
Heterogenous Clinical Landscape in a Consanguineous Malonic Aciduria Family

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
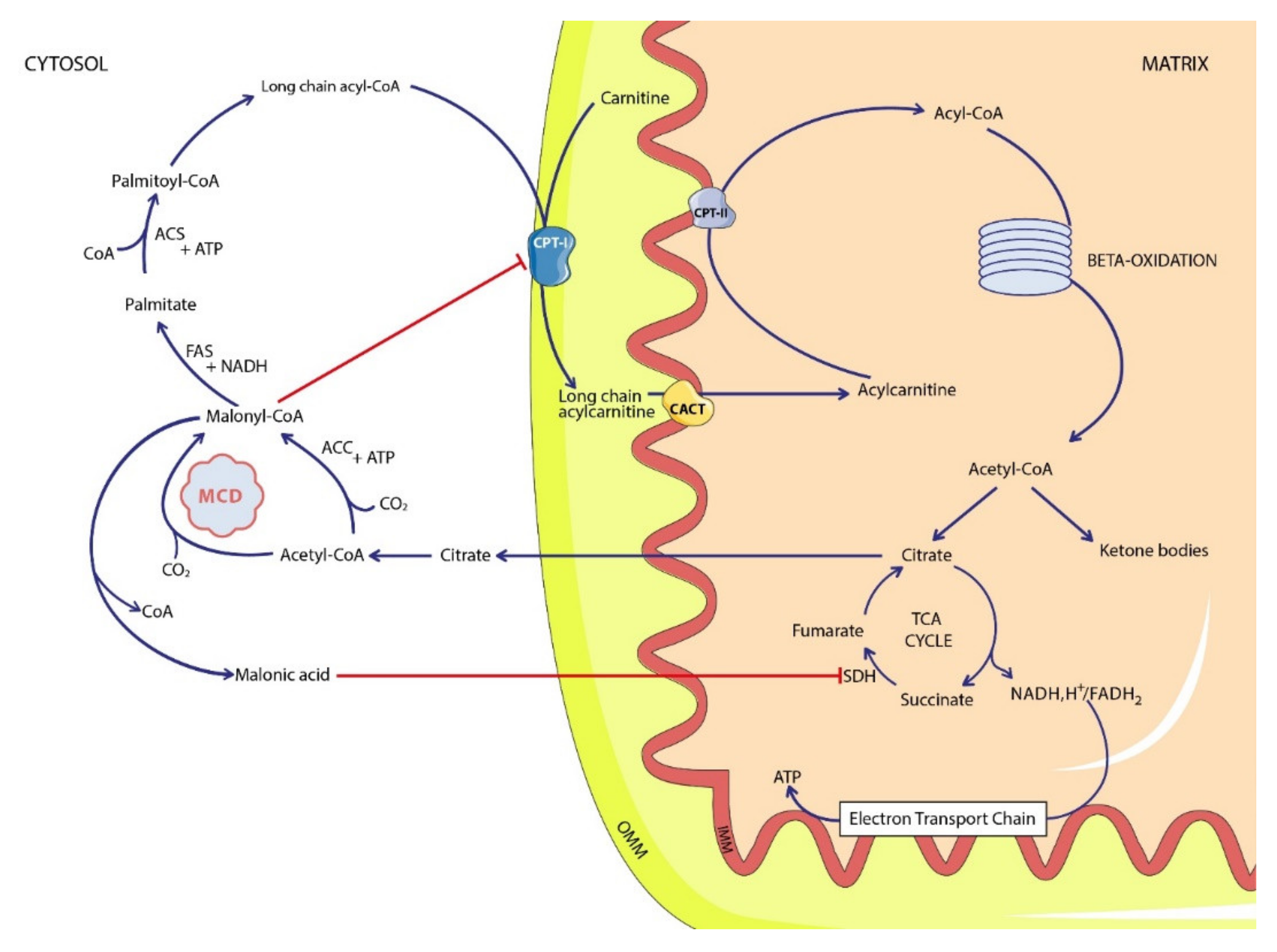
:1. Introduction
2. Patients and Methods
2.1. Clinical Presentations
2.1.1. Case 1
2.1.2. Case 2
2.2. Metabolic Investigations
2.2.1. Acylcarninite Profile
2.2.2. Urinary Organic Acids
2.3. Molecular Analysis
2.4. Literature Review
3. Results
3.1. Metabolic Investigations
3.1.1. Case 1
3.1.2. Case 2
3.2. Molecular Analysis
3.3. Patients Follow-Up
3.3.1. Case 1
3.3.2. Case 2
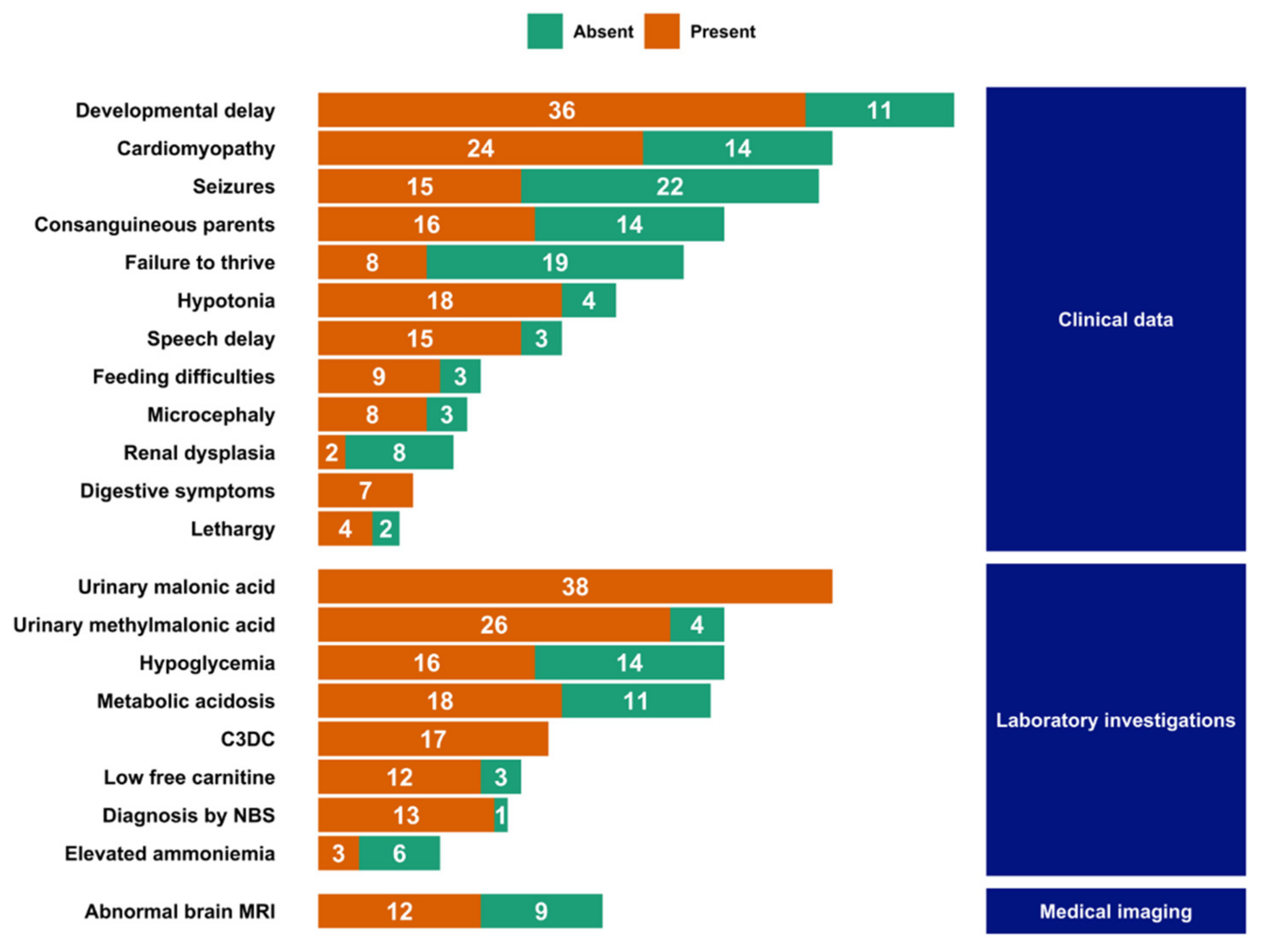
3.4. Literature Review
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- FitzPatrick, D.R.; Hill, A.; Tolmie, J.L.; Thorburn, D.R.; Christodoulou, J. The molecular basis of malonyl-CoA decarboxylase deficiency. Am. J. Hum. Genet. 1999, 65, 318–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, G.; German, N.J.; Saha, A.K.; de Boer, V.C.J.; Davies, M.; Koves, T.R.; Dephoure, N.; Fischer, F.; Boanca, G.; Vaitheesvaran, B.; et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol. Cell 2013, 50, 686–698. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Yuen, P.; Chu, D.; Thon, V.; McConnell, S.; Brown, S.; Tsang, A.; Pena, M.; Russell, A.; Cheng, J.F.; et al. Expression, purification, and characterization of human malonyl-CoA decarboxylase. Protein Expr. Purif. 2004, 34, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, D.; Perez-Luque, R.; Carpena, X.; Díaz, M.; Ferrer, J.C.; Loewen, P.C.; Fita, I. Structural asymmetry and disulfide bridges among subunits modulate the activity of human malonyl-CoA decarboxylase. J. Biol. Chem. 2013, 288, 11907–11919. [Google Scholar] [CrossRef] [Green Version]
- Froese, D.S.; Forouhar, F.; Tran, T.H.; Vollmar, M.; Kim, Y.S.; Lew, S.; Neely, H.; Seetharaman, J.; Shen, Y.; Xiao, R.; et al. Crystal structures of malonyl-coenzyme a decarboxylase provide insights into its catalytic mechanism and disease-causing mutations. Structure 2013, 21, 1182–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacksteder, K.A.; Morrell, J.C.; Wanders, R.J.A.; Matalon, R.; Gould, S.J. MCD encodes peroxisomal and cytoplasmic forms of malonyl-CoA decarboxylase and is mutated in malonyl-CoA decarboxylase deficiency. J. Biol. Chem. 1999, 274, 24461–24468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouzakri, K.; Austin, R.; Rune, A.; Lassman, M.E.; Garcia-Roves, P.M.; Berger, J.P.; Krook, A.; Chibalin, A.V.; Zhang, B.B.; Zierath, J.R. Malonyl coenzymeA decarboxylase regulates lipid and glucose metabolism in human skeletal muscle. Diabetes 2008, 57, 1508–1516. [Google Scholar] [CrossRef] [Green Version]
- El-Gharbawy, A.; Vockley, J. Inborn Errors of Metabolism with Myopathy: Defects of Fatty Acid Oxidation and the Carnitine Shuttle System. Pediatr. Clin. N. Am. 2018, 65, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Hayaishi, O. Participation of ATP and coenzyme A in the enzymatic decarboxylation of malonic acid. J. Am. Chem. Soc. 1953, 75, 4367. [Google Scholar] [CrossRef]
- Brown, G.K.; Scholem, R.D.; Bankier, A.; Danks, D.M. Malonyl coenzyme a decarboxylase deficiency. J. Inherit. Metab. Dis. 1984, 7, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Grotto, S.; Sudrie-Arnaud, B.; Drouin-Garraud, V.; Nafeh-Bizet, C.; Chadefaux-Vekemans, B.; Gobin, S.; Bekri, S.; Tebani, A. Dilated Cardiomyopathy and Premature Ovarian Failure Unveiling Propionic Aciduria. Clin. Chem. 2018, 64, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Salomons, G.S.; Jakobs, C.; Pope, L.; Errami, A.; Potter, M.; Nowaczyk, M.; Olpin, S.; Manning, N.; Raiman, J.A.J.; Slade, T.; et al. Clinical, enzymatic and molecular characterization of nine new patients with malonyl-coenzyme A decarboxylase deficiency. J. Inherit. Metab. Dis. 2007, 30, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Ozand, P.T.; Nyhan, W.L.; Al Aqeel, A.; Christodoulou, J. Malonic aciduria. Brain Dev. 1994, 16, 7–11. [Google Scholar] [CrossRef]
- Richards, C.S.; Bale, S.; Bellissimo, D.B.; Das, S.; Grody, W.W.; Hegde, M.R.; Lyon, E.; Ward, B.E. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 2008, 10, 294–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haan, E.A.; Scholem, R.D.; Croll, H.B.; Brown, G.K. Malonyl coenzyme A decarboxylase deficiency. Clinical and biochemical findings in a second child with a more severe enzyme defect. Eur. J. Pediatr. 1986, 144, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Wightman, P.J.; Santer, R.; Ribes, A.; Dougherty, F.; McGill, N.; Thorburn, D.R.; FitzPatrick, D.R. MLYCD mutation analysis: Evidence for protein mistargeting as a cause of MLYCD deficiency. Hum. Mutat. 2003, 22, 288–300. [Google Scholar] [CrossRef]
- Ficicioglu, C.; Chrisant, M.R.K.; Payan, I.; Chace, D.H. Cardiomyopathy and hypotonia in a 5-month-old infant with malonyl-CoA decarboxylase deficiency: Potential for preclinical diagnosis with expanded newborn screening. Pediatr. Cardiol. 2005, 26, 881–883. [Google Scholar] [CrossRef] [PubMed]
- De Wit, M.C.Y.; De Coo, I.F.M.; Verbeek, E.; Schot, R.; Schoonderwoerd, G.C.; Duran, M.; De Klerk, J.B.C.; Huijmans, J.G.M.; Lequin, M.H.; Verheijen, F.W.; et al. Brain abnormalities in a case of malonyl-CoA decarboxylase deficiency. Mol. Genet. Metab. 2006, 87, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Malvagia, S.; Papi, L.; Morrone, A.; Donati, M.A.; Ciani, F.; Pasquini, E.; la Marca, G.; Scholte, H.R.; Genuardi, M.; Zammarchi, E. Fatal malonyl coa decarboxylase deficiency due to maternal uniparental isodisomy of the telomeric end of chromosome 16. Ann. Hum. Genet. 2007, 71, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Footitt, E.J.; Stafford, J.; Dixon, M.; Burch, M.; Jakobs, C.; Salomons, G.S.; Cleary, M.A. Use of a long-chain triglyceride-restricted/medium-chain triglyceride-supplemented diet in a case of malonyl-CoA decarboxylase deficiency with cardiomyopathy. J. Inherit. Metab. Dis. 2010, 33, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Prada, C.E.; Jefferies, J.L.; Grenier, M.A.; Huth, C.M.; Page, K.I.; Spicer, R.L.; Towbin, J.A.; Leslie, N.D. Malonyl coenzyme a decarboxylase deficiency: Early dietary restriction and time course of cardiomyopathy. Pediatrics 2012, 130, e456–e460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Peng, J.; Zhou, M.; Zhong, L.; Yin, F.; Liang, D.; Wu, L. Novel compound heterozygous mutation of MLYCD in a Chinese patient with malonic aciduria. Mol. Genet. Metab. 2012, 105, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Celato, A.; Mitola, C.; Tolve, M.; Giannini, M.T.; De Leo, S.; Carducci, C.; Carducci, C.; Leuzzi, V. A new case of malonic aciduria with a presymptomatic diagnosis and an early treatment. Brain Dev. 2013, 35, 675–680. [Google Scholar] [CrossRef]
- Baertling, F.; Mayatepek, E.; Thimm, E.; Schlune, A.; Kovacevic, A.; Distelmaier, F.; Salomons, G.S.; Meissner, T. Malonic aciduria: Long-term follow-up of new patients detected by newborn screening. Eur. J. Pediatr. 2014, 173, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- MacPhee, G.B.; Logan, R.W.; Mitchell, J.S.; Howells, D.W.; Tsotsis, E.; Thorburn, D.R. Malonyl coenzyme A decarboxylase deficiency. Arch. Dis. Child. 1993, 69, 433–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polinati, P.P.; Valanne, L.; Tyni, T. Malonyl-CoA decarboxylase deficiency: Long-term follow-up of a patient new clinical features and novel mutations. Brain Dev. 2015, 37, 107–113. [Google Scholar] [CrossRef]
- Liu, H.; Tan, D.; Han, L.; Ye, J.; Qiu, W.; Gu, X.; Zhang, H. A new case of malonyl-CoA decarboxylase deficiency with mild clinical features. Am. J. Med. Genet. Part A 2016, 170, 1347–1351. [Google Scholar] [CrossRef]
- Ersoy, M.; Akyol, M.B.; Ceylaner, S.; Çakır Biçer, N. A novel frameshift mutation of malonyl-CoA decarboxylase deficiency: Clinical signs and therapy response of a late-diagnosed case. Clin. Case Rep. 2017, 5, 1284–1288. [Google Scholar] [CrossRef]
- Chapel-Crespo, C.; Gavrilov, D.; Sowa, M.; Myers, J.; Day-Salvatore, D.L.; Lynn, H.; Regier, D.; Starin, D.; Steenari, M.; Schoonderwoerd, K.; et al. Clinical, biochemical and molecular characteristics of malonyl-CoA decarboxylase deficiency and long-term follow-up of nine patients. Mol. Genet. Metab. 2019, 128, 113–121. [Google Scholar] [CrossRef]
- Lee, S.H.; Ko, J.M.; Song, M.K.; Song, J.; Park, K.S. A Korean child diagnosed with malonic aciduria harboring a novel start codon mutation following presentation with dilated cardiomyopathy. Mol. Genet. Genom. Med. 2020, 8, 3–7. [Google Scholar] [CrossRef]
- Matalon, R.; Michaels, K.; Kaul, R.; Whitman, V.; Rodriguez-Novo, J.; Goodman, S.; Thorburn, D. Malonic aciduria and cardiomyopathy. J. Inherit. Metab. Dis. 1993, 16, 571–573. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Sweetman, L.; Thorburn, D.R.; Mofidi, S.; Williams, J.C. A new case of malonyl coenzyme A decarboxylase deficiency presenting with cardiomyopathy. Eur. J. Pediatr. 1997, 156, 382–383. [Google Scholar] [CrossRef] [PubMed]
- Buyukgebiz, B.; Jakobs, C.; Scholte, H.R.; Huijmans, J.G.M.; Kleijer, W.J. Fatal neonatal malonic aciduria. J. Inherit. Metab. Dis. 1998, 21, 76–77. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.J.; Evans, C.E.; Kumar, V.; Pourfarzam, M. Malonic aciduria presenting with developmental delay, malonyl-carnitine inscreased in bloodspots. J. Inherit. Metab. Dis. 1998, 21, 53. [Google Scholar]
- Gao, J.; Waber, L.; Bennett, M.J.; Gibson, K.M.; Cohen, J.C. Cloning and mutational analysis of human malonyl-coenzyme A decarboxylase. J. Lipid Res. 1999, 40, 178–182. [Google Scholar] [CrossRef]
- Krishnamoorthy, K.S.; Vianey-Sabin, C.; Shih, V.E. Malonic aciduria due to mitochondrial malonyl coenzyme A decarboxylase deficiency: A rare inborn error of metabolism. J. Inherit. Metab. Dis. 1999, 22, 93. [Google Scholar]
- Krawinkel, M.B.; Oldigs, H.D.; Santer, R.; Lehnert, W.; Wendel, U.; Schaub, J. Association of malonyl-CoA decarboxylase deficiency and heterozygote state for haemoglobin C disease. J. Inherit. Metab. Dis. 1994, 17, 636–637. [Google Scholar] [CrossRef]
- Kasapkara, C.S.; Civelek Urey, B.; Ceylan, A.C.; Unal Uzun, O.; Cetin, I.I. Malonyl coenzyme A decarboxylase deficiency with a novel mutation. Cardiol. Young 2021, 31, 1535–1537. [Google Scholar] [CrossRef]
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.; Adams, J. Current status of newborn screening worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef] [Green Version]
- Sloan, J.L.; Johnston, J.J.; Manoli, I.; Chandler, R.J.; Krause, C.; Carrillo-Carrasco, N.; Chandrasekaran, S.D.; Sysol, J.R.; O’Brien, K.; Hauser, N.S.; et al. Exome sequencing identifies ACSF3 as a cause of combined malonic and methylmalonic aciduria. Nat. Genet. 2011, 43, 883–886. [Google Scholar] [CrossRef] [Green Version]
- Levtova, A.; Waters, P.J.; Buhas, D.; Lévesque, S.; Auray-Blais, C.; Clarke, J.T.R.; Laframboise, R.; Maranda, B.; Mitchell, G.A.; Brunel-Guitton, C.; et al. Combined malonic and methylmalonic aciduria due to ACSF3 mutations: Benign clinical course in an unselected cohort. J. Inherit. Metab. Dis. 2019, 42, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Tucci, S. Brain metabolism and neurological symptoms in combined malonic and methylmalonic aciduria. Orphanet J. Rare Dis. 2020, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Reindl, B.A.; Lynch, D.W.; Ramirez, M.; Valbracht, M.; Davis-Keppen, L.; Tams, K.C.; Groeneveld, S. Sani-cloth wipe mimics rare enzyme deficiency malonic aciduria on newborn screen. Pediatrics 2012, 130, e1363–e1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snanoudj, S.; Torre, S.; Sudrié-Arnaud, B.; Abily-Donval, L.; Goldenberg, A.; Salomons, G.S.; Marret, S.; Bekri, S.; Tebani, A. Heterogenous Clinical Landscape in a Consanguineous Malonic Aciduria Family. Int. J. Mol. Sci. 2021, 22, 12633. https://doi.org/10.3390/ijms222312633
Snanoudj S, Torre S, Sudrié-Arnaud B, Abily-Donval L, Goldenberg A, Salomons GS, Marret S, Bekri S, Tebani A. Heterogenous Clinical Landscape in a Consanguineous Malonic Aciduria Family. International Journal of Molecular Sciences. 2021; 22(23):12633. https://doi.org/10.3390/ijms222312633
Chicago/Turabian StyleSnanoudj, Sarah, Stéphanie Torre, Bénédicte Sudrié-Arnaud, Lenaig Abily-Donval, Alice Goldenberg, Gajja S. Salomons, Stéphane Marret, Soumeya Bekri, and Abdellah Tebani. 2021. "Heterogenous Clinical Landscape in a Consanguineous Malonic Aciduria Family" International Journal of Molecular Sciences 22, no. 23: 12633. https://doi.org/10.3390/ijms222312633