
MyD88 in Macrophages Enhances Liver Fibrosis by Activation of NLRP3 Inflammasome in HSCs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
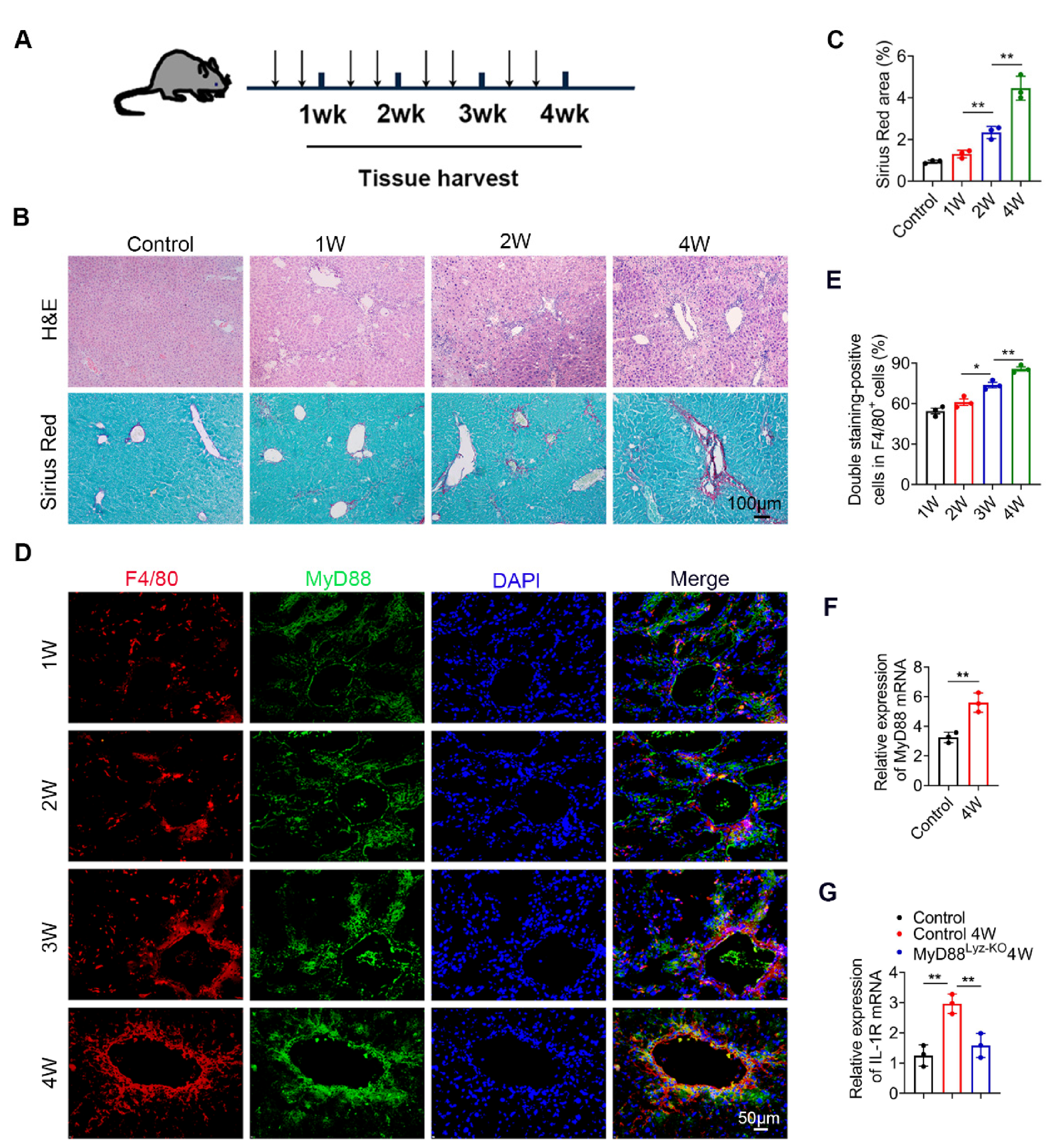
2.1. MyD88 Expression in Macrophages Is Upregulated during the Progression of Liver Fibrosis
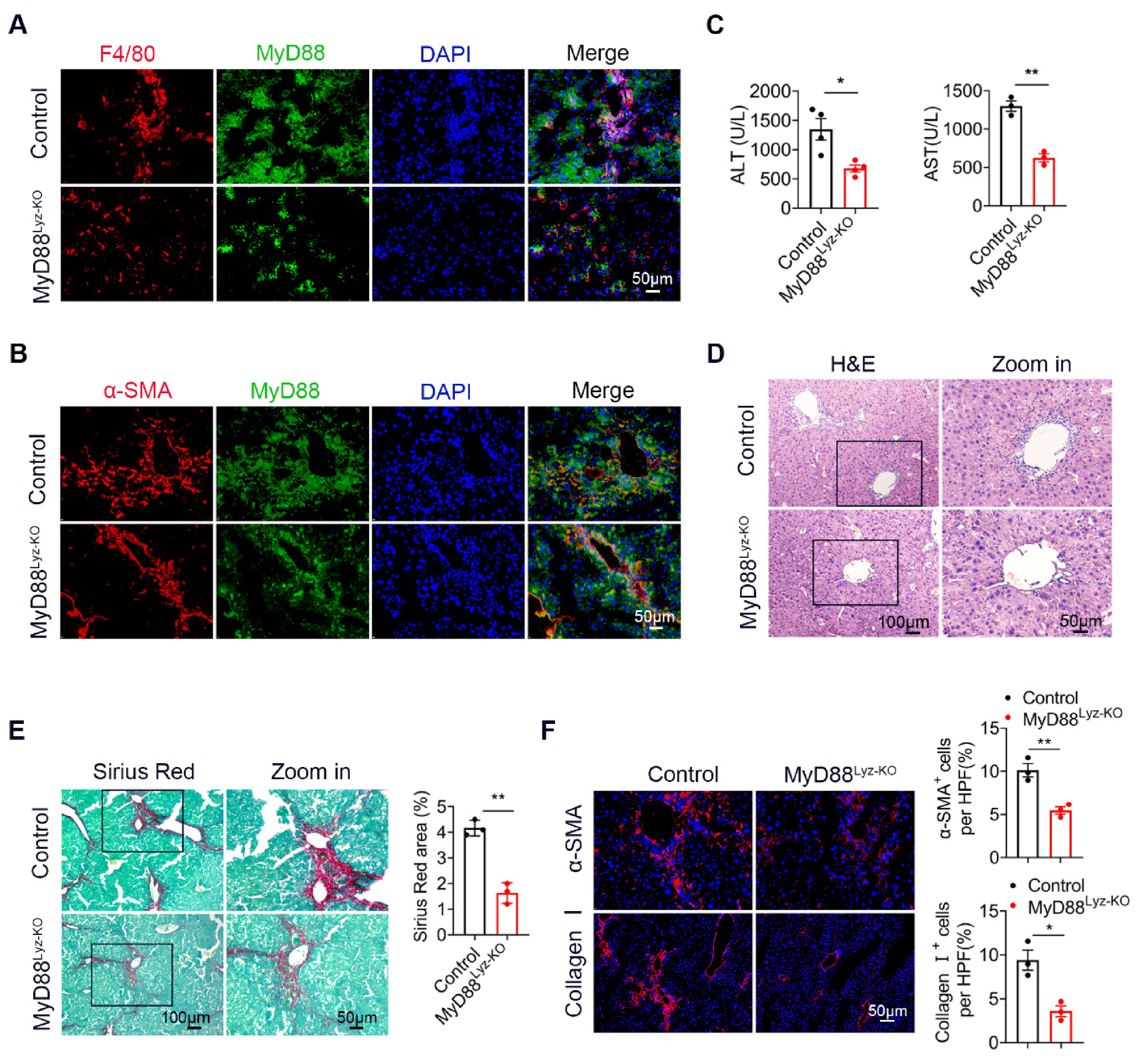
2.2. MyD88 Deficiency in Myeloid Cells Attenuates Liver Fibrosis in Mice
2.3. MyD88 Deficiency in Myeloid Cells Decreases Inflammatory Cell Infiltration in the Liver

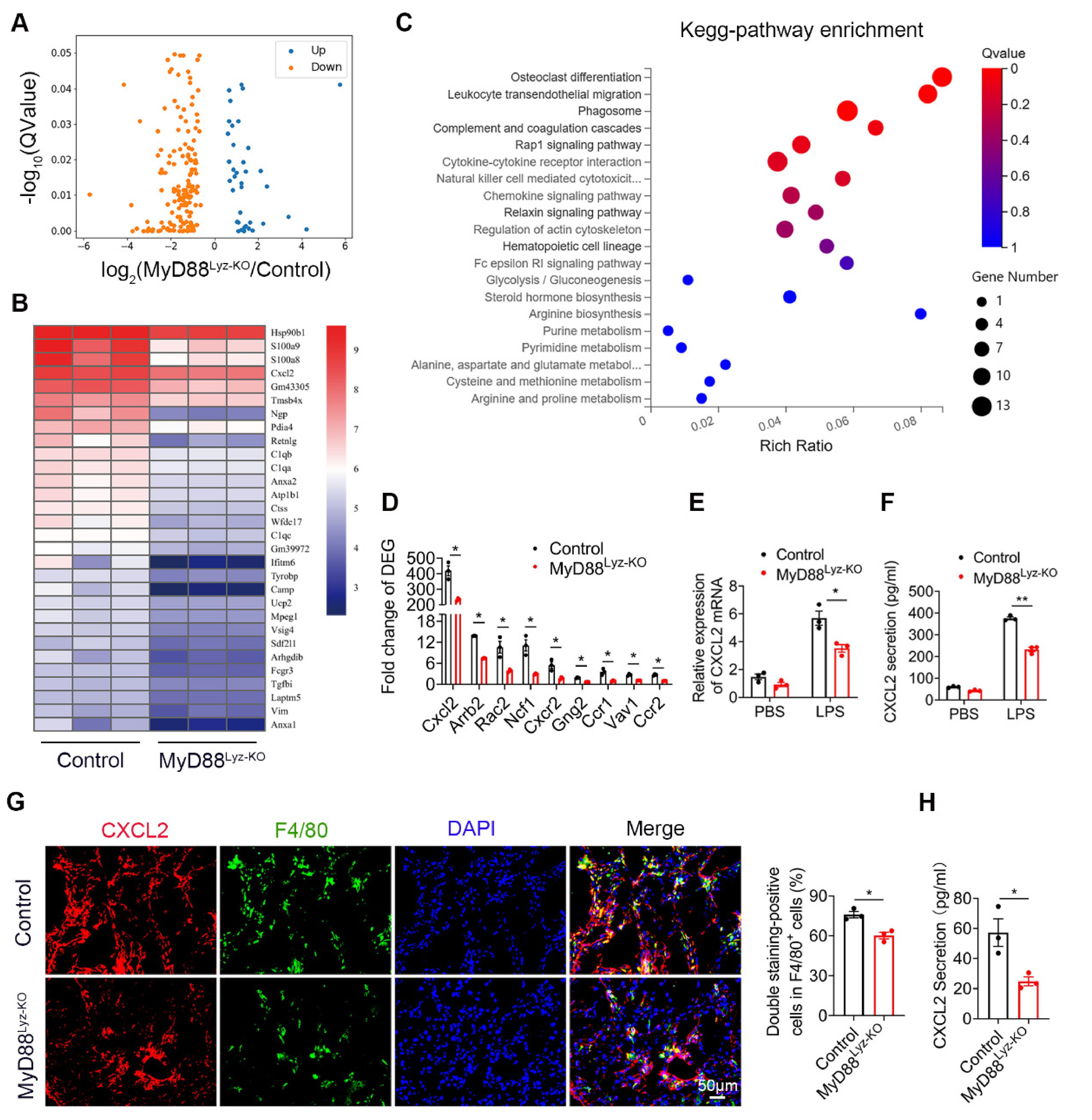
2.4. Specific Genetic Deletion of MyD88 in Macrophages Reduces CXCL2 Secretion
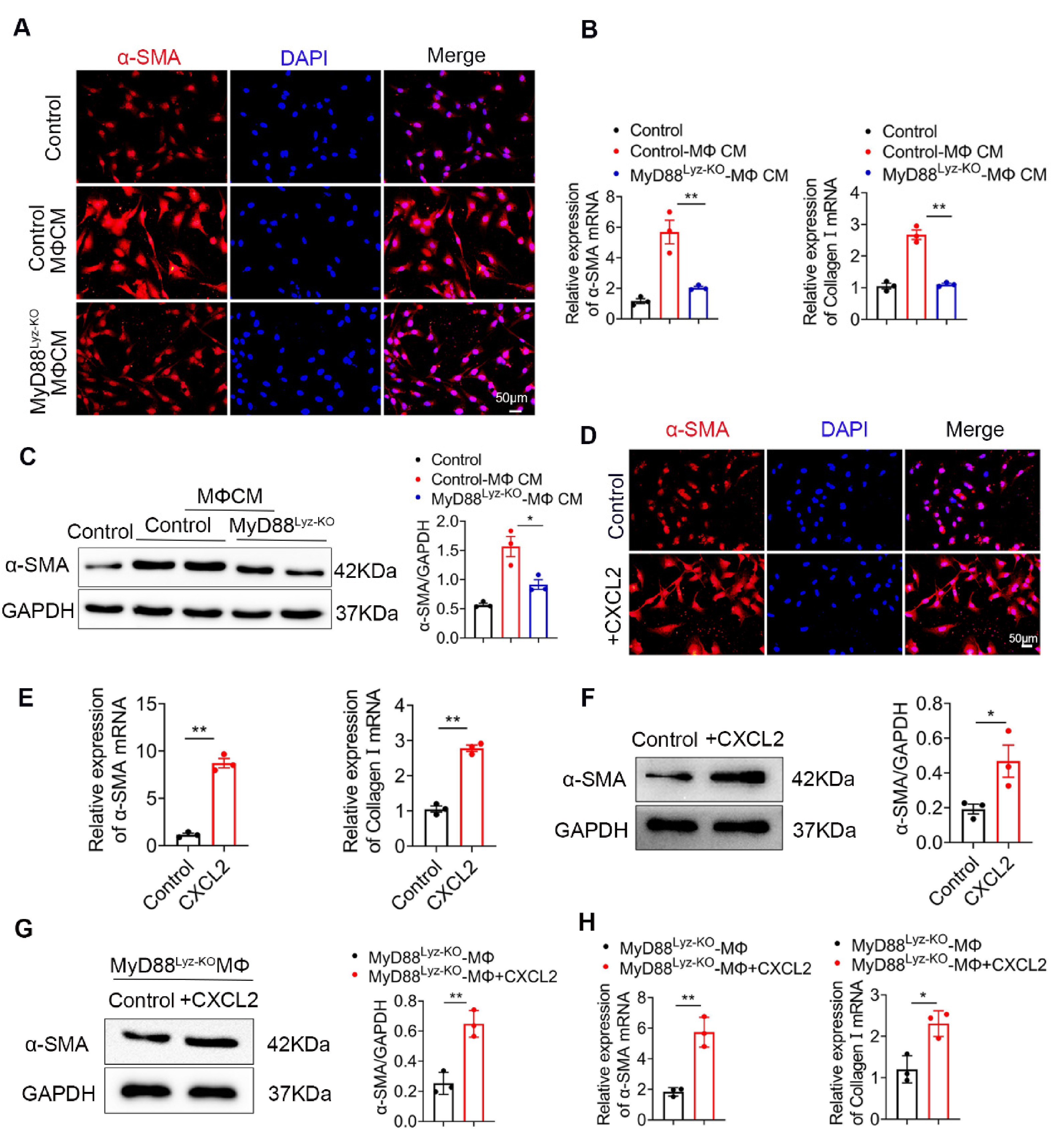
2.5. Macrophages Promote the Activation of HSCs by Secreting CXCL2
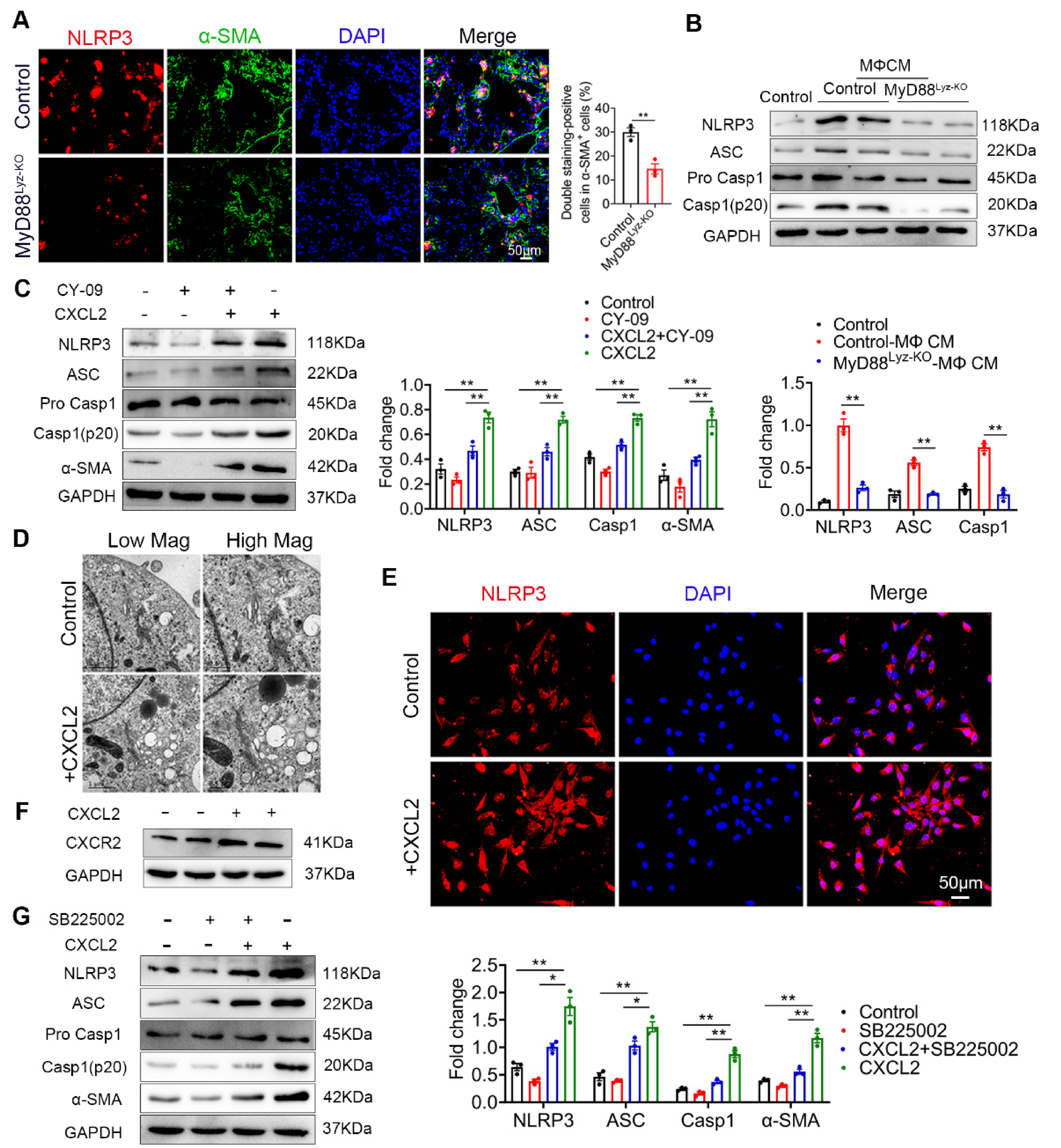
2.6. CXCL2 Induced NLRP3 Inflammasome Activation in HSCs
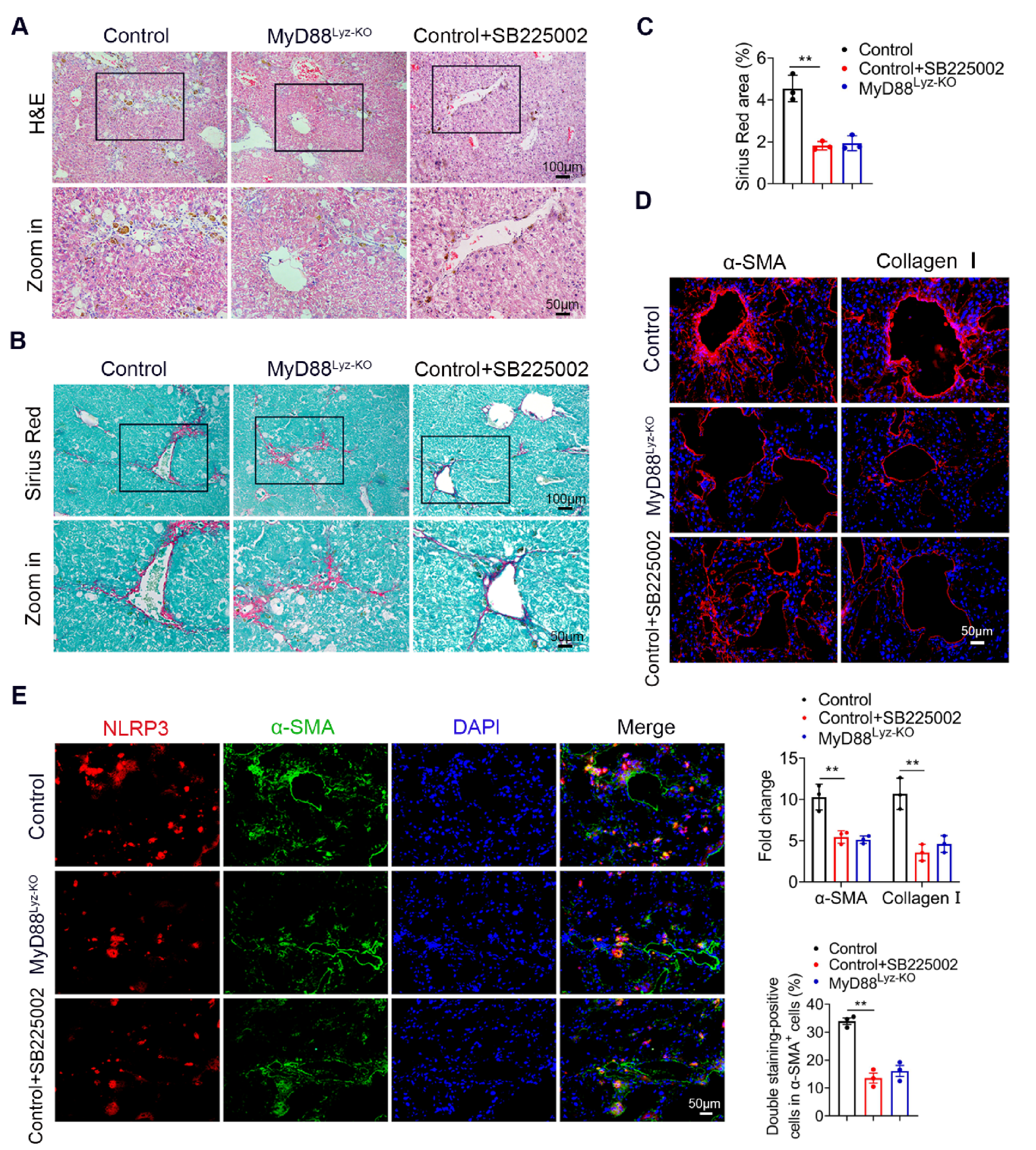
2.7. Targeting CXCL2 with a CXCR2 Inhibitor Effectively Attenuates CCl4-Induced Liver Fibrosis
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. CCl4-Induced Liver Fibrosis
4.3. Cell Lines and Treatments
4.4. Blood Biochemical Assays
4.5. Histology and Immunostaining
4.6. Isolation of Monocyte-Derived Macrophages
4.7. Collection of Conditioned Media
4.8. Cellular Immunofluorescence
4.9. Flow Cytometry Analysis
4.10. Transmission Electron Microscopy
4.11. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
4.12. Western Blot Analysis
4.13. ELISA
4.14. RNA Sequencing Analysis
4.15. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ginès, P.; Krag, A.; Abraldes, J.G.; Solà, E.; Fabrellas, N.; Kamath, P.S. Liver cirrhosis. Lancet 2021, 398, 1359–1376. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, G.; Morabito, A.; D’Amico, M.; Pasta, L.; Malizia, G.; Rebora, P.; Valsecchi, M.G. New concepts on the clinical course and stratification of compensated and decompensated cirrhosis. Hepatol. Int. 2018, 12 (Suppl. 1), 34–43. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef]
- Barcena-Varela, M.; Colyn, L.; Fernandez-Barrena, M.G. Epigenetic Mechanisms in Hepatic Stellate Cell Activation During Liver Fibrosis and Carcinogenesis. Int. J. Mol. Sci. 2019, 20, 10. [Google Scholar] [CrossRef] [Green Version]
- Lua, I.; Li, Y.; Zagory, J.A.; Wang, K.S.; French, S.W.; Sevigny, J.; Asahina, K. Characterization of hepatic stellate cells, portal fibroblasts, and mesothelial cells in normal and fibrotic livers. J. Hepatol. 2016, 64, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Schuppan, D.; Kim, Y.O. Evolving therapies for liver fibrosis. J. Clin. Investig. 2013, 123, 1887–1901. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, X.; Xu, W.; Wang, S.; Hu, Z.; Zhang, Q.; Deng, X.; Wang, J.; Zhang, J.; Guo, C. Antifibrotic effects of luteolin on hepatic stellate cells and liver fibrosis by targeting AKT/mTOR/p70S6K and TGFbeta/Smad signalling pathways. Liver Int. 2015, 35, 1222–1233. [Google Scholar] [CrossRef]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef]
- Collins, A.R.; Ji, R.; Ramirez, M.R.; Minze, L.J.; Liu, J.Z.; Arredondo, M.; Ren, Y.; Deng, T.; Wang, J.; Lyon, C.J.; et al. Myeloid deletion ofnuclear factor erythroid 2-related factor 2 increases atherosclerosis and liver injury. Arter. Thromb. Vasc. Biol. 2012, 32, 2839–2846. [Google Scholar] [CrossRef] [Green Version]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 123–147. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014, 59, 2034–2042. [Google Scholar] [CrossRef]
- Huang, E.; Peng, N.; Xiao, F.; Hu, D.; Wang, X.; Lu, L. The Roles of Immune Cells in the Pathogenesis of Fibrosis. Int. J. Mol. Sci 2020, 21, 15. [Google Scholar] [CrossRef]
- Li, H.; Zheng, H.W.; Chen, H.; Xing, Z.Z.; You, H.; Cong, M.; Jia, J.D. Hepatitis B virus particles preferably induce Kupffer cells to produce TGF-beta1 over pro-inflammatory cytokines. Dig. Liver Dis. 2012, 44, 328–333. [Google Scholar] [CrossRef]
- Neil, C.; Henderson, A.C.M.; Farnworth, S.L.; Poirier, F.; Russo, F.P.; Iredale, J.P.; Haslett, C.; Simpson, K.J.; Sethi, T. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 5060–5065. [Google Scholar]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Palsson-McDermott, E.M.; Bowie, A.G.; Jefferies, C.A.; Mansell, A.S.; Brady, G.; Brint, E.; Dunne, A.; Gray, P.; Harte, M.T.; et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 2001, 413, 78–83. [Google Scholar] [CrossRef]
- Layland, L.E.; Wagner, H.; da Costa, C.U. Lack of antigen-specific Th1 response alters granuloma formation and composition in Schistosoma mansoni-infected MyD88-/- mice. Eur. J. Immunol. 2005, 35, 3248–3257. [Google Scholar] [CrossRef]
- Thapa, M.; Chinnadurai, R.; Velazquez, V.M.; Tedesco, D.; Elrod, E.; Han, J.H.; Sharma, P.; Ibegbu, C.; Gewirtz, A.; Anania, F.; et al. Liver fibrosis occurs through dysregulation of MyD88-dependent innate B-cell activity. Hepatology 2015, 61, 2067–2079. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Xu, Y.; Han, X.; Yin, L.; Xu, L.; Qi, Y.; Zhao, Y.; Liu, K.; Peng, J. Dioscin alleviates alcoholic liver fibrosis by attenuating hepatic stellate cell activation via the TLR4/MyD88/NF-kappaB signaling pathway. Sci. Rep. 2015, 5, 18038. [Google Scholar] [CrossRef] [Green Version]
- Gieling, R.G.; Wallace, K.; Han, Y.P. Interleukin-1 participates in the progression from liver injury to fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1324–G1331. [Google Scholar] [CrossRef]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [Green Version]
- Wree, A.; McGeough, M.D.; Inzaugarat, M.E.; Eguchi, A.; Schuster, S.; Johnson, C.D.; Pena, C.A.; Geisler, L.J.; Papouchado, B.G.; Hoffman, H.M.; et al. NLRP3 inflammasome driven liver injury and fibrosis: Roles of IL-17 and TNF in mice. Hepatology 2018, 67, 736–749. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564, 71–76. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Wang, W.; Qin, W.; Cheng, K.; Coulup, S.; Chavez, S.; Jiang, S.; Raparia, K.; De Almeida, L.M.V.; Stehlik, C.; et al. TLR4-dependent fibroblast activation drives persistent organ fibrosis in skin and lung. JCI Insight 2018, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Huebener, P.; Schwabe, R.F. Regulation of wound healing and organ fibrosis by toll-like receptors. Biochim. Biophys. Acta 2013, 1832, 1005–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Seki, E. Toll-like receptors in liver fibrosis: Cellular crosstalk and mechanisms. Front. Physiol. 2012, 3, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cengiz, M.; Ozenirler, S.; Elbeg, S. Role of serum toll-like receptors 2 and 4 in non-alcoholic steatohepatitis and liver fibrosis. J. Gastroenterol. Hepatol. 2015, 30, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Wang, S.; Friedman, S.L. The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells. Cell Metab. 2021, 33, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Tacke, F. Roles for chemokines in liver disease. Gastroenterology 2014, 147, 577–594. [Google Scholar] [CrossRef]
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [Green Version]
- Karthikeyan, S.; Potter, J.J.; Geschwind, J.F.; Sur, S.; Hamilton, J.P.; Vogelstein, B.; Kinzler, K.W.; Mezey, E.; Ganapathy-Kanniappan, S. Deregulation of energy metabolism promotes antifibrotic effects in human hepatic stellate cells and prevents liver fibrosis in a mouse model. Biochem. Biophys. Res. Commun. 2016, 469, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Wehr, A.; Baeck, C.; Heymann, F.; Niemietz, P.M.; Hammerich, L.; Martin, C.; Zimmermann, H.W.; Pack, O.; Gassler, N.; Hittatiya, K.; et al. Chemokine receptor CXCR6-dependent hepatic NK T Cell accumulation promotes inflammation and liver fibrosis. J. Immunol. 2013, 190, 5226–5236. [Google Scholar] [CrossRef] [Green Version]
- Blengio, F.; Raggi, F.; Pierobon, D.; Cappello, P.; Eva, A.; Giovarelli, M.; Varesio, L.; Bosco, M.C. The hypoxic environment reprograms the cytokine/chemokine expression profile of human mature dendritic cells. Immunobiology 2013, 218, 76–89. [Google Scholar] [CrossRef]
- Subat, S.; Mogushi, K.; Yasen, M.; Kohda, T.; Ishikawa, Y.; Tanaka, H. Identification of genes and pathways, including the CXCL2 axis, altered by DNA methylation in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2019, 145, 675–684. [Google Scholar] [CrossRef]
- Miura, K.; Matsuo, J.; Rahman, M.A.; Kumagai, Y.; Li, X.; Rikihisa, Y. Ehrlichia chaffeensis induces monocyte inflammatory responses through MyD88, ERK, and NF-kappaB but not through TRIF, interleukin-1 receptor 1 (IL-1R1)/IL-18R1, or toll-like receptors. Infect. Immun. 2011, 79, 4947–4956. [Google Scholar] [CrossRef] [Green Version]
- Haraguchi, K.; Kawamoto, A.; Isami, K.; Maeda, S.; Kusano, A.; Asakura, K.; Shirakawa, H.; Mori, Y.; Nakagawa, T.; Kaneko, S. TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J. Neurosci. 2012, 32, 3931–3941. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Wang, J.; Wang, J.; Zhou, Q.; Yang, B.; He, Q.; Weng, Q. Intercellular crosstalk of hepatic stellate cells in liver fibrosis: New insights into therapy. Pharm. Res. 2020, 155, 104720. [Google Scholar] [CrossRef]
- Ezhilarasan, D. Hepatic stellate cells in the injured liver: Perspectives beyond hepatic fibrosis. J. Cell. Physiol. 2021, 1–14. [Google Scholar] [CrossRef]
- Wree, A.; McGeough, M.D.; Pena, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [Green Version]
- Artlett, C.M.; Thacker, J.D. Molecular activation of the NLRP3 Inflammasome in fibrosis: Common threads linking divergent fibrogenic diseases. Antioxid. Redox. Signal. 2015, 22, 1162–1175. [Google Scholar] [CrossRef]
- Boaru, S.G.; Borkham-Kamphorst, E.; Tihaa, L.; Haas, U.; Weiskirchen, R. Expression analysis of inflammasomes in experimental models of inflammatory and fibrotic liver disease. J. Inflamm. 2012, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Duan, N.N.; Liu, X.J.; Wu, J. Palmitic acid elicits hepatic stellate cell activation through inflammasomes and hedgehog signaling. Life Sci. 2017, 176, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Zhang, Y.; Zheng, J.H.; Li, X.; Yao, Y.L.; Wu, Y.L.; Song, S.Z.; Sun, P.; Nan, J.X.; Lian, L.H. Potentiation of hepatic stellate cell activation by extracellular ATP is dependent on P2X7R-mediated NLRP3 inflammasome activation. Pharm. Res. 2017, 117, 82–93. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, F.; Xiong, X.; Lu, C.; Lian, N.; Lu, Y.; Zheng, S. Tetramethylpyrazine reduces inflammation in liver fibrosis and inhibits inflammatory cytokine expression in hepatic stellate cells by modulating NLRP3 inflammasome pathway. IUBMB Life 2015, 67, 312–321. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Zhang, J.; Dai, C.; Liu, X.; Wang, J.; Gao, Z.; Guo, H.; Wang, R.; Lu, S.; et al. S100A4 promotes liver fibrosis via activation of hepatic stellate cells. J. Hepatol. 2015, 62, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Gu, J.; Zhang, J.; Liu, S.; Wang, Q.; Tian, T.; Chen, Z.; Zhang, J. MyD88 in myofibroblasts enhances colitis-associated tumorigenesis via promoting macrophage M2 polarization. Cell Rep. 2021, 34, 108724. [Google Scholar] [CrossRef]
- Baeck, C.; Wei, X.; Bartneck, M.; Fech, V.; Heymann, F.; Gassler, N.; Hittatiya, K.; Eulberg, D.; Luedde, T.; Trautwein, C.; et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology 2014, 59, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Tacke, F.; Angeli, V.; Bogunovic, M.; Loubeau, M.; Dai, X.M.; Stanley, E.R.; Randolph, G.J.; Merad, M. Langerhans cells arise from monocytes in vivo. Nat. Immunol. 2006, 7, 265–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Q.; Hou, S.; Zhai, J.; Tian, T.; Wu, Y.; Wu, Z.; He, J.; Chen, Z.; Zhang, J. S100A4 promotes inflammation but suppresses lipid accumulation via the STAT3 pathway in chronic ethanol-induced fatty liver. J. Mol. Med. 2019, 97, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Tian, T.; Qi, D.; Sun, K.; Yuan, Q.; Wang, Z.; Qin, Z.; Wu, Z.; Chen, Z.; Zhang, J. S100A4 promotes lung tumor development through beta-catenin pathway-mediated autophagy inhibition. Cell Death Dis. 2018, 9, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, S.; Yang, W.; Chen, H.; Yuan, Q.; Liu, S.; Zhao, Y.; Zhang, J. MyD88 in Macrophages Enhances Liver Fibrosis by Activation of NLRP3 Inflammasome in HSCs. Int. J. Mol. Sci. 2021, 22, 12413. https://doi.org/10.3390/ijms222212413
Ge S, Yang W, Chen H, Yuan Q, Liu S, Zhao Y, Zhang J. MyD88 in Macrophages Enhances Liver Fibrosis by Activation of NLRP3 Inflammasome in HSCs. International Journal of Molecular Sciences. 2021; 22(22):12413. https://doi.org/10.3390/ijms222212413
Chicago/Turabian StyleGe, Shuang, Wei Yang, Haiqiang Chen, Qi Yuan, Shi Liu, Yongxiang Zhao, and Jinhua Zhang. 2021. "MyD88 in Macrophages Enhances Liver Fibrosis by Activation of NLRP3 Inflammasome in HSCs" International Journal of Molecular Sciences 22, no. 22: 12413. https://doi.org/10.3390/ijms222212413
APA StyleGe, S., Yang, W., Chen, H., Yuan, Q., Liu, S., Zhao, Y., & Zhang, J. (2021). MyD88 in Macrophages Enhances Liver Fibrosis by Activation of NLRP3 Inflammasome in HSCs. International Journal of Molecular Sciences, 22(22), 12413. https://doi.org/10.3390/ijms222212413