
Pathophysiological Potentials of NRF3-Regulated Transcriptional Axes in Protein and Lipid Homeostasis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Assembly of the Ubiquitin-Independent 20S Proteasome
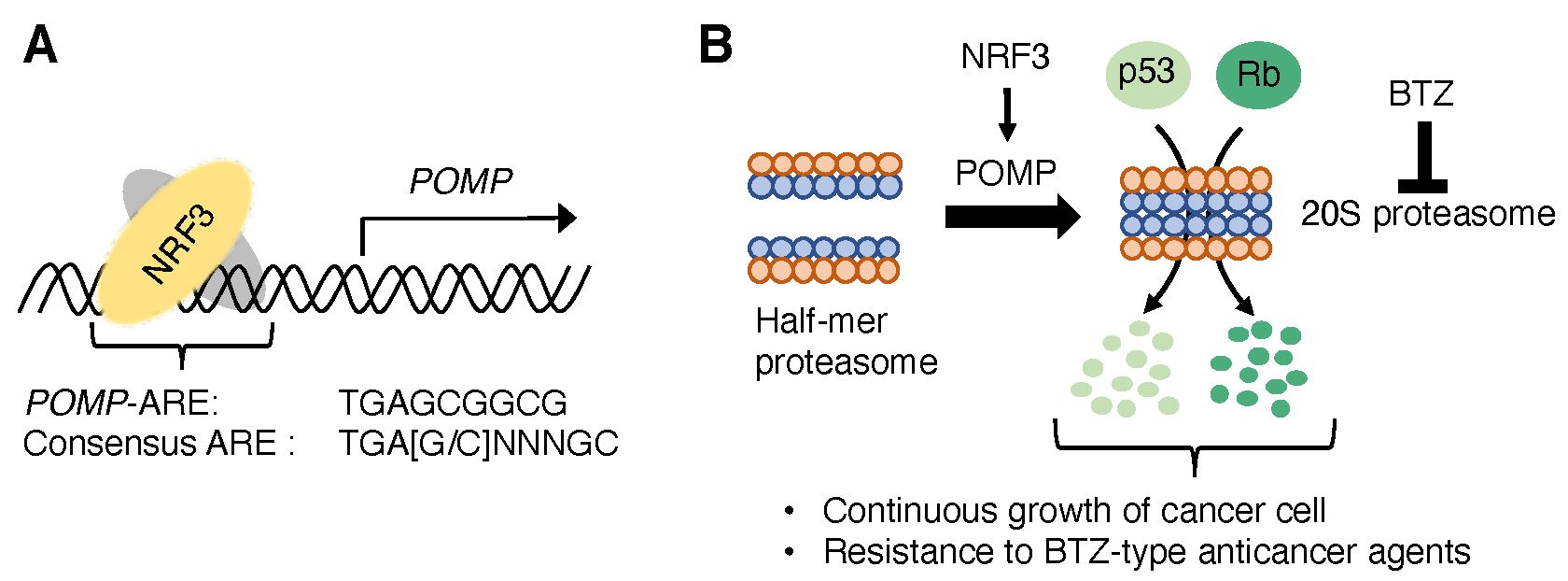
2.1. POMP, a 20S Proteasome Assembly Factor
2.2. NRF3-POMP-20S Proteasome Assembly Axis for Cancer Development
3. Complementary Maintenance of Proteasome with NRF1
3.1. CPEB3, a Translational Repressor of NRF1
3.2. Clinical Significance of the NRF3-CPEB3-NRF1 Translational Repression Axis
4. Reprogramming of Lipid Metabolism
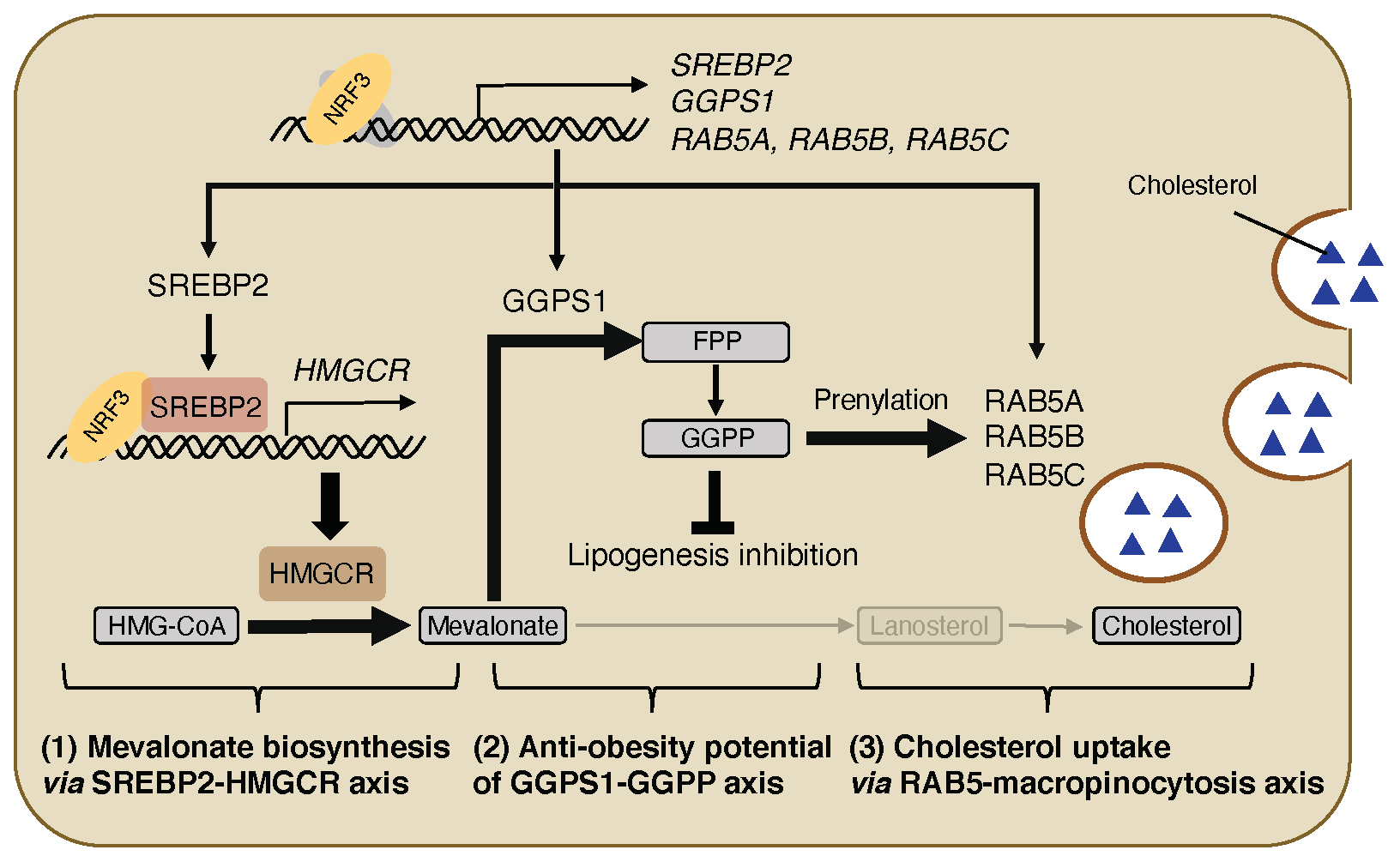
4.1. NRF3-SREBP2-HMGCR Axis for Mevalonate Biosynthesis
4.2. NRF3-GGPS1-GGPP Production Axis for Lipogenesis Inhibition
4.3. NRF3-RAB5-Macropincytosis Induction Axis for Cholesterol Uptake
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guang, M.H.Z.; Kavanagh, E.; Dunne, L.; Dowling, P.; Zhang, L.; Lindsay, S.; Bazou, D.; Goh, C.; Hanley, C.; Bianchi, G.; et al. Targeting Proteotoxic Stress in Cancer: A Review of the Role that Protein Quality Control Pathways Play in Oncogenesis. Cancers 2019, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Su, K.-H.; Dai, C. Metabolic control of the proteotoxic stress response: Implications in diabetes mellitus and neurodegenerative disorders. Cell. Mol. Life Sci. 2016, 73, 4231–4248. [Google Scholar] [CrossRef] [Green Version]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef]
- Yadav, R.S.; Tiwari, N.K. Lipid Integration in Neurodegeneration: An Overview of Alzheimer’s Disease. Mol. Neurobiol. 2014, 50, 168–176. [Google Scholar] [CrossRef]
- Kobayashi, A.; Ito, E.; Toki, T.; Kogame, K.; Takahashi, S.; Igarashi, K.; Hayashi, N.; Yamamoto, M. Molecular Cloning and Functional Characterization of a New Cap’n’ Collar Family Transcription Factor Nrf3. J. Biol. Chem. 1999, 274, 6443–6452. [Google Scholar] [CrossRef] [Green Version]
- Chénais, B.; Derjuga, A.; Massrieh, W.; Red-Horse, K.; Bellingard, V.; Fisher, S.J.; Blank, V. Functional and Placental Expression Analysis of the Human NRF3 Transcription Factor. Mol. Endocrinol. 2005, 19, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription Factor Nrf1 Mediates the Proteasome Recovery Pathway after Proteasome Inhibition in Mammalian Cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.Y.; Kwong, M.; Lu, R.; Chang, J.; Wang, B.; Yen, T.; Kan, Y.W. Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J. 1998, 17, 1779–1787. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Tsukide, T.; Miyasaka, T.; Morita, T.; Mizoroki, T.; Saito, Y.; Ihara, Y.; Takashima, A.; Noguchi, N.; Fukamizu, A.; et al. Central nervous system-specific deletion of transcription factor Nrf1 causes progressive motor neuronal dysfunction. Genes Cells 2011, 16, 692–703. [Google Scholar] [CrossRef]
- Kim, J.; Xing, W.; Wergedal, J.; Chan, J.Y.; Mohan, S. Targeted disruption of nuclear factor erythroid-derived 2-like 1 in osteoblasts reduces bone size and bone formation in mice. Physiol. Genom. 2010, 40, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Hu, T.; Nguyen, J.M.; Zhang, J.; Martin, M.V.; Vawter, M.P.; Huang, E.J.; Chan, J.Y. Loss of nuclear factor E2-related factor 1 in the brain leads to dysregulation of proteasome gene expression and neurodegeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 8408–8413. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Chen, L.; Leung, L.; Yen, T.S.B.; Lee, C.; Chan, J.Y. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc. Natl. Acad. Sci. USA 2005, 102, 4120–4125. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological Significance of Reactive Cysteine Residues of Keap1 in Determining Nrf2 Activity. Mol. Cell. Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef] [Green Version]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; Leboeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef]
- Derjuga, A.; Gourley, T.S.; Holm, T.M.; Heng, H.H.Q.; Shivdasani, R.A.; Ahmed, R.; Andrews, N.; Blank, V. Complexity of CNC Transcription Factors As Revealed by Gene Targeting of the Nrf3 Locus. Mol. Cell. Biol. 2004, 24, 3286–3294. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Ohta, T.; Yamamoto, M. Unique Function of the Nrf2–Keap1 Pathway in the Inducible Expression of Antioxidant and Detoxifying Enzymes. Methods Enzymol. 2004, 378, 273–286. [Google Scholar] [CrossRef]
- Chevillard, G.; Blank, V. NFE2L3 (NRF3): The Cinderella of the Cap‘n’Collar transcription factors. Cell. Mol. Life Sci. 2011, 68, 3337–3348. [Google Scholar] [CrossRef]
- Bury, M.; Le Calvé, B.; Lessard, F.; Maso, T.D.; Saliba, J.; Michiels, C.; Ferbeyre, G.; Blank, V. NFE2L3 Controls Colon Cancer Cell Growth through Regulation of DUX4, a CDK1 Inhibitor. Cell Rep. 2019, 29, 1469–1481.e9. [Google Scholar] [CrossRef]
- Wang, H.; Zhan, M.; Yang, R.; Shi, Y.; Liu, Q.; Wang, J. Elevated expression of NFE2L3 predicts the poor prognosis of pancreatic cancer patients. Cell Cycle 2018, 17, 2164–2174. [Google Scholar] [CrossRef]
- Siegenthaler, B.; Defila, C.; Muzumdar, S.; Beer, H.-D.; Meyer, M.; Tanner, S.; Bloch, W.; Blank, V.; Schäfer, M.; Werner, S. Nrf3 promotes UV-induced keratinocyte apoptosis through suppression of cell adhesion. Cell Death Differ. 2018, 25, 1749–1765. [Google Scholar] [CrossRef] [Green Version]
- Aono, S.; Hatanaka, A.; Hatanaka, A.; Gao, Y.; Hippo, Y.; Taketo, M.M.; Waku, T.; Kobayashi, A. β-Catenin/TCF4 Complex-Mediated Induction of the NRF3 (NFE2L3) Gene in Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3344. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, A.M.M.A.; Katoh, H.; Hatanaka, A.; Iwanari, H.; Nakamura, N.; Hamakubo, T.; Natsume, T.; Waku, T.; Kobayashi, A. Multiple regulatory mechanisms of the biological function of NRF3 (NFE2L3) control cancer cell proliferation. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. eLife 2016, 5, 1–10. [Google Scholar] [CrossRef]
- Zhang, Y.; Kobayashi, A.; Yamamoto, M.; Hayes, J. The Nrf3 Transcription Factor Is a Membrane-bound Glycoprotein Targeted to the Endoplasmic Reticulum through Its N-terminal Homology Box 1 Sequence. J. Biol. Chem. 2009, 284, 3195–3210. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Motohashi, H.; O’Connor, T.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 2002, 294, 1–12. [Google Scholar] [CrossRef]
- Liu, P.; Kerins, M.J.; Tian, W.; Neupane, D.; Zhang, D.D.; Ooi, A. Differential and overlapping targets of the transcriptional regulators NRF1, NRF2, and NRF3 in human cells. J. Biol. Chem. 2019, 294, 18131–18149. [Google Scholar] [CrossRef]
- Waku, T.; Nakamura, N.; Koji, M.; Watanabe, H.; Katoh, H.; Tatsumi, C.; Tamura, N.; Hatanaka, A.; Hirose, S.; Katayama, H.; et al. NRF3-POMP-20S Proteasome Assembly Axis Promotes Cancer Development via Ubiquitin-Independent Proteolysis of p53 and Retinoblastoma Protein. Mol. Cell. Biol. 2020, 40. [Google Scholar] [CrossRef] [Green Version]
- Waku, T.; Katayama, H.; Hiraoka, M.; Hatanaka, A.; Nakamura, N.; Tanaka, Y.; Tamura, N.; Watanabe, A.; Kobayashi, A. NFE2L1 and NFE2L3 Complementarily Maintain Basal Proteasome Activity in Cancer Cells through CPEB3-Mediated Translational Repression. Mol. Cell. Biol. 2020, 40. [Google Scholar] [CrossRef]
- Waku, T.; Hagiwara, T.; Tamura, N.; Atsumi, Y.; Urano, Y.; Suzuki, M.; Iwami, T.; Sato, K.; Yamamoto, M.; Noguchi, N.; et al. NRF3 upregulates gene expression in SREBP2-dependent mevalonate pathway with cholesterol uptake and lipogenesis inhibition. iScience 2021, 24, 103180. [Google Scholar] [CrossRef]
- Coux, O.; Tanaka, K.; Goldberg, A.L. Structure and functions of the 20s and 26s proteasomes. Annu. Rev. Biochem. 1996, 65, 801–847. [Google Scholar] [CrossRef]
- Murata, S.; Yashiroda, H.; Tanaka, K. Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell Biol. 2009, 10, 104–115. [Google Scholar] [CrossRef]
- Gallastegui, N.; Groll, M. The 26S proteasome: Assembly and function of a destructive machine. Trends Biochem. Sci. 2010, 35, 634–642. [Google Scholar] [CrossRef]
- Witt, E.; Zantopf, D.; Schmidt, M.; Kraft, R.; Kloetzel, P.-M.; Krüger, E. Characterisation of the newly identified human Ump1 homologue POMP and analysis of LMP7(β5i) incorporation into 20 S proteasomes. J. Mol. Biol. 2000, 301, 1–9. [Google Scholar] [CrossRef]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S Proteasome Ubiquitin-Independent Degradation Pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [Green Version]
- Hyer, M.L.; A Milhollen, M.; Ciavarri, J.; Fleming, P.; Traore, T.; Sappal, D.; Huck, J.; Shi, J.; Gavin, J.; Brownell, J.; et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat. Med. 2018, 24, 186–193. [Google Scholar] [CrossRef]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Waldman, T.; Kinzler, K.W.; Vogelstein, B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995, 55. [Google Scholar]
- Nakano, K.; Vousden, K.H. PUMA, a Novel Proapoptotic Gene, Is Induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Groll, M.; Berkers, C.R.; Ploegh, H.L.; Ovaa, H. Crystal Structure of the Boronic Acid-Based Proteasome Inhibitor Bortezomib in Complex with the Yeast 20S Proteasome. Structure 2006, 14, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A. Roles of NRF3 in the Hallmarks of Cancer: Proteasomal Inactivation of Tumor Suppressors. Cancers 2020, 12, 2681. [Google Scholar] [CrossRef]
- Fernández-Miranda, G.; Méndez, R. The CPEB-family of proteins, translational control in senescence and cancer. Ageing Res. Rev. 2012, 11, 460–472. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Xue, L.; Qi, H.; Zhang, H.; Ding, L.; Huang, Q.; Zhao, D.; Wu, B.J.; Li, X. Targeting SREBP-2-Regulated Mevalonate Metabolism for Cancer Therapy. Front. Oncol. 2020, 10, 1510. [Google Scholar] [CrossRef]
- Landt, S.G.; Marinov, G.; Kundaje, A.; Kheradpour, P.; Pauli, F.; Batzoglou, S.; Bernstein, B.E.; Bickel, P.; Brown, J.B.; Cayting, P.; et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012, 22, 1813–1831. [Google Scholar] [CrossRef] [Green Version]
- Bertolio, R.; Napoletano, F.; Mano, M.; Maurer-Stroh, S.; Fantuz, M.; Zannini, A.; Bicciato, S.; Sorrentino, G.; Del Sal, G. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yeh, Y.-S.; Goto, T.; Takahashi, N.; Egawa, K.; Takahashi, H.; Jheng, H.-F.; Kim, Y.-I.; Kawada, T. Geranylgeranyl pyrophosphate performs as an endogenous regulator of adipocyte function via suppressing the LXR pathway. Biochem. Biophys. Res. Commun. 2016, 478, 1317–1322. [Google Scholar] [CrossRef]
- Lamiquiz-Moneo, I.; Mateo-Gallego, R.; Bea, A.M.; Dehesa-García, B.; Pérez-Calahorra, S.; Marco-Benedí, V.; Baila-Rueda, L.; Laclaustra, M.; Civeira, F.; Cenarro, A. Genetic predictors of weight loss in overweight and obese subjects. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Monda, K.L.; Chen, G.K.; Taylor, K.C.; Palmer, C.; Edwards, T.L.; A Lange, L.; Ng, M.C.Y.; A Adeyemo, A.; A Allison, M.; et al.; NABEC Consortium A meta-analysis identifies new loci associated with body mass index in individuals of African ancestry. Nat. Genet. 2013, 45, 690–696. [Google Scholar] [CrossRef] [Green Version]
- Steck, T.L.; Lange, Y. Cell cholesterol homeostasis: Mediation by active cholesterol. Trends Cell Biol. 2010, 20, 680–687. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.L.; Brown, M.S. The LDL Receptor. Arter. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Zeigerer, A.; Gilleron, J.; Bogorad, R.L.; Marsico, G.; Nonaka, H.; Seifert, S.; Epstein-Barash, H.; Kuchimanchi, S.; Peng, C.G.; Ruda, V.M.; et al. Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. Nat. Cell Biol. 2012, 485, 465–470. [Google Scholar] [CrossRef]
- Kruth, H.S.; Jones, N.L.; Huang, W.; Zhao, B.; Ishii, I.; Chang, J.; Combs, C.A.; Malide, D.; Zhang, W.-Y. Macropinocytosis Is the Endocytic Pathway That Mediates Macrophage Foam Cell Formation with Native Low Density Lipoprotein. J. Biol. Chem. 2005, 280, 2352–2360. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, M.A.; Hoffenberg, S.; Roberts, R.; Mukhopadhyay, A.; Pomrehn, A.; Dickey, B.F.; Stahl, P.D. Evidence for a Symmetrical Requirement for Rab5-GTP in in Vitro Endosome-Endosome Fusion. J. Biol. Chem. 1998, 273, 25850–25855. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Casey, P. Protein prenylation: Unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 2016, 17, 110–122. [Google Scholar] [CrossRef]
- Commisso, C.; Flinn, R.J.; Bar-Sagi, D. Determining the macropinocytic index of cells through a quantitative image-based assay. Nat. Protoc. 2014, 9, 182–192. [Google Scholar] [CrossRef] [Green Version]
- Sankaranarayanan, K.; Jaiswal, A.K. Nrf3 Negatively Regulates Antioxidant-response Element-mediated Expression and Antioxidant Induction of NAD(P)H:Quinone Oxidoreductase1 Gene. J. Biol. Chem. 2004, 279, 50810–50817. [Google Scholar] [CrossRef] [Green Version]
- Lecerf, J.-M.; de Lorgeril, M. Dietary cholesterol: From physiology to cardiovascular risk. Br. J. Nutr. 2011, 106, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Nelson, R.H. Hyperlipidemia as a Risk Factor for Cardiovascular Disease. Prim. Care Clin. Off. Pr. 2013, 40, 195–211. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Tian, Z. Dyslipidemia and colorectal cancer risk: A meta-analysis of prospective studies. Cancer Causes Control. 2015, 26, 257–268. [Google Scholar] [CrossRef]
- Vourakis, M.; Mayer, G.; Rousseau, G. The Role of Gut Microbiota on Cholesterol Metabolism in Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 8074. [Google Scholar] [CrossRef]
- Kenny, D.J.; Plichta, D.R.; Shungin, D.; Koppel, N.; Hall, A.B.; Fu, B.; Vasan, R.S.; Shaw, S.Y.; Vlamakis, H.; Balskus, E.P.; et al. Cholesterol Metabolism by Uncultured Human Gut Bacteria Influences Host Cholesterol Level. Cell Host Microbe 2020, 28, 245–257.e6. [Google Scholar] [CrossRef]
- Le Roy, T.; Lécuyer, E.; Chassaing, B.; Rhimi, M.; Lhomme, M.; Boudebbouze, S.; Ichou, F.; Haro Barceló, J.; Huby, T.; Guerin, M.; et al. The Intestinal Microbiota Regulates Host Cholesterol Homeostasis. BMC Biol. 2019, 17, 94. [Google Scholar] [CrossRef] [Green Version]
- Bianchini, F.; Kaaks, R.; Vainio, H. Overweight, obesity, and cancer risk. Lancet Oncol. 2002, 3, 565–574. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [Green Version]
- Khandekar, M.J.; Cohen, P.; Spiegelman, B.M. Molecular mechanisms of cancer development in obesity. Nat. Rev. Cancer 2011, 11, 886–895. [Google Scholar] [CrossRef]
- Guest, C.B.; Chakour, K.S.; Freund, G.G. Macropinocytosis is decreased in diabetic mouse macrophages and is regulated by AMPK. BMC Immunol. 2008, 9, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, B.; Long, L.; Luo, P.; Xiang, W.; Li, X.; Wang, H.; Jiang, Q.; Tan, X.; Luo, S.; et al. Improvement of obesity-associated disorders by a small-molecule drug targeting mitochondria of adipose tissue macrophages. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Jayashankar, V.; Edinger, A.L. Macropinocytosis confers resistance to therapies targeting cancer anabolism. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yang, F.; Fu, R.; Li, X.; French, R.; Mose, E.; Pu, X.; Trinh, B.; Kumar, A.; Liu, J.; et al. Cancer cells escape autophagy inhibition via NRF2-induced macropinocytosis. Cancer Cell 2021, 39, 678–693.e11. [Google Scholar] [CrossRef]
- Widenmaier, S.; Snyder, N.A.; Nguyen, T.B.; Arduini, A.; Lee, G.Y.; Arruda, A.P.; Saksi, J.; Bartelt, A.; Hotamisligil, G.S. NRF1 Is an ER Membrane Sensor that Is Central to Cholesterol Homeostasis. Cell 2017, 171, 1094–1109.e15. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waku, T.; Kobayashi, A. Pathophysiological Potentials of NRF3-Regulated Transcriptional Axes in Protein and Lipid Homeostasis. Int. J. Mol. Sci. 2021, 22, 12686. https://doi.org/10.3390/ijms222312686
Waku T, Kobayashi A. Pathophysiological Potentials of NRF3-Regulated Transcriptional Axes in Protein and Lipid Homeostasis. International Journal of Molecular Sciences. 2021; 22(23):12686. https://doi.org/10.3390/ijms222312686
Chicago/Turabian StyleWaku, Tsuyoshi, and Akira Kobayashi. 2021. "Pathophysiological Potentials of NRF3-Regulated Transcriptional Axes in Protein and Lipid Homeostasis" International Journal of Molecular Sciences 22, no. 23: 12686. https://doi.org/10.3390/ijms222312686