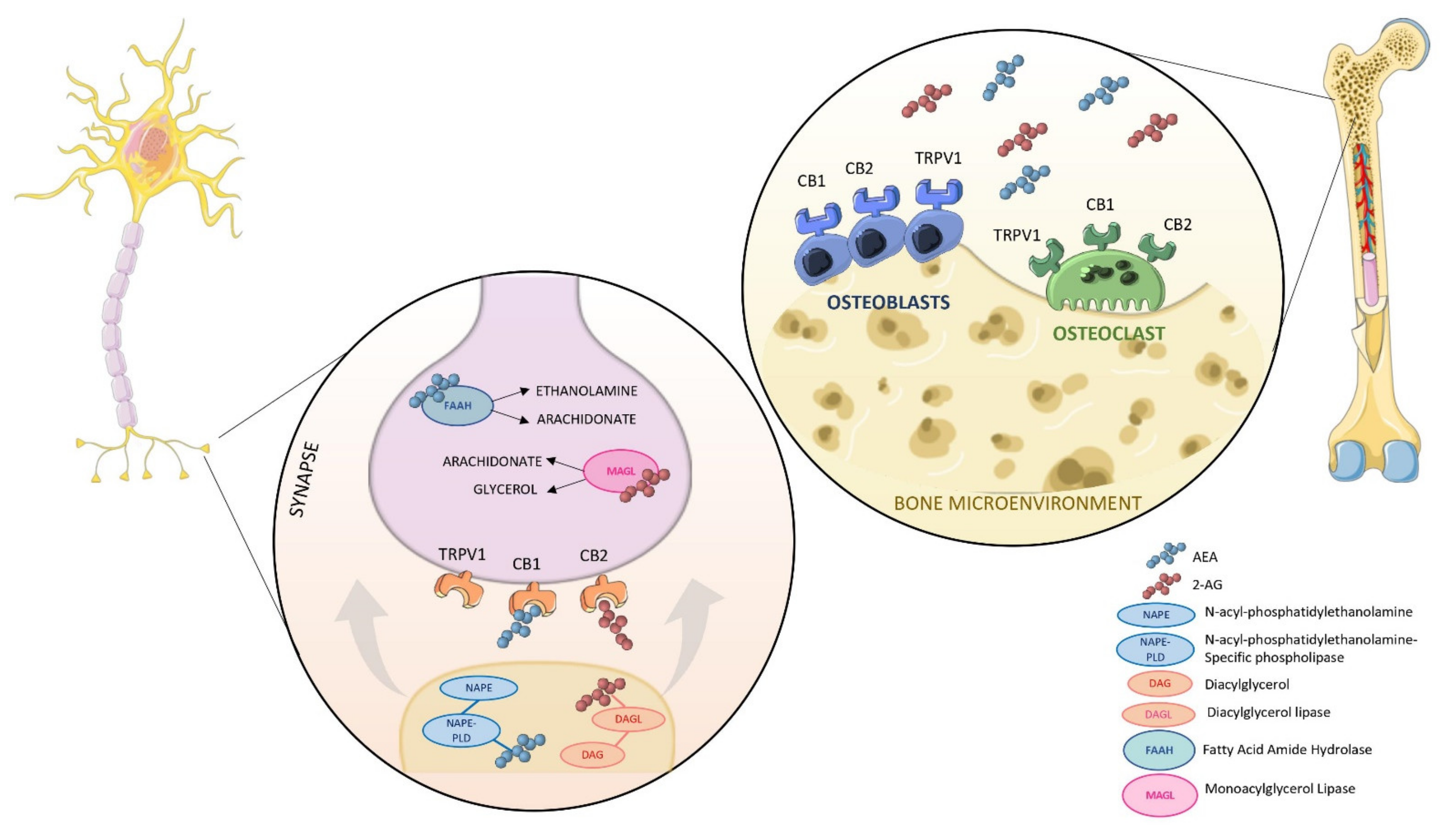
2. An Overview of the Endocannabinoids System (ECS)
3. Detection of ECs in Biological Fluids, Hair, and Tissues
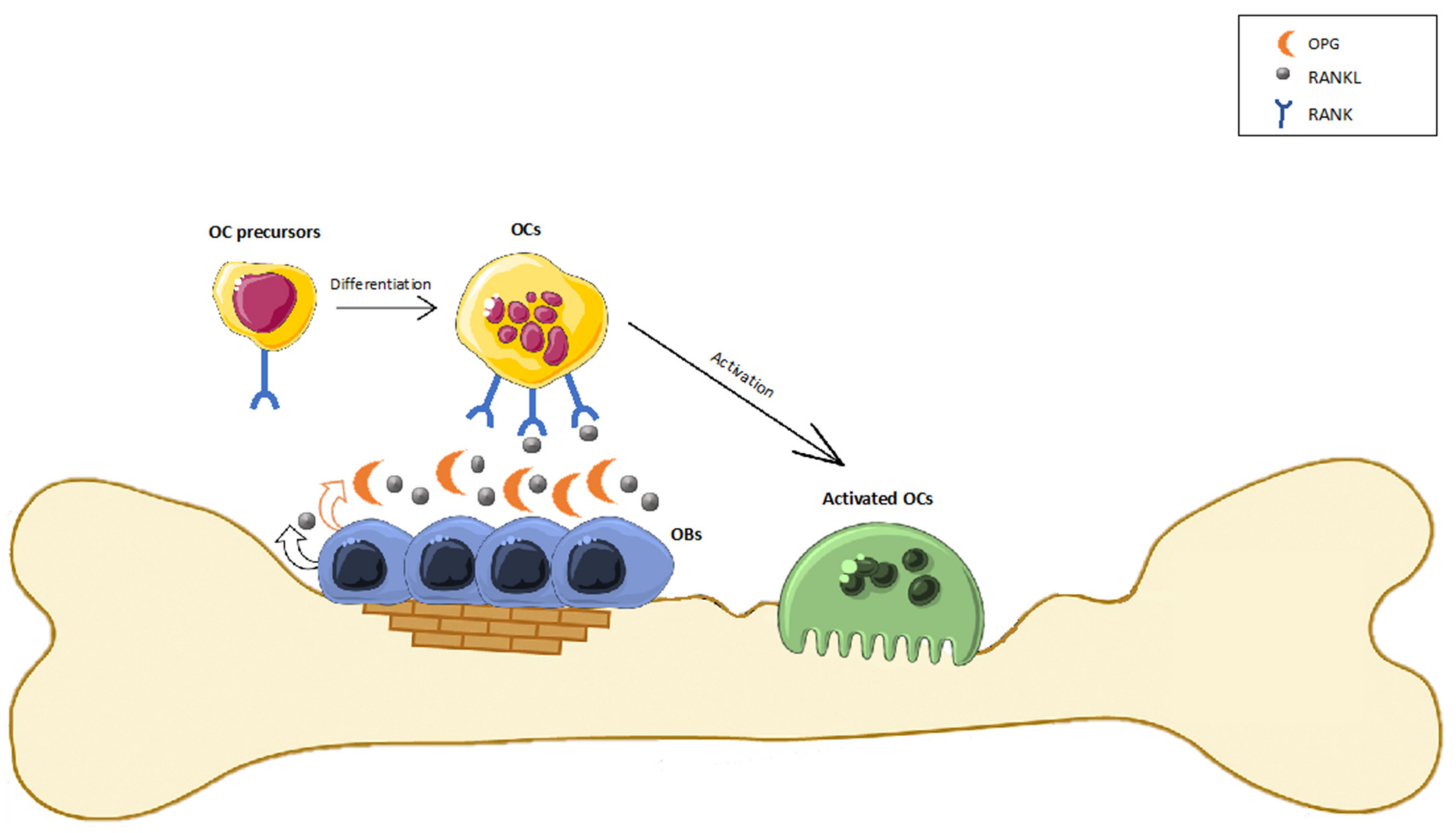
4. The Involvement of the ECS in Bone Metabolism
4.1. In Vitro Evidence of a Possible Modulation of ECS in Bone
4.2. Animal Studies Provide Further Insight in the Role of CBRs in Bone Biology
5. Cannabinoid Receptors as Therapeutic Target for Bone Diseases
5.1. Osteoporosis
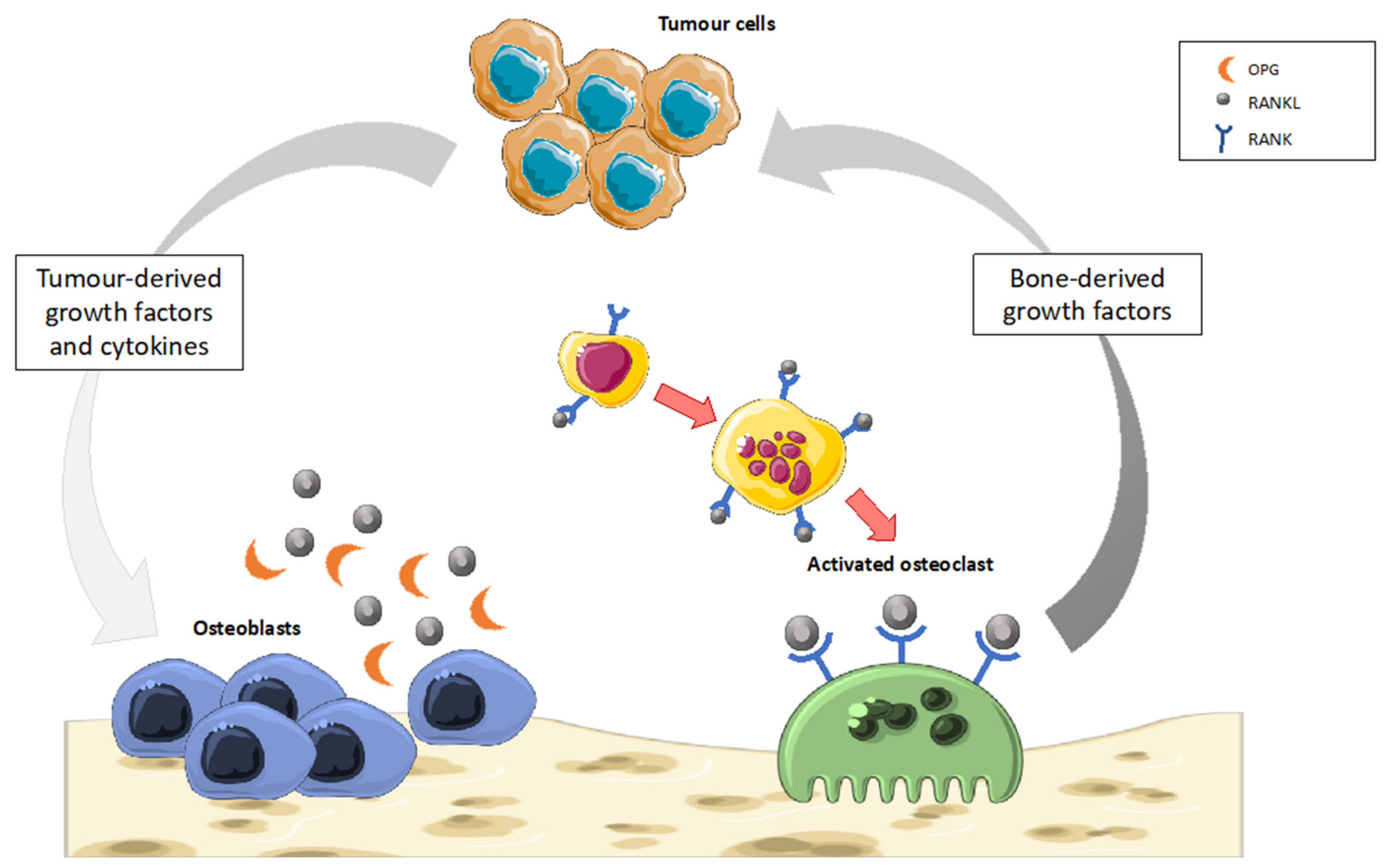
5.2. Bone Cancer
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eriksen, E.F. Cellular Mechanisms of Bone Remodeling. Rev. Endocr. Metab. Disord. 2010, 11, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.; Branco, J.C. Rank/Rankl/Opg: Literature Review. Acta Reumatol. Port. 2011, 36, 209–218. [Google Scholar]
- Raggatt, L.J.; Partridge, N.C. Cellular and Molecular Mechanisms of Bone Remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef]
- Hadjidakis, D.J.; Androulakis, I.I. Bone Remodeling. Ann. N. Y. Acad. Sci. 2006, 1092, 385–396. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in Bone Modeling and Remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast Differentiation and Activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Brylka, L.J.; Schinke, T. Chemokines in Physiological and Pathological Bone Remodeling. Front. Immunol. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Siddiqui, J.A.; Partridge, N.C. Physiological Bone Remodeling: Systemic Regulation and Growth Factor Involvement. Physiology 2016, 31, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, J.; Horowitz, M.; Choi, Y. Osteoimmunology: Interactions of the Bone and Immune System. Endocr. Rev. 2008, 29, 403–440. [Google Scholar] [CrossRef]
- Nagy, V.; Penninger, J.M. The RANKL-RANK Story. Gerontology 2015, 61, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Idris, A.I.; Ralston, S.H. Role of Cannabinoids in the Regulation of Bone Remodeling. Front. Endocrin. 2012, 3, 136. [Google Scholar] [CrossRef]
- Rossi, R.; Tortora, C.; Punzo, F.; Bellini, G.; Argenziano, M.; Di Paola, A.; Torella, M.; Perrotta, S. The Endocannabinoid/Endovanilloid System in Bone: From Osteoporosis to Osteosarcoma. Int. J. Mol. Sci. 2019, 20, 1919. [Google Scholar] [CrossRef]
- Idris, A.I.; Sophocleous, A.; Landao-Bassonga, E.; van’t Hof, R.J.; Ralston, S.H. Regulation of Bone Mass, Osteoclast Function, and Ovariectomy-Induced Bone Loss by the Type 2 Cannabinoid Receptor. Endocrinology 2008, 149, 5619–5626. [Google Scholar] [CrossRef]
- Alger, B.E. Getting High on the Endocannabinoid System. Cerebrum 2013, 2013, 14. [Google Scholar] [PubMed]
- Lambert, D.M.; Fowler, C.J. The Endocannabinoid System: Drug Targets, Lead Compounds, and Potential Therapeutic Applications. J. Med. Chem. 2005, 48, 5059–5087. [Google Scholar] [CrossRef]
- Di Marzo, V. New Approaches and Challenges to Targeting the Endocannabinoid System. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular Characterization of a Peripheral Receptor for Cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Irving, A.; Abdulrazzaq, G.; Chan, S.L.F.; Penman, J.; Harvey, J.; Alexander, S.P.H. Cannabinoid Receptor-Related Orphan G Protein-Coupled Receptors. Adv. Pharmacol. 2017, 80, 223–247. [Google Scholar] [CrossRef]
- Iannotti, F.A.; Di Marzo, V.; Petrosino, S. Endocannabinoids and Endocannabinoid-Related Mediators: Targets, Metabolism and Role in Neurological Disorders. Prog. Lipid Res. 2016, 62, 107–128. [Google Scholar] [CrossRef]
- Borgelt, L.M.; Franson, K.L.; Nussbaum, A.M.; Wang, G.S. The Pharmacologic and Clinical Effects of Medical Cannabis. Pharmacotherapy 2013, 33, 195–209. [Google Scholar] [CrossRef]
- Zou, S.; Kumar, U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; González-Mariscal, I.; Egan, J.M.; Moaddel, R. Targeted proteomics of cannabinoid receptor CB1 and the CB1b isoform. J. Pharm. Biomed. Anal. 2017, 144, 154–158. [Google Scholar] [CrossRef] [PubMed]
- González-Mariscal, I.; Krzysik-Walker, S.M.; Doyle, M.E.; Liu, Q.R.; Cimbro, R.; Santa-Cruz Calvo, S.; Ghosh, S.; Cieśla, Ł.; Moaddel RCarlson, O.D.; Witek, R.P.; et al. Human CB1 Receptor Isoforms, present in Hepatocytes and β-cells, are Involved in Regulating Metabolism. Sci. Rep. 2016, 6, 33302. [Google Scholar] [CrossRef]
- Stella, N. Cannabinoid and Cannabinoid-like Receptors in Microglia, Astrocytes, and Astrocytomas. Glia 2010, 58, 1017–1030. [Google Scholar] [CrossRef]
- Liu, Q.R.; Pan, C.H.; Hishimoto, A.; Li, C.Y.; Xi, Z.X.; Llorente-Berzal, A.; Viveros, M.P.; Ishiguro, H.; Arinami, T.; Onaivi, E.S.; et al. Species differences in cannabinoid receptor 2 (CNR2 gene): Identification of novel human and rodent CB2 isoforms, differential tissue expression and regulation by cannabinoid receptor ligands. Genes Brain Behav. 2009, 5, 519–530. [Google Scholar] [CrossRef]
- Malfitano, A.M.; Basu, S.; Maresz, K.; Bifulco, M.; Dittel, B.N. What We Know and Do Not Know about the Cannabinoid Receptor 2 (CB2). Semin. Immunol. 2014, 26, 369–379. [Google Scholar] [CrossRef]
- Lu, H.-C.; Mackie, K. Review of the Endocannabinoid System. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2021, 6, 607–615. [Google Scholar] [CrossRef]
- Lu, H.-C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef]
- Basavarajappa, B. Critical Enzymes Involved in Endocannabinoid Metabolism. Protein Pept. Lett. 2007, 14, 237–246. [Google Scholar] [CrossRef]
- Nomura, D.K.; Morrison, B.E.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.G.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid Hydrolysis Generates Brain Prostaglandins That Promote Neuroinflammation. Science 2011, 334, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Zoerner, A.A.; Gutzki, F.-M.; Batkai, S.; May, M.; Rakers, C.; Engeli, S.; Jordan, J.; Tsikas, D. Quantification of Endocannabinoids in Biological Systems by Chromatography and Mass Spectrometry: A Comprehensive Review from an Analytical and Biological Perspective. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2011, 1811, 706–723. [Google Scholar] [CrossRef] [PubMed]
- Marchioni, C.; de Souza, I.D.; Acquaro Junior, V.R.; de Souza Crippa, J.A.; Tumas, V.; Queiroz, M.E.C. Recent advances in LC-MS/MS methods to determine endocannabinoids in biological samples: Application in neurodegen-erative diseases. Anal. Chim. Acta 2018, 1044, 12–28. [Google Scholar] [CrossRef]
- Sempio, C.; Klawitter, J.; Jackson, M.; Freni, F.; Shillingburg, R.; Hutchison, K.; Bidwell, L.C.; Christians, U.; Klawitter, J. Analysis of 14 endocannabinoids and endocannabinoid congeners in human plasma using column switching high-performance atmospheric pressure chemical ionization liquid chromatography–mass spectrometry. Anal. Bioanal. Chem. 2021, 413, 3381–3392. [Google Scholar] [CrossRef]
- Röhrig, W.; Achenbach, S.; Deutsch, B.; Pischetsrieder, M. Quantification of 24 Circulating Endocannabinoids, Endocannabinoid-Related Compounds, and Their Phospholipid Precursors in Human Plasma by UHPLC-MS/MS. J. Lipid Res. 2019, 60, 1475–1488. [Google Scholar] [CrossRef]
- Heumann, K.G. Isotope Dilution Mass Spectrometry of Inorganic and Organic Substances. Fresenius’ Z. Fuer Anal. Chem. 1986, 325, 661–666. [Google Scholar] [CrossRef]
- Gross, J.H. Inorganic Mass Spectrometry. In Mass Spectrometry; Springer: Cham, Switerland, 2017; pp. 889–925. [Google Scholar] [CrossRef]
- Mwanza, C.; Chen, Z.; Zhang, Q.; Chen, S.; Wang, W.; Deng, H. Simultaneous HPLC-APCI-MS/MS Quantification of Endogenous Cannabinoids and Glucocorticoids in Hair. J. Chromatogr. B Biomed. Appl. 2016, 1028, 1–10. [Google Scholar] [CrossRef]
- Battista, N.; Sergi, M.; Montesano, C.; Napoletano, S.; Compagnone, D.; Maccarrone, M. Analytical Approaches for the Determination of Phytocannabinoids and Endocannabinoids in Human Matrices: Phytocannabinoids and Endocannabinoids in Human Matrices. Drug Test. Anal. 2014, 6, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Zoerner, A.A.; Batkai, S.; Suchy, M.-T.; Gutzki, F.-M.; Engeli, S.; Jordan, J.; Tsikas, D. Simultaneous UPLC–MS/MS Quantification of the Endocannabinoids 2-Arachidonoyl Glycerol (2AG), 1-Arachidonoyl Glycerol (1AG), and Anandamide in Human Plasma: Minimization of Matrix-Effects, 2AG/1AG Isomerization and Degradation by Toluene Solvent Extraction. J. Chromatogr. B Biomed. Appl. 2012, 883–884, 161–171. [Google Scholar] [CrossRef]
- Fanelli, F.; Di Lallo, V.D.; Belluomo, I.; De Iasio, R.; Baccini, M.; Casadio, E.; Gasparini, D.I.; Colavita, M.; Gambineri, A.; Grossi, G.; et al. Estimation of Reference Intervals of Five Endocannabinoids and Endocannabinoid Related Compounds in Human Plasma by Two Dimensional-LC/MS/MS. J. Lipid Res. 2012, 53, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Koal, T.; Deters, M.; Casetta, B.; Kaever, V. Simultaneous Determination of Four Immunosuppressants by Means of High Speed and Robust On-Line Solid Phase Extraction–High Performance Liquid Chromatography–Tandem Mass Spectrometry. J. Chromatogr. B Biomed. Appl. 2004, 805, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Saba, A.; Raffaelli, A.; Cupisti, A.; Petri, A.; Marcocci, C.; Salvadori, P. Recent Advances in the Assessment of the Ratios of Cortisol to Cortisone and of Some of Their Metabolites in Urine by LC-MS-MS: Cortisol, Cortisone and Their Metabolites in Urine. J. Mass Spectrom. 2009, 44, 541–548. [Google Scholar] [CrossRef]
- Ayrton, J.; Dear, G.J.; Leavens, W.J.; Mallet, D.N.; Plumb, R.S. The Use of Turbulent Flow Chromatography/Mass Spectrometry for the Rapid, Direct Analysis of a Novel Pharmaceutical Compound in Plasma. Rapid Commun. Mass Spectrom. 1997, 11, 1953–1958. [Google Scholar] [CrossRef]
- Kantae, V.; Ogino, S.; Noga, M.; Harms, A.C.; Van Dongen, R.M.; Onderwater, G.L.J.; Van den Maagdenberg, A.M.J.M.; Terwindt, G.M.; van der Stelt, M.; Ferrari, M.D.; et al. Quantitative Profiling of Endocannabinoids and Related N-Acylethanolamines in Human CSF Using Nano LC-MS/MS. J. Lipid Res. 2017, 58, 615–624. [Google Scholar] [CrossRef]
- Ho, W.-S.; Barrett, D.A.; Randall, M.D. ‘Entourage’ Effects of N-Palmitoylethanolamide and N-Oleoylethanolamide on Vasorelaxation to Anandamide Occur through TRPV1 Receptors: Effect of Congeners on Anandamide Vasorelaxation. Br. J. Pharmacol. 2008, 155, 837–846. [Google Scholar] [CrossRef]
- Gachet, M.S.; Rhyn, P.; Bosch, O.G.; Quednow, B.B.; Gertsch, J. A Quantitiative LC-MS/MS Method for the Measurement of Arachidonic Acid, Prostanoids, Endocannabinoids, N-Acylethanolamines and Steroids in Human Plasma. J. Chromatogr. B 2015, 976, 6–18. [Google Scholar] [CrossRef]
- Ney, L.J.; Felmingham, K.L.; Bruno, R.; Matthews, A.; Nichols, D.S. Simultaneous Quantification of Endocannabinoids, Oleoylethanolamide and Steroid Hormones in Human Plasma and Saliva. J. Chromatogr. B 2020, 1152, 122252. [Google Scholar] [CrossRef]
- Ney, L.J.; Felmingham, K.L.; Bruno, R.; Matthews, A.; Nichols, D.S. Chloroform-Based Liquid-Liquid Extraction and LC–MS/MS Quantification of Endocannabinoids, Cortisol and Progesterone in Human Hair. J. Pharm. Biomed. Anal. 2021, 201, 114103. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane-Stanley, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.C.; Nielsen, A.; Briley, E.M.; Palkovits, M.; Priller, J.; Axelrod, J.; Nguyen, D.N.; Richardson, J.M.; Riggin, R.M.; Koppel, G.A.; et al. Isolation and Measurement of the Endogenous Cannabinoid Receptor Agonist, Anandamide, in Brain and Peripheral Tissues of Human and Rat. FEBS Lett. 1996, 393, 231–235. [Google Scholar] [CrossRef]
- Ita, M.; Kelly, J. Characterization of Cerebral Cortical Endocannabinoid Levels in a Rat Inguinal Surgery Model Using Liquid Chromatography–Tandem Mass Spectrometry (LC–MS/MS). Ir. J. Psychol. Med. 2019, 36, 1–10. [Google Scholar] [CrossRef]
- Di Marzo, V.; Goparaju, S.K.; Wang, L.; Liu, J.; Batkai, S.; Jarai, Z.; Fezza, F.; Miura, G.I.; Palmiter, R.D.; Sugiura, T.; et al. Lep-tin-regulated endocannabinoids are involved in maintaining food intake. Nature 2001, 410, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Dimitri, P.; Rosen, C. The Central Nervous System and Bone Metabolism: An Evolving Story. Calcif. Tissue Int. 2017, 100, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Wildburger, R.; Zarkovic, N.; Tonkovic, G.; Škoric, T.; Frech, S.; Hartleb, M.; Loncaric, I.; Zarkovic, K. Post-Traumatic Hormonal Disturbances: Prolactin as a Link between Head Injury and Enhanced Osteogenesis. J. Endocrinol. Investig. 1998, 21, 78–86. [Google Scholar] [CrossRef]
- Panikashvili, D.; Simeonidou, C.; Ben-Shabat, S.; Hanuš, L.; Breuer, A.; Mechoulam, R.; Shohami, E. An Endogenous Cannabinoid (2-AG) Is Neuroprotective after Brain Injury. Nature 2001, 413, 527–531. [Google Scholar] [CrossRef]
- Melamed, E.; Robinson, D.; Halperin, N.; Wallach, N.; Keren, O.; Groswasser, Z. Brain Injury-Related Heterotopic Bone Formation: Treatment Strategy and Results. Am. J. Phys. Med. Rehabil. 2002, 81, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.; Gabet, Y. The Skeletal Endocannabinoid System: Clinical and Experimental Insights. J. Basic Clin. Physiol. Pharmacol. 2016, 27, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.-W.; Lin, T.-P.; Ko, J.-Y.; Yeh, D.-W.; Chen, M.-W.; Ke, H.-C.; Wu, S.-L.; Wang, F.-S. Cannabinoid Receptor 1 Regulates ERK and GSK-3β-Dependent Glucocorticoid Inhibition of Osteoblast Differentiation in Murine MC3T3-E1 Cells. Bone 2011, 49, 1255–1263. [Google Scholar] [CrossRef]
- Niu, F.; Zhao, S.; Xu, C.Y.; Sha, H.; Bi, G.B.; Chen, L.; Ye, L.; Gong, P.; Nie, T.H. Potentiation of the Antitumor Activity of Adriamycin against Osteosarcoma by Cannabinoid WIN-55,212-2. Oncol. Lett. 2015, 10, 2415–2421. [Google Scholar] [CrossRef]
- Dunn, S.L.; Wilkinson, J.M.; Crawford, A.; Le Maitre, C.L.; Bunning, R.A.D. Cannabinoid WIN-55,212-2 Mesylate Inhibits Interleukin-1β Induced Matrix Metalloproteinase and Tissue Inhibitor of Matrix Metalloproteinase Expression in Human Chondrocytes. Osteoarthr. Cartil. 2014, 22, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Idris, A.I.; Sophocleous, A.; Landao-Bassonga, E.; Canals, M.; Milligan, G.; Baker, D.; van’t Hof, R.J.; Ralston, S.H. Cannabinoid Receptor Type 1 Protects against Age- Related Osteoporosis by Regulating Osteoblast and Adipocyte Differentiation in Marrow Stromal Cells. Cell Metab. 2009, 10, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Whyte, L.S.; Ford, L.; Ridge, S.A.; Cameron, G.A.; Rogers, M.J.; Ross, R.A. Cannabinoids and Bone: Endocannabinoids Modulate Human Osteoclast Function in Vitro. Br. J. Pharm. 2012, 165, 2584–2597. [Google Scholar] [CrossRef]
- Ofek, O.; Karsak, M.; Leclerc, N.; Fogel, M.; Frenkel, B.; Wright, K.; Tam, J.; Attar-Namdar, M.; Kram, V.; Shohami, E.; et al. Peripheral Cannabinoid Receptor, CB2, Regulates Bone Mass. Proc. Natl. Acad. Sci. USA 2006, 103, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Sophocleous, A.; Landao-Bassonga, E.; van‘t Hof, R.J.; Idris, A.I.; Ralston, S.H. The Type 2 Cannabinoid Receptor Regulates Bone Mass and Ovariectomy-Induced Bone Loss by Affecting Osteoblast Differentiation and Bone Formation. Endocrinology 2011, 152, 2141–2149. [Google Scholar] [CrossRef]
- Sophocleous, A.; Marino, S.; Logan, J.G.; Mollat, P.; Ralston, S.H.; Idris, A.I. Bone Cell-Autonomous Contribution of Type 2 Cannabinoid Receptor to Breast Cancer-Induced Osteolysis. J. Biol. Chem. 2015, 290, 22049–22060. [Google Scholar] [CrossRef] [PubMed]
- Smoum, R.; Baraghithy, S.; Chourasia, M.; Breuer, A.; Mussai, N.; Attar-Namdar, M.; Kogan, N.M.; Raphael, B.; Bolognini, D.; Cascio, M.G.; et al. CB2 Cannabinoid Receptor Agonist Enantiomers HU-433 and HU-308: An Inverse Relationship between Binding Affinity and Biological Potency. Proc. Natl. Acad. Sci. USA 2015, 112, 8774–8779. [Google Scholar] [CrossRef] [PubMed]
- Punzo, F.; Tortora, C.; Di Pinto, D.; Manzo, I.; Bellini, G.; Casale, F.; Rossi, F. Anti-Proliferative, pro-Apoptotic and Anti-Invasive Effect of EC/EV System in Human Osteosarcoma. Oncotarget 2017, 8, 54459–54471. [Google Scholar] [CrossRef]
- Idris, A.I.; Van’t Hof, R.J.; Greig, I.R.; Ridge, S.A.; Baker, D.; Ross, R.A.; Ralston, S.H. Regulation of Bone Mass, Bone Loss and Osteoclast Activity by Cannabinoid Receptors. Nat. Med. 2005, 11, 774–779. [Google Scholar] [CrossRef]
- Yang, P.; Wang, L.; Feng, R.; Almehizia, A.A.; Tong, Q.; Myint, K.-Z.; Ouyang, Q.; Alqarni, M.H.; Wang, L.; Xie, X.-Q. Novel Triaryl Sulfonamide Derivatives as Selective Cannabinoid Receptor 2 Inverse Agonists and Osteoclast Inhibitors: Discovery, Optimization, and Biological Evaluation. J. Med. Chem. 2013, 56, 2045–2058. [Google Scholar] [CrossRef]
- Marino, S.; Carrasco, G.; Li, B.; Shah, K.M.; Lath, D.L.; Sophocleous, A.; Lawson, M.A.; Idris, A.I. JZL184, A Monoacylglycerol Lipase Inhibitor, Induces Bone Loss in a Multiple Myeloma Model of Immunocompetent Mice. Calcif. Tissue Int. 2020, 107, 72–85. [Google Scholar] [CrossRef]
- Liu, H.; Zhou, C.; Qi, D.; Gao, Y.; Zhu, M.; Tao, T.; Sun, X.; Xiao, J. Inhibiting Monoacylglycerol Lipase Suppresses RANKL-Induced Osteoclastogenesis and Alleviates Ovariectomy-Induced Bone Loss. Front. Cell Dev. Biol. 2021, 9, 640867. [Google Scholar] [CrossRef]
- Ihn, H.-J.; Kim, Y.-S.; Lim, S.; Bae, J.-S.; Jung, J.-C.; Kim, Y.-H.; Park, J.-W.; Wang, Z.; Koh, J.-T.; Bae, Y.-C.; et al. PF-3845, a Fatty Acid Amide Hydrolase Inhibitor, Directly Suppresses Osteoclastogenesis through ERK and NF-ΚB Pathways In Vitro and Alveolar Bone Loss In Vivo. Int. J. Mol. Sci. 2021, 22, 1915. [Google Scholar] [CrossRef] [PubMed]
- Bab, I.; Ofek, O.; Tam, J.; Rehnelt, J.; Zimmer, A. Endocannabinoids and the Regulation of Bone Metabolism. J. Neuroendocrinol. 2008, 20, 69–74. [Google Scholar] [CrossRef]
- Tam, J.; Trembovler, V.; Di Marzo, V.; Petrosino, S.; Leo, G.; Alexandrovich, A.; Regev, E.; Casap, N.; Shteyer, A.; Ledent, C.; et al. The Cannabinoid CB1 Receptor Regulates Bone Formation by Modulating Adrenergic Signaling. FASEB J. 2008, 22, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Ofek, O.; Attar-Namdar, M.; Kram, V.; Dvir-Ginzberg, M.; Mechoulam, R.; Zimmer, A.; Frenkel, B.; Shohami, E.; Bab, I. CB2 Cannabinoid Receptor Targets Mitogenic Gi Protein-Cyclin D1 Axis in Osteoblasts. J. Bone Miner. Res. 2011, 26, 308–316. [Google Scholar] [CrossRef]
- Rossi, F.; Bellini, G.; Tortora, C.; Bernardo, M.E.; Luongo, L.; Conforti, A.; Starc, N.; Manzo, I.; Nobili, B.; Locatelli, F.; et al. CB2 and TRPV1 Receptors Oppositely Modulate In Vitro Human Osteoblast Activity. Pharmacol. Res. 2015, 99, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, H.L.; Li, Y.; Hannon, K.; Watkins, B.A. Eicosapentaenoic Acid Decreases Expression of Anandamide Synthesis Enzyme and Cannabinoid Receptor 2 in Osteoblast-like Cells. J. Nutr. Biochem. 2011, 22, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Kostrzewa, M.; Mahmoud, A.M.; Verde, R.; Scotto di Carlo, F.; Gianfrancesco, F.; Piscitelli, F.; Ligresti, A. Modulation of Endocannabinoid Tone in Osteoblastic Differentiation of MC3T3-E1 Cells and in Mouse Bone Tissue over Time. Cells 2021, 10, 1199. [Google Scholar] [CrossRef]
- Bab, I.; Zimmer, A.; Melamed, E. Cannabinoids and the Skeleton: From Marijuana to Reversal of Bone Loss. Ann. Med. 2009, 41, 560–567. [Google Scholar] [CrossRef]
- Rayman, N.; Lam, K.H.; Laman, J.D.; Simons, P.J.; Löwenberg, B.; Sonneveld, P.; Delwel, R. Distinct Expression Profiles of the Peripheral Cannabinoid Receptor in Lymphoid Tissues Depending on Receptor Activation Status. J. Immunol. 2004, 172, 2111–2117. [Google Scholar] [CrossRef]
- Beamer, W.G.; Donahue, L.R.; Rosen, C.J.; Baylink, D.J. Genetic Variability in Adult Bone Density among Inbred Strains of Mice. Bone 1996, 18, 397–403. [Google Scholar] [CrossRef]
- Raphael-Mizrahi, B.; Gabet, Y. The Cannabinoids Effect on Bone Formation and Bone Healing. Curr. Osteoporos. Rep. 2020, 18, 433–438. [Google Scholar] [CrossRef]
- Malinowska, B.; Piszcz, J.; Koneczny, B.; Hryniewicz, A.; Schlicker, E. Modulation of the Cardiac Autonomic Transmission of Pithed Rats by Presynaptic Opioid OP 4 and Cannabinoid CB 1 Receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2001, 364, 233–241. [Google Scholar] [CrossRef]
- Sophocleous, A.; Sims, A.H.; Idris, A.I.; Ralston, S.H. Modulation of Strain-Specific Differences in Gene Expression by Cannabinoid Type 2 Receptor Deficiency. Calcif. Tissue Int. 2014, 94, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Sophocleous, A.; Idris, A.I.; Ralston, S.H. Genetic Background Modifies the Effects of Type 2 Cannabinoid Receptor Deficiency on Bone Mass and Bone Turnover. Calcif. Tissue Int. 2014, 94, 259–268. [Google Scholar] [CrossRef]
- Sophocleous, A.; Marino, S.; Kabir, D.; Ralston, S.H.; Idris, A.I. Combined Deficiency of the Cnr1 and Cnr2 Receptors Protects against Age-Related Bone Loss by Osteoclast Inhibition. Aging Cell 2017, 16, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Ofek, O.; Fride, E.; Ledent, C.; Gabet, Y.; Müller, R.; Zimmer, A.; Mackie, K.; Mechoulam, R.; Shohami, E.; et al. Involvement of Neuronal Cannabinoid Receptor CB1 in Regulation of Bone Mass and Bone Remodeling. Mol. Pharmacol. 2006, 70, 786–792. [Google Scholar] [CrossRef]
- Black, D.M.; Rosen, C.J. Clinical Practice. Postmenopausal Osteoporosis. N. Engl. J. Med. 2016, 358, 494–501. [Google Scholar] [CrossRef]
- Sunyecz, J.A. The use of calcium and vitamin D in the management of osteoporosis. Ther. Clin. Risk Manag. 2008, 4, 827–836. [Google Scholar] [CrossRef]
- Aspray, T.J.; Hill, T.R. Osteoporosis and the Ageing Skeleton. Subcell Biochem. 2019, 91, 453–476. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, A.; Olsson, T.; Nordstrom, P. Bone gained from physical activity and lost through detraining: A longitudinal study in young males. Osteoporos. Int. 2005, 16, 835–841. [Google Scholar] [CrossRef]
- Nordstrom, A.; Olsson, T.; Nordstrom, P. Sustained benefits from previous physical activity on bone mineral density in males. J. Clin. Endocrinol. Metab. 2006, 91, 2600–2604. [Google Scholar] [CrossRef]
- Daly, R.M.; Bass, S.L. Lifetime sport and leisure activity participation is associated with greater bone size, quality and strength in older men. Osteoporos. Int. 2006, 17, 1258–1267. [Google Scholar] [CrossRef]
- Carter, M.I.; Hinton, P.S. Physical Activity and Bone Health. Mo. Med. 2014, 111, 59–64. [Google Scholar]
- Gkastaris, K.; Goulis, D.G.; Potoupnis, M.; Anastasilakis, A.D.; Kapetanos, G. Obesity, osteoporosis and bone metabolism. J. Musculoskelet. Neuronal Interact. 2020, 20, 372–381. [Google Scholar]
- Cetani, F.; Saponaro, F.; Borsari, S.; Marcocci, C. Familial and Hereditary Forms of Primary Hyperparathyroidism. Front. Hor. Res. 2019, 51, 40–51. [Google Scholar] [CrossRef]
- Marcocci, C.; Saponaro, F. Epidemiology, Pathogenesis of Primary Hyperparathyroidism: Current Data. Ann. Endocrinol. 2015, 76, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Colangelo, L.; Biamonte, F.; Pepe, J.; Cipriani, C.; Minisola, S. Understanding and Managing Secondary Osteoporosis. Expert Rev. Endocrinol. Metab. 2019, 14, 111–122. [Google Scholar] [CrossRef]
- Bhutani, G.; Gupta, M. Emerging Therapies for the Treatment of Osteoporosis. J. Mid-Life Health 2013, 4, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Bab, I.; Zimmer, A. Cannabinoid Receptors and the Regulation of Bone Mass. Br. J. Pharm. 2008, 153, 182–188. [Google Scholar] [CrossRef]
- Devoto, M.; Shimoya, K.; Caminis, J.; Ott, J.; Tenenhouse, A.; Whyte, M.P.; Sereda, L.; Hall, S.; Considine, E.; Williams, C.J.; et al. First-Stage Autosomal Genome Screen in Extended Pedigrees Suggests Genes Predisposing to Low Bone Mineral Density on Chromosomes 1p, 2p and 4q. Eur. J. Hum. Genet. 1998, 6, 151–157. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Devoto, M.; Specchia, C.; Li, H.H.; Caminis, J.; Tenenhouse, A.; Rodriguez, H.; Spotila, L.D. Variance Component Linkage Analysis Indicates a QTL for Femoral Neck Bone Mineral Density on Chromosome 1p36. Hum. Mol. Genet. 2001, 10, 2447–2452. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Devoto, M.; Spotila, L.D.; Stabley, D.L.; Wharton, G.N.; Rydbeck, H.; Korkko, J.; Kosich, R.; Prockop, D.; Tenenhouse, A.; Sol-Church, K. Univariate and Bivariate Variance Component Linkage Analysis of a Whole-Genome Scan for Loci Contributing to Bone Mineral Density. Eur. J. Hum. Genet. 2005, 13, 781–788. [Google Scholar] [CrossRef]
- Karsak, M.; Cohen-Solal, M.; Freudenberg, J.; Ostertag, A.; Morieux, C.; Kornak, U.; Essig, J.; Erxlebe, E.; Bab, I.; Kubisch, C.; et al. Cannabinoid Receptor Type 2 Gene Is Associated with Human Osteoporosis. Hum. Mol. Genet. 2005, 14, 3389–3396. [Google Scholar] [CrossRef]
- Karsak, M.; Malkin, I.; Tolia, M.R.; Kubisch, C.; Nürnberg, P.; Zimmer, A.; Livshits, G. The Cannabinoid Receptor Type 2 (CNR2) Gene Is Associated with Hand Bone Strength Phenotypes in an Ethnically Homogeneous Family Sample. Hum. Genet. 2009, 126, 629–636. [Google Scholar] [CrossRef]
- Yamada, Y.; Ando, F.; Shimokata, H. Association of Candidate Gene Polymorphisms with Bone Mineral Density in Community-Dwelling Japanese Women and Men. Int. J. Mol. Med. 2007, 19, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.Y.; Li, G.H.Y.; Kung, A.W.C. Multiple Osteoporosis Susceptibility Genes on Chromosome 1p36 in Chinese. Bone 2009, 44, 984–988. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.H.; Kim, H.; Kim, J.H.; Kim, J.G. Cannabinoid Receptor Gene Polymorphisms and Bone Mineral Density in Korean Postmenopausal Women. Menopause 2015, 22, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Idris, A.I. Emerging Therapeutic Targets in Cancer Induced Bone Disease: A Focus on the Peripheral Type 2 Cannabinoid Receptor. Pharm. Res. 2017, 119, 391–403. [Google Scholar] [CrossRef]
- Sterling, J.A.; Edwards, J.R.; Martin, T.J.; Mundy, G.R. Advances in the Biology of Bone Metastasis: How the Skeleton Affects Tumor Behavior. Bone 2011, 48, 6–15. [Google Scholar] [CrossRef]
- Maurizi, A.; Rucci, N. The Osteoclast in Bone Metastasis: Player and Target. Cancers 2018, 10, 218. [Google Scholar] [CrossRef]
- Esposito, M.; Guise, T.; Kang, Y. The Biology of Bone Metastasis. Cold Spring Harb. Perspect. Med. 2018, 8, a031252. [Google Scholar] [CrossRef]
- Bussard, K.M.; Gay, C.V.; Mastro, A.M. The Bone Microenvironment in Metastasis; What Is Special about Bone? Cancer Metastasis Rev. 2008, 27, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; Stenzl, A.; Tombal, B. Advances in the Therapy of Prostate Cancer–Induced Bone Disease: Current Insights and Future Perspectives on the RANK/RANKL Pathways. Eur. Urol. Suppl. 2009, 8, 747–752. [Google Scholar] [CrossRef]
- Roodman, G.D. Mechanisms of Bone Metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Baud’huin, M.; Duplomb, L.; Velasco, C.R.; Fortun, Y.; Heymann, D.; Padrines, M. Key Roles of the OPG–RANK–RANKL System in Bone Oncology. Expert Rev. Anticancer Ther. 2007, 7, 221–232. [Google Scholar] [CrossRef]
- Ramer, R.; Merkord, J.; Rohde, H.; Hinz, B. Cannabidiol Inhibits Cancer Cell Invasion via Upregulation of Tissue Inhibitor of Matrix Metalloproteinases-1. Biochem. Pharmacol. 2010, 79, 955–966. [Google Scholar] [CrossRef]
- Ramer, R.; Bublitz, K.; Freimuth, N.; Merkord, J.; Rohde, H.; Haustein, M.; Borchert, P.; Schmuhl, E.; Linnebacher, M.; Hinz, B. Cannabidiol Inhibits Lung Cancer Cell Invasion and Metastasis via Intercellular Adhesion Molecule-1. FASEB J. 2012, 26, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Casanova, M.L.; Planas, A.; Del Pulgar, T.G.; Villanueva, C.; Fernández-Aceñero, M.J.; Aragonés, J.; Huffman, J.W.; Jorcano, J.L.; Guzmán, M. Inhibition of Tumor Angiogenesis by Cannabinoids. FASEB J. 2003, 17, 1–16. [Google Scholar] [CrossRef]
- Punzo, F.; Tortora, C.; Di Pinto, D.; Pota, E.; Argenziano, M.; Di Paola, A.; Casale, F.; Rossi, F. Bortezomib and Endocannabinoid/Endovanilloid System: A Synergism in Osteosarcoma. Pharmacol. Res. 2018, 137, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Ondoua, A.N.; Hanlon, K.E.; Symons-Liguori, A.M.; Largent-Milnes, T.M.; Havelin, J.J.; Ferland, H.L.; Chandramouli, A.; Owusu-Ankomah, M.; Nikolich-Zugich, T.; Bloom, A.P.; et al. Disease Modification of Breast Cancer-Induced Bone Remodeling by Cannabinoid 2 Receptor Agonists. J. Bone Miner. Res. 2013, 28, 92–107. [Google Scholar] [CrossRef]
- Hsu, S.S.; Huang, C.J.; Cheng, H.H.; Chou, C.T.; Lee, H.Y.; Wang, J.L.; Chen, I.S.; Liu, S.I.; Lu, Y.C.; Chang, H.T.; et al. Anandamide-Induced Ca2+ Elevation Leading to P38 MAPK Phosphorylation and Subsequent Cell Death via Apoptosis in Human Osteosarcoma Cells. Toxicology 2007, 231, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Notaro, A.; Sabella, S.; Pellerito, O.; Di Fiore, R.; De Blasio, A.; Vento, R.; Calvaruso, G.; Giuliano, M. Involvement of PAR-4 in Cannabinoid-Dependent Sensitization of Osteosarcoma Cells to TRAIL-Induced Apoptosis. Int. J. Biol. Sci. 2014, 10, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Ondoua, A.N.; Wright, C.; Vardanyan, A.; King, T.; Largent-Milnes, T.M.; Nelson, M.; Jimenez-Andrade, J.M.; Mantyh, P.W.; Vanderah, T.W. A Cannabinoid 2 Receptor Agonist Attenuates Bone Cancer-Induced Pain and Bone Loss. Life Sci. 2010, 86, 646–653. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
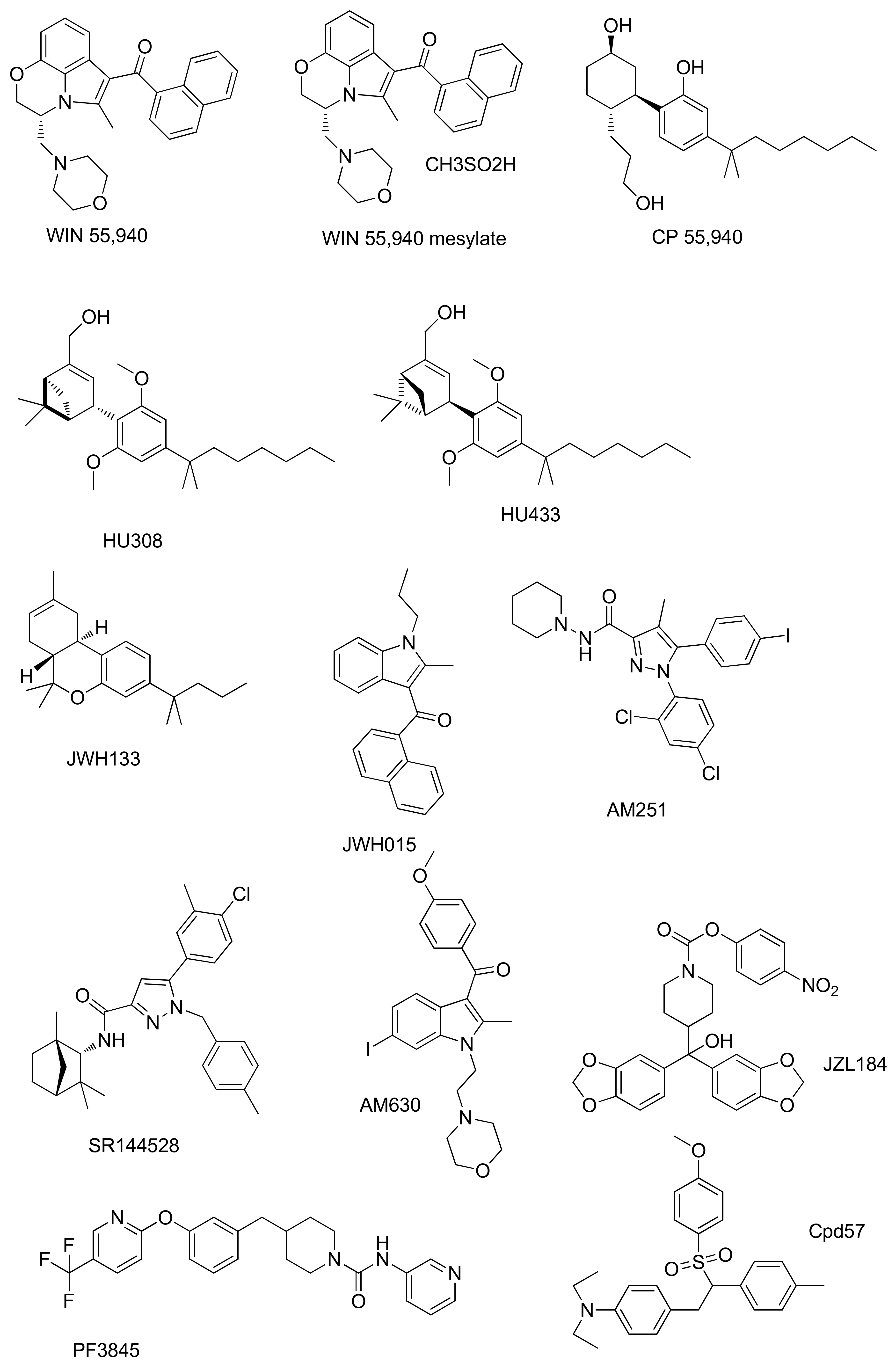
| Compound | Activity | Experimental Model | Effect on Bone | References |
|---|---|---|---|---|
| WIN55,212 | CB1R agonism | Murine MC3T3-E1 osteoblasts. | Glucocorticoid-induced inhibition of mineralization in MC3T3-E1 osteoblasts | [60] |
| CBR agonism | MG-63 human osteosarcoma cell line. | Antitumor activity and its combined effect with adriamycin against the MG-63 human osteosarcoma cell line | [61] | |
| WIN55,212-2 mesylate | CBR agonism | Human chondrocytes. | Prevention of cartilage breakdown in arthritis | [62] |
| CP55940 | CB1R agonism | Bone marrow mouse cell. | Stimulation of bone nodule formation | [63] |
| CB2R agonism | Human osteoclasts. | Inhibition of human and mouse osteoclast formation Stimulation of human osteoclast polarization and resorption | [64] | |
| HU308 | CB2R agonism | Bone-marrow-derived osteoblasts/stromal cells. Osteoblasts isolated from mouse calvarial bones | Increased osteoblast differentiation and activity | [65,66] |
| OVX C3H mouse model of postmenopausal osteoporosis. | Prevention of osteoporosis in ovariectomized rats | [14,65,66] | ||
| M-CSF-generated osteoclasts | At low concentration stimulation of osteoclast formation. At high concentrations of inhibition of osteoclast formation | [14] | ||
| 4T1 mice cell line and MDA-MB-231 human cell line | Reduction in osteotropic breast cancer cells proliferation | [67] | ||
| HU433 | CB2R agonism | Newborn mouse calvarial osteoblasts | Stimulation of osteoblast proliferation | [68] |
| Osteoclastogenic cultures from bone marrow-derived monocytes | Increase in osteoclast apoptosis | |||
| Ovariectomized-mouse model | Prevention of osteoporosis in ovariectomized mouse | |||
| JWH 133 | CB2R agonism | M-CSF-generated osteoclasts | Stimulation of osteoclast formation | [14] |
| Six different OS cell lines | Anti-proliferative, pro-apoptotic, anti-invasive effect. | [69] | ||
| 4T1 mice cell line and MDA-MB-231 human cell line | Reduction in osteotropic breast cancer cells proliferation | [67] | ||
| JWH015 | CB2R agonism | Human osteoclasts | Stimulation of human osteoclast polarization and resorption | [64] |
| AM251 | CB1R antagonism | Mouse osteoclast cultures | Inhibition of osteoclast formation | [70] |
| Ovariectomized-mouse model | Protection against ovariectomy induced bone loss | |||
| SR144528 | CB2R antagonism | Mouse osteoclast cultures | Inhibition of osteoclast formation | [70] |
| Ovariectomized-mouse model | Protection against ovariectomy induced bone loss | |||
| AM630 | CB2R antagonism | Mouse osteoclast cultures | Inhibition of osteoclast formation | [70] |
| Cpd 57 | CB2R inverse agonism | Osteoclast from RAW 264.7 cells | Inhibition of osteoclast formation | [71] |
| JZL184 | MAGL inhibition | Osteoclast from RAW 264.7 cells | At low concentration stimulates osteoclast formation. At high concentrations inhibits osteoclast area | [72] |
| Human osteoblast-like cells Saos-2 | Inhibition of osteoblastic bone formation No effect to mature or form bone nodules | |||
| Bone marrow macrophages (BMMs) | Suppression of osteoclast differentiation and function | [73] | ||
| C57BL/6 mice | Reduction OVX-Induced Bone Loss | |||
| PF-3845 | FAAH inhibition | Osteoclast from mouse bone marrow macrophages | Suppression of osteoclast differentiation. Anti-resorptive activity prevents alveolar bone loss | [74] |
| male C57BL/6 mice | Prevention alveolar bone loss |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saponaro, F.; Ferrisi, R.; Gado, F.; Polini, B.; Saba, A.; Manera, C.; Chiellini, G. The Role of Cannabinoids in Bone Metabolism: A New Perspective for Bone Disorders. Int. J. Mol. Sci. 2021, 22, 12374. https://doi.org/10.3390/ijms222212374
Saponaro F, Ferrisi R, Gado F, Polini B, Saba A, Manera C, Chiellini G. The Role of Cannabinoids in Bone Metabolism: A New Perspective for Bone Disorders. International Journal of Molecular Sciences. 2021; 22(22):12374. https://doi.org/10.3390/ijms222212374
Chicago/Turabian StyleSaponaro, Federica, Rebecca Ferrisi, Francesca Gado, Beatrice Polini, Alessandro Saba, Clementina Manera, and Grazia Chiellini. 2021. "The Role of Cannabinoids in Bone Metabolism: A New Perspective for Bone Disorders" International Journal of Molecular Sciences 22, no. 22: 12374. https://doi.org/10.3390/ijms222212374
APA StyleSaponaro, F., Ferrisi, R., Gado, F., Polini, B., Saba, A., Manera, C., & Chiellini, G. (2021). The Role of Cannabinoids in Bone Metabolism: A New Perspective for Bone Disorders. International Journal of Molecular Sciences, 22(22), 12374. https://doi.org/10.3390/ijms222212374