
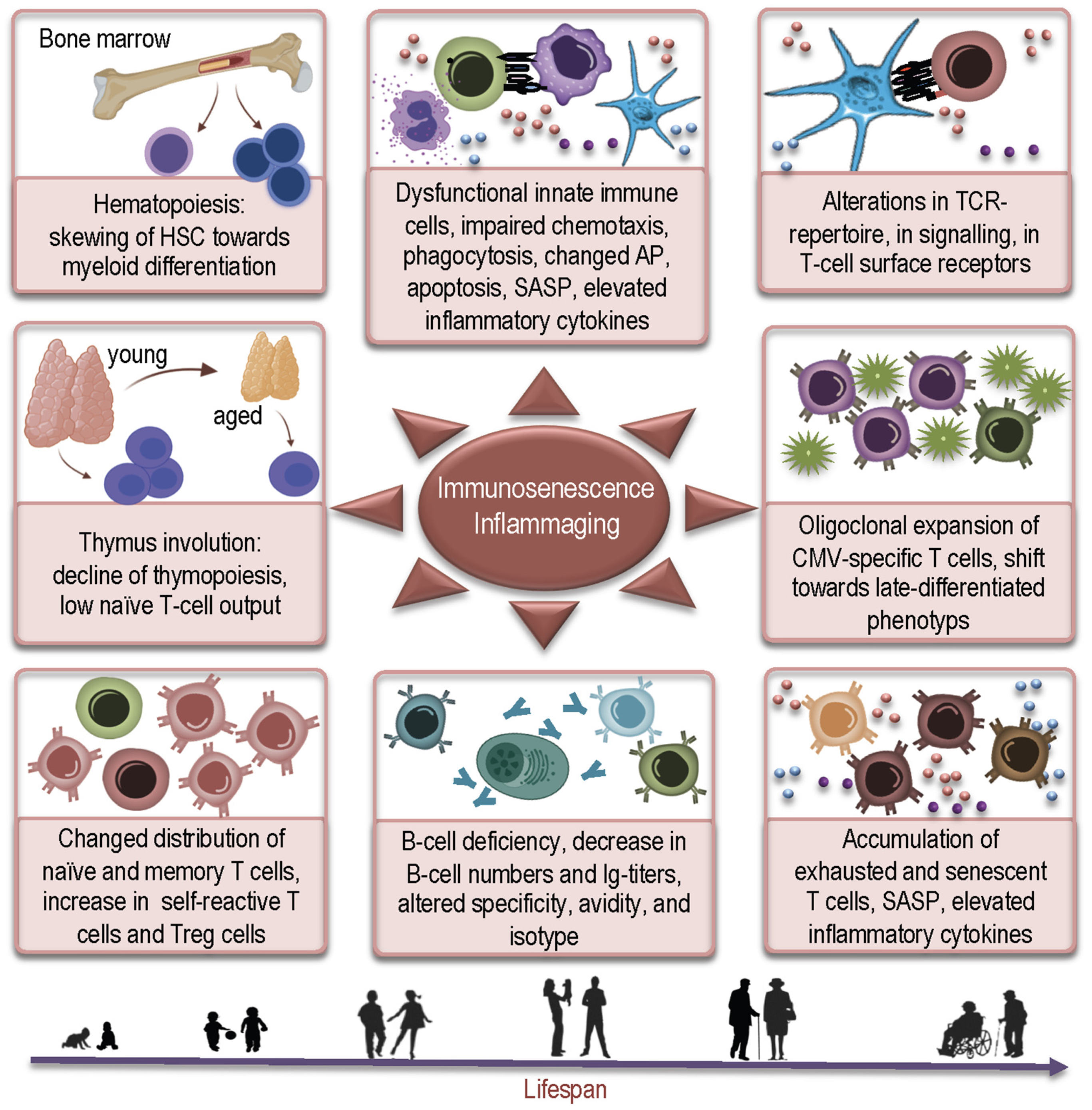
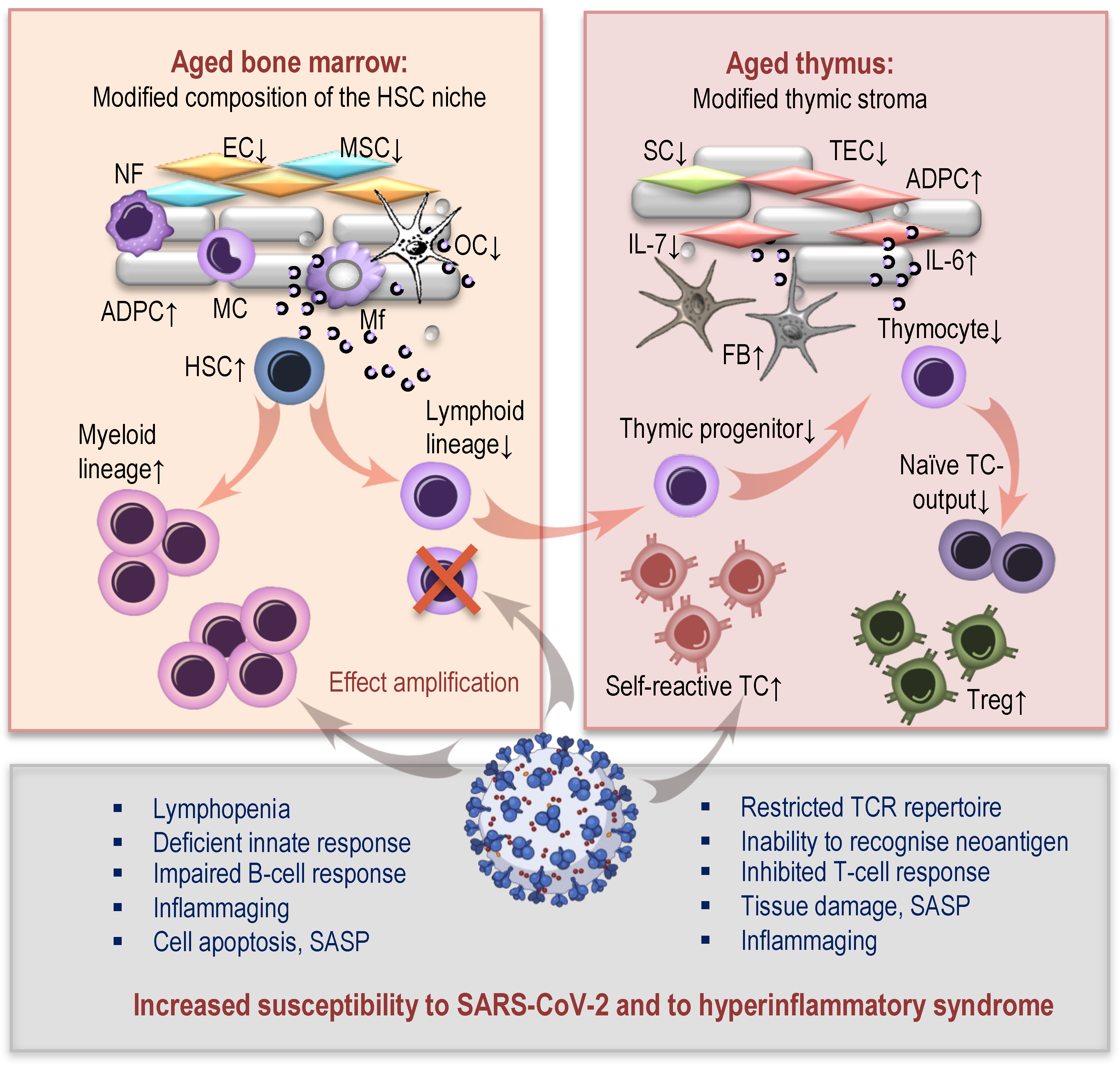
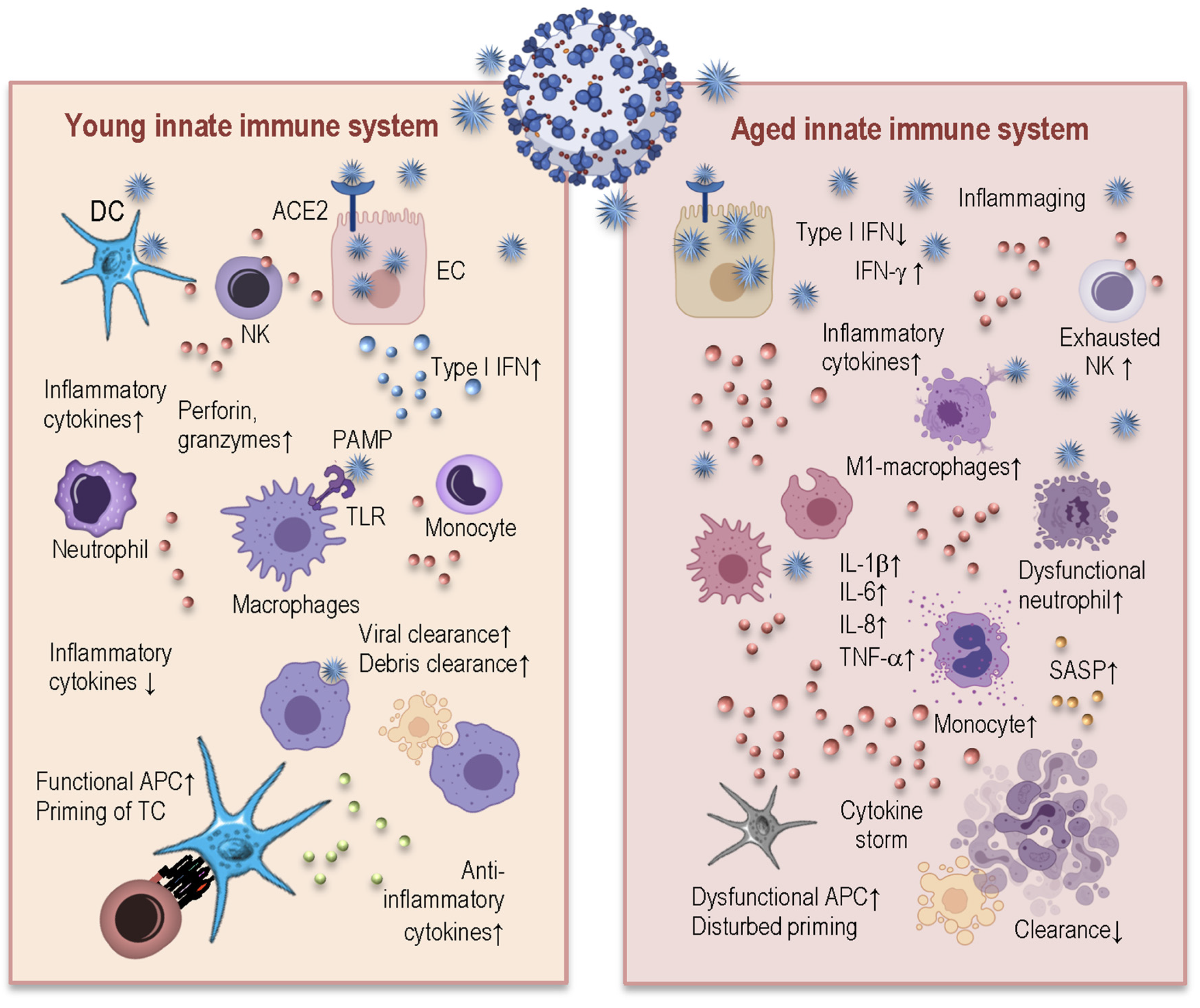
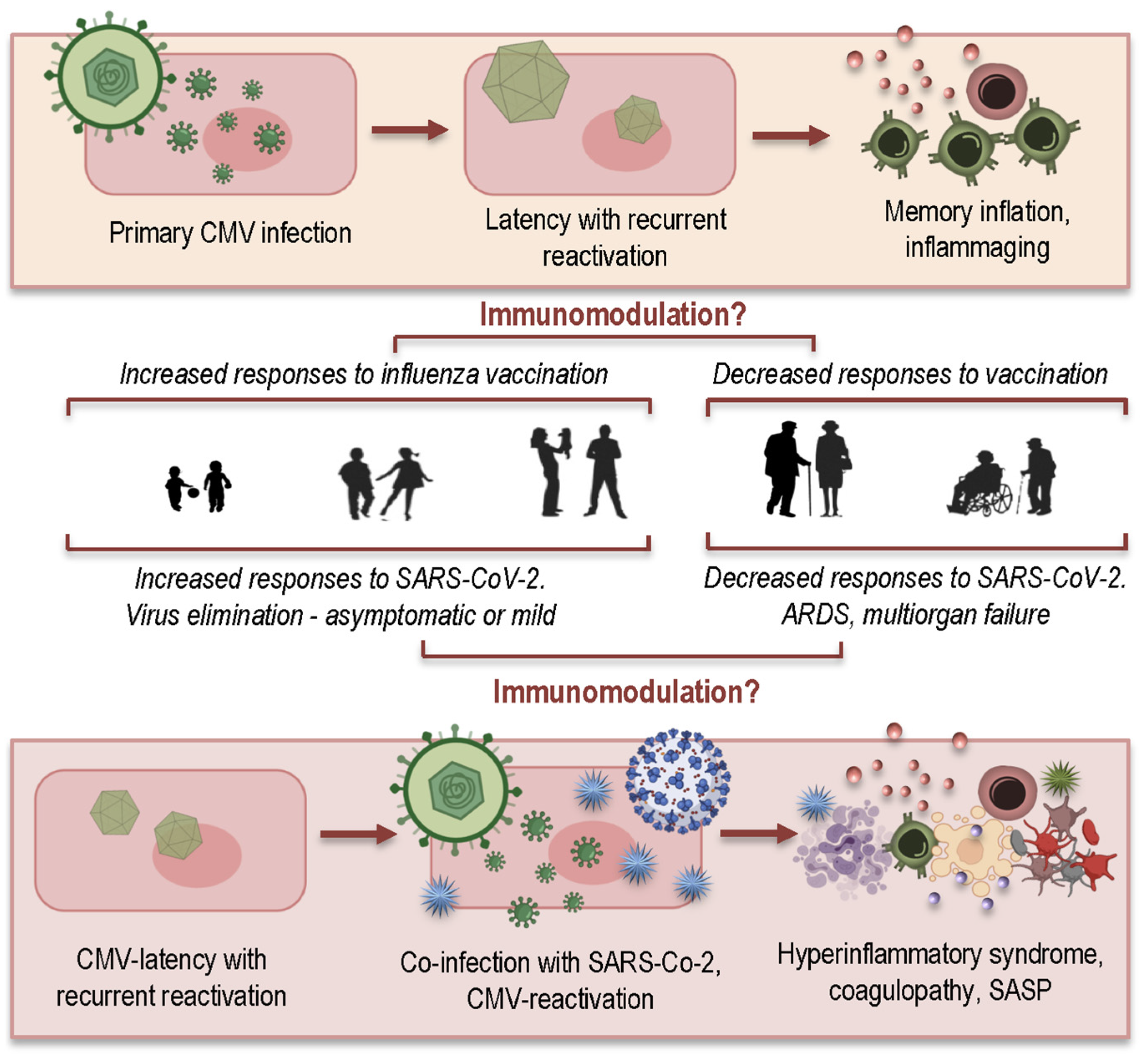
How Immunosenescence and Inflammaging May Contribute to Hyperinflammatory Syndrome in COVID-19

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Share and Cite
Müller, L.; Di Benedetto, S. How Immunosenescence and Inflammaging May Contribute to Hyperinflammatory Syndrome in COVID-19. Int. J. Mol. Sci. 2021, 22, 12539. https://doi.org/10.3390/ijms222212539
Müller L, Di Benedetto S. How Immunosenescence and Inflammaging May Contribute to Hyperinflammatory Syndrome in COVID-19. International Journal of Molecular Sciences. 2021; 22(22):12539. https://doi.org/10.3390/ijms222212539
Chicago/Turabian StyleMüller, Ludmila, and Svetlana Di Benedetto. 2021. "How Immunosenescence and Inflammaging May Contribute to Hyperinflammatory Syndrome in COVID-19" International Journal of Molecular Sciences 22, no. 22: 12539. https://doi.org/10.3390/ijms222212539
APA StyleMüller, L., & Di Benedetto, S. (2021). How Immunosenescence and Inflammaging May Contribute to Hyperinflammatory Syndrome in COVID-19. International Journal of Molecular Sciences, 22(22), 12539. https://doi.org/10.3390/ijms222212539