
CRISPR Screen Contributes to Novel Target Discovery in Prostate Cancer

Abstract
:1. Introduction
2. Methodology of CRISPR Screen
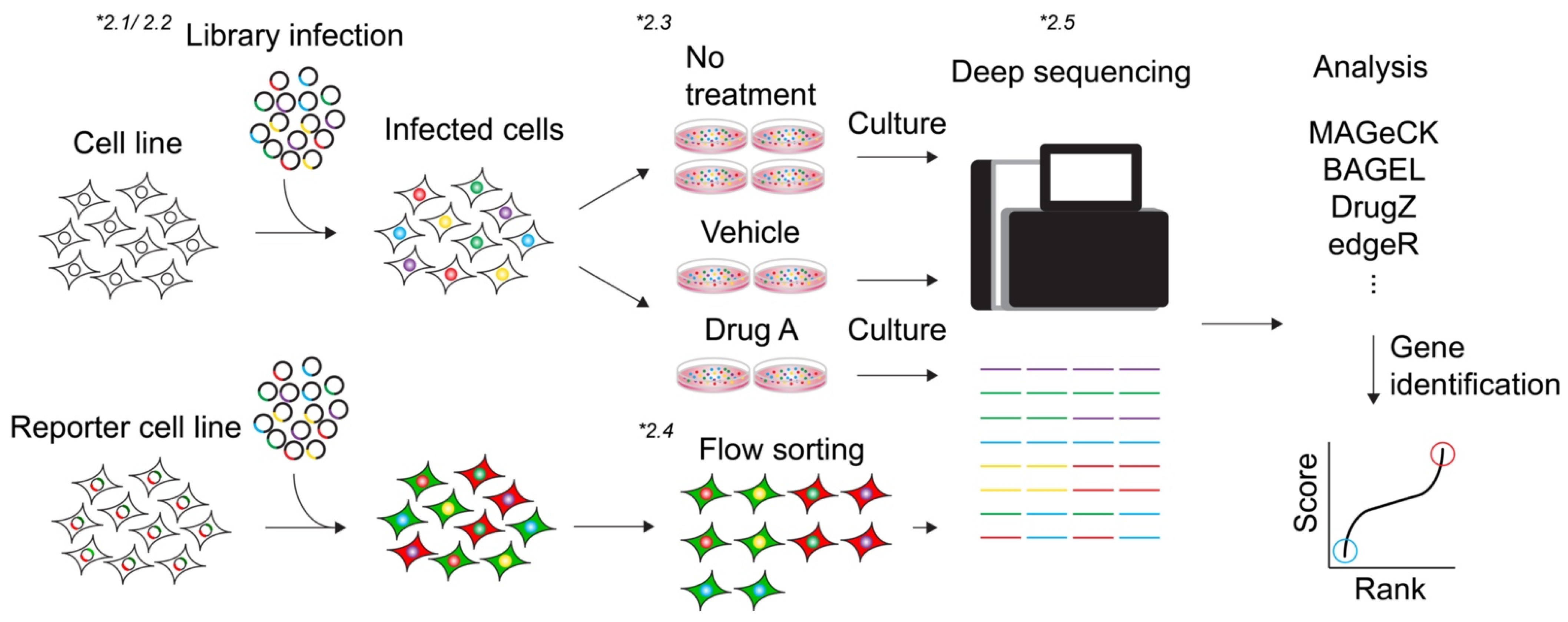
2.1. Library
2.2. Viral Packaging of Library and Transduction
2.3. Viability-Based Screens
2.4. Marker Selection Screen
2.5. Analysis (Algorithms)
2.6. Validation
3. CRISPR Screens in PCa
3.1. Discovery of Potential Target

{kind=link}
| Study | Screen Type | Library (Number of Genes) | Cell Line | Algorithm | Biomarkers | Results |
|---|---|---|---|---|---|---|
| Fei et al. (2017) [57] | Knockout | GeCKO v2 (19,050) | LNCaP | MAGeCK, MAGeCK-VISPR | HNRNPL | HNRNPL and its RNA clients as players in PCa growth and potential therapeutic targets. |
| Li et al. (2018) [58] | Knockout | Nuclear proteins sgRNA sub-pool library (3733) | DU145, 17p loss-DU145 | edgeR | RBX1 | Heterozygous deletion of 17p confers a selective dependence on RBX1. |
| Aquirre et al. (2016) [59] analyzed by Yoshiyama et al. (2021) [60] | Knockout | GeCKO v2 (19,050) | LNCaP, PC3 | BAGEL | JMJD1C | JMJD1C depletion leads to specific growth suppression of AR-negative cells via activation of the TNFα network. |
| Das et al. (2021) [62] | Knockdown | Human CRISPRi v2 Top5 sgRNA library (18,905) | LNCaP, C4-2B | ScreenProcessing | KIF4A, WDR62 | KIF4A and WDR62 drive aggressive prostate cancer phenotypes irrespective of AR-status. |
| Jiang et al. (2021) [63] | Knockout | E3 ubiquitin ligase contained CRISPR/Cas9 library (943) | EGFP-PDK1 reporter HEK293 | Not shown | SPOP | PDK1 underwent SPOP-mediated ubiquitination and subsequent proteasome-dependent degradation, which suppresses AKT kinase activity and oncogenic functions. |
| Palit et al. (2019) [67] | Knockout | GeCKO library A (19,052) | LNCaP | MAGeCK | TLE3 | Loss of TLE3 confers resistance to AR antagonists apalutamide and enzalutamide. |
| Palit et al. (2021) [68] | Knockout | NKI Human Kinome CRISPR pooled sgRNA library (578) | CWR-R1 | MAGeCK | BRAF | BRAF contribute to resistance ton AR targeted therapy in PCa. BRAF mutated patients is candidate for AR inhibitors. |
| Lei et al. (2021) [69] | Knockout | kinome CRISPR library (507) | C4-2 | MAGeCK | CDK12 | CDK12 is a conservative vulnerability of PCa cells. The synergy of THZ531 and AR antagonists suggests a potential combination therapy for PCa. |
| Zimmermann et al. (2018) [70] | Knockout | TKOv1 (17,661) | Hela, RPE1-hTERT, SUM149PT | DrugZ, MAGeCK | RNASEH2 | Mutations in all three genes encoding RNASEH2 sensitized cells to PARP inhibition. |
| Wang et al. (2019) [71] | Knockout | TKOv3 (18,053) | 293A, HCT116, MCF10A | BAGEL | RNASEH2 | RNASEH2 deficiency is synthetic lethal with ATR inhibition both in vitro and in vivo. |
| Chen et al. (2020) [72] | Activation | CRISPR/Cas9 Synergistic Activation Mediator (SAM) pooled library (23,430) | DU145, PC-3 | Not shown | RAD9A | The activation of RAD9A contributed to in vitro resistance to metformin. |
| Chu et al. (2021) [73] | Knockout | GeCKO v2 library A (19,050) | M231-ADIR (ADI resistant MDA-MB) | Subread aligner, DESeq2. | TRME1/CCL2 | TREM1/CCL2 activation, in addition to restored ASS1 expression, as a key pathway involved in full ADI-resistance in breast and prostate cancer models. |
3.2. Discovery of Drug-Induced Synthetic Lethality Targets and Resistance Mechanisms
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PCa | Prostate cancer |
| ADT | androgen deprivation therapy |
| mCRPC | metastatic castration-resistant prostate cancer |
| AR | androgen receptor |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| RNAi | RNA interference |
| WT | wild-type |
| dCas9 | dead Cas9 |
| CRISPRa | CRISPR activation |
| CRISPRi | CRISPR interference |
| CRPC | castration-resistant prostate cancer |
| sgRNA | single guide RNAs |
| MOI | multiplicity of infection |
| FACS | fluorescence-activated cell sorting |
| NGS | next-generation sequencing |
| MAGeCK | Model based Analysis of Genome-wide CRISPR/Cas9 Knockout |
| BAGEL | Bayesian Analysis of Gene EssentiaLity |
| caRpools | CRISPR AnalyzeR for Pooled Screens |
| PinAPL-Py | Platform-independent Analysis of Pooled Screens using Python |
| DepMaP | Cancer Dependency Map |
| TNFα | tumor necrosis factor alpha |
| KIF4A | Kinesin Family Member 4A |
| WDR62 | WD Repeat Domain 62 |
| TLE3 | transducin-like enhancer of split 3 |
| GR | glucocorticoid receptor |
| CDK12 | cyclin-dependent kinase 12 |
| CDK12-ISTs | CDK12 inhibition-sensitive transcripts |
| TIME | tumor immune microenvironment |
| ADI | arginine deiminase |
| PARP | poly(ADP-ribose) polymerase |
References
- Pernar, C.H.; Ebot, E.M.; Wilson, K.M.; Mucci, L.A. The epidemiology of prostate cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030361. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massie, C.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [Green Version]
- Attard, G.; Parker, C.; Eeles, R.A.; Schröder, F.; Tomlins, S.A.; Tannock, I.; Drake, C.G.; de Bono, J.S. Prostate cancer. Lancet 2016, 387, 70–82. [Google Scholar] [CrossRef]
- Komura, K.; Sweeney, C.J.; Inamoto, T.; Ibuki, N.; Azuma, H.; Kantoff, P.W. Current treatment strategies for advanced prostate cancer. Int. J. Urol. 2018, 25, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Nuhn, P.; De Bono, J.S.; Fizazi, K.; Freedland, S.J.; Grilli, M.; Kantoff, P.W.; Sonpavde, G.; Sternberg, C.N.; Yegnasubramanian, S.; Antonarakis, E.S. Update on Systemic Prostate Cancer Therapies: Management of Metastatic Castration-resistant Prostate Cancer in the Era of Precision Oncology. Eur. Urol. 2019, 75, 88–99. [Google Scholar] [CrossRef] [PubMed]
- de Wit, R.; de Bono, J.; Sternberg, C.N.; Fizazi, K.; Tombal, B.; Wülfing, C.; Kramer, G.; Eymard, J.-C.; Bamias, A.; Carles, J.; et al. Cabazitaxel versus Abiraterone or Enzalutamide in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 2506–2518. [Google Scholar] [CrossRef]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men with Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Stopsack, K.H. Efficacy of PARP Inhibition in Metastatic Castration-resistant Prostate Cancer is Very Different with Non-BRCA DNA Repair Alterations: Reconstructing Prespecified Endpoints for Cohort B from the Phase 3 PROfound Trial of Olaparib. Eur. Urol. 2021, 79, 442–445. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Fizazi, K.; et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): An open-label, phase 2 trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Berns, K.; Hijmans, M.; Mullenders, J.; Brummelkamp, T.R.; Velds, A.; Heimerikx, M.; Kerkhoven, R.M.; Madiredjo, M.; Nijkamp, W.; Weigelt, B.; et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nat. Cell Biol. 2004, 428, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nat. Cell Biol. 2013, 504, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Ai, N.; Wang, S.; Bhattacharya, N.; Vrbanac, V.; Collins, M.; Signoretti, S.; Hu, Y.; Boyce, F.M.; Gravdal, K.; et al. GRK3 is essential for metastatic cells and promotes prostate tumor progression. Proc. Natl. Acad. Sci. USA 2014, 111, 1521–1526. [Google Scholar] [CrossRef] [Green Version]
- Abdulkadir, S.; Meer, R.V.; Roh, M. Abstract 5185: RNAi screen identifies a synthetic lethal interaction between Pim1 overexpression and Plk1 inhibition. Mol. Cell. Biol. 2013, 20, 3211–3221. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Aach, J.; Stranges, P.; Esvelt, K.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef]
- Larson, M.H.; Gilbert, L.; Wang, X.; Lim, W.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef] [Green Version]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Xiao, T.; Chen, C.-H.; Li, W.; Meyer, C.A.; Wu, Q.; Wu, D.; Cong, L.; Zhang, F.; Liu, J.S.; et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015, 25, 1147–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Brigham, M.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.; Habib, N.; Gootenberg, J.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nat. Cell Biol. 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joung, J.; Konermann, S.; Gootenberg, J.S.; Abudayyeh, O.; Platt, R.J.; Brigham, M.D.; Sanjana, N.; Zhang, F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017, 12, 828–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, T.; Li, W.; Peng, J.; Xiao, T.; Chen, C.-H.; Wu, A.; Huang, J.; Zang, C.; Liu, X.S.; Brown, M. Deciphering essential cistromes using genome-wide CRISPR screens. Proc. Natl. Acad. Sci. USA 2019, 116, 25186–25195. [Google Scholar] [CrossRef] [Green Version]
- Munoz, D.; Stegmeier, F.P.; Schlabach, M. Abstract B21: CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false-positive hits for highly amplified genomic regions. Precis. Med. 2017, 6, 900–913. [Google Scholar] [CrossRef]
- Miles, L.A.; Garippa, R.J.; Poirier, J.T. Poirier, Design, execution, and analysis of pooled in vitro CRISPR/Cas9 screens. FEBS J. 2016, 283, 3170–3180. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Cheung, H.W.; Subramanian, A.; Sharifnia, T.; Okamoto, M.; Yang, X.; Hinkle, G.; Boehm, J.; Beroukhim, R.; Weir, B.A.; et al. Highly parallel identification of essential genes in cancer cells. Proc. Natl. Acad. Sci. USA 2008, 105, 20380–20385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Yu, H.; Hughes, N.W.; Liu, B.; Kendirli, A.; Klein, K.; Chen, W.; Lander, E.S.; Sabatini, D.M. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 2017, 168, 890–903.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, E.R., III; Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, 577–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzelepis, K.; Koike-Yusa, H.; De Braekeleer, E.; Li, Y.; Metzakopian, E.; Dovey, O.M.; Grinkevich, V.; Mupo, A.; Li, M.; Mazan, M.; et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016, 17, 1193–1205. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Alexe, G.; Dharia, N.; Ross, L.; Iniguez, A.B.; Conway, A.S.; Wang, E.J.; Veschi, V.; Lam, N.; Qi, J.; et al. CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J. Clin. Investig. 2017, 128, 446–462. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, N.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, L.; Gambino, V.; Raaijmakers, J.; Schlicker, A.; Fumagalli, A.; Russo, M.; Villanueva, A.; Beerling, E.; Bartolini, A.; Mollevi, D.G.; et al. A Vulnerability of a Subset of Colon Cancers with Potential Clinical Utility. Cell 2016, 165, 317–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orchard, R.C.; Wilen, C.B.; Doench, J.G.; Baldridge, M.T.; McCune, B.T.; Lee, Y.-C.J.; Lee, S.; Pruett-Miller, S.M.; Nelson, C.A.; Fremont, D.H.; et al. Discovery of a proteinaceous cellular receptor for a norovirus. Science 2016, 353, 933–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marceau, C.D.; Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Brewer, S.M.; Fuchs, G.; Swaminathan, K.; Mata, M.A.; Elias, J.E.; Sarnow, P.; et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nat. Cell Biol. 2016, 535, 159–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, R.; Wang, T.; Koundakjian, D.; Hultquist, J.; Lamothe-Molina, P.; Monel, B.; Schumann, K.; Yu, H.; Krupzcak, K.M.; Garcia-Beltran, W.; et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 2017, 49, 193–203. [Google Scholar] [CrossRef]
- Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Carette, J.E. A CRISPR toolbox to study virus–host interactions. Nat. Rev. Genet. 2017, 15, 351–364. [Google Scholar] [CrossRef]
- Wei, J.; Alfajaro, M.M.; DeWeirdt, P.C.; Hanna, R.E.; Lu-Culligan, W.J.; Cai, W.L.; Strine, M.S.; Zhang, S.-M.; Graziano, V.R.; Schmitz, C.O.; et al. Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell 2021, 184, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Deans, R.M.; Morgens, D.W.; Ökesli, A.; Pillay, S.; Horlbeck, M.A.; Kampmann, M.; Gilbert, L.A.; Li, A.; Mateo, R.; Smith, M.; et al. Parallel shRNA and CRISPR-Cas9 screens enable antiviral drug target identification. Nat. Chem. Biol. 2016, 12, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Parnas, O.; Jovanovic, M.; Eisenhaure, T.M.; Herbst, R.; Dixit, A.; Ye, C.J.; Przybylski, D.; Platt, R.; Tirosh, I.; Sanjana, N.; et al. A Genome-wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks. Cell 2015, 162, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Köster, J.; Xu, H.; Chen, C.H.; Xiao, T.; Liu, J.S.; Brown, M.; Liu, X.S. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 2015, 16, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wang, M.; Zhang, W.; Xiao, T.; Chen, C.-H.; Wu, A.; Wu, F.; Traugh, N.; Wang, X.; Li, Z.; et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc. 2019, 14, 756–780. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Sheridan, J.M.; Gearing, L.J.; Moore, D.L.; Su, S.; Wormald, S.; Wilcox, S.; Dickins, R.A.; Blewitt, M.E.; Ritchie, M.E.; et al. edgeR: A versatile tool for the analysis of shRNA-seq and CRISPR-Cas9 genetic screens. F1000Research 2014, 3, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, T.; Moffat, J. BAGEL: A computational framework for identifying essential genes from pooled library screens. BMC Bioinform. 2016, 17, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, J.; Breinig, M.; Heigwer, F.; Brügemann, D.; Leible, S.; Pelz, O.; Zhan, T.; Boutros, M. caRpools: An R package for exploratory data analysis and documentation of pooled CRISPR/Cas9 screens. Bioinformatics 2016, 32, 632–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spahn, P.N.; Bath, T.; Weiss, R.J.; Kim, J.; Esko, J.D.; Lewis, N.E.; Harismendy, O. PinAPL-Py: A comprehensive web-application for the analysis of CRISPR/Cas9 screens. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colic, M.; Wang, G.; Zimmermann, M.; Mascall, K.; McLaughlin, M.; Bertolet, L.; Lenoir, W.; Moffat, J.; Angers, S.; Durocher, D.; et al. Identifying chemogenetic interactions from CRISPR screens with drugZ. Genome Med. 2019, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, D.B.; Root, D.E. Resources for the design of CRISPR gene editing experiments. Genome Biol. 2015, 16, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, T.; Chen, Y.; Xiao, T.; Li, W.; Cato, L.; Zhang, P.; Cotter, M.B.; Bowden, M.; Lis, R.T.; Zhao, S.G.; et al. Genome-wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc. Natl. Acad. Sci. USA 2017, 114, E5207–E5215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, Y.; Xu, H.; Jiang, G.; Van Der Jeught, K.; Fang, Y.; Zhou, Z.; Zhang, L.; Frieden, M.; Wang, L.; et al. Heterozygous deletion of chromosome 17p renders prostate cancer vulnerable to inhibition of RNA polymerase II. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, A.J.; Meyers, R.; Weir, B.A.; Vazquez, F.; Zhang, C.-Z.; Ben-David, U.; Cook, A.; Ha, G.; Harrington, W.F.; Doshi, M.B.; et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 2016, 6, 914–929. [Google Scholar] [CrossRef] [Green Version]
- Yoshihama, Y.; LaBella, K.A.; Kim, E.; Bertolet, L.; Colic, M.; Li, J.; Shang, X.; Wu, C.-J.; Spring, D.J.; Wang, Y.A.; et al. AR-negative prostate cancer is vulnerable to loss of JMJD1C demethylase. Proc. Natl. Acad. Sci. USA 2021, 118, 4118. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Sjöström, M.; Shrestha, R.; Yogodzinski, C.; Egusa, E.A.; Chesner, L.N.; Chen, W.S.; Chou, J.; Dang, D.K.; Swinderman, J.T.; et al. An integrated functional and clinical genomics approach reveals genes driving aggressive metastatic prostate cancer. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Zheng, N.; Bu, L.; Zhang, X.; Zhang, X.; Wu, Y.; Su, Y.; Wang, L.; Zhang, X.; Ren, S.; et al. SPOP-mediated ubiquitination and degradation of PDK1 suppresses AKT kinase activity and oncogenic functions. Mol. Cancer 2021, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Stransky, N.; Nickerson, E.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.; et al. Mutational landscape and significance across 12 major cancer types. Nat. Cell Biol. 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palit, S.A.; Vis, D.; Stelloo, S.; Lieftink, C.; Prekovic, S.; Bekers, E.; Hofland, I.; Sustic, T.; Wolters, L.; van der Heijden, M.S.; et al. TLE3 loss confers AR inhibitor resistance by facilitating GR-mediated human prostate cancer cell growth. eLife 2019, 8, e47430. [Google Scholar] [CrossRef] [PubMed]
- Palit, S.A.L.; van Dorp, J.; Vis, D.; Lieftink, C.; Linder, S.; Beijersbergen, R.; Bergman, A.M.; Zwart, W.; van der Heijden, M.S. A kinome-centered CRISPR-Cas9 screen identifies activated BRAF to modulate enzalutamide resistance with potential therapeutic implications in BRAF-mutated prostate cancer. Sci. Rep. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Lei, H.; Wang, Z.; Jiang, D.; Liu, F.; Liu, M.; Lei, X.; Yang, Y.; He, B.; Yan, M.; Huang, H.; et al. CRISPR screening identifies CDK12 as a conservative vulnerability of prostate cancer. Cell Death Dis. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Zimmermann, M.; Murina, O.; Reijns, M.; Agathanggelou, A.; Challis, R.; Tarnauskaitė, Ž.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nat. Cell Biol. 2018, 559, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, G.; Feng, X.; Shepherd, P.; Zhang, J.; Tang, M.; Chen, Z.; Srivastava, M.; McLaughlin, M.E.; Navone, N.M.; et al. Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene 2019, 38, 2451–2463. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, Y.; Tang, Z.; Li, M.; Ling, X.; Liao, J.; Zhou, X.; Fang, S.; Zhao, H.; Zhong, W.; et al. Genome-Scale CRISPR-Cas9 Transcriptional Activation Screening in Metformin Resistance Related Gene of Prostate Cancer. Front. Cell Dev. Biol. 2021, 8, 1726. [Google Scholar] [CrossRef]
- Chu, C.-Y.; Lee, Y.-C.; Hsieh, C.-H.; Yeh, C.-T.; Chao, T.-Y.; Chen, P.-H.; Lin, I.-H.; Hsieh, T.-H.; Shih, J.-W.; Cheng, C.-H.; et al. Genome-wide CRISPR/Cas9 knockout screening uncovers a novel inflammatory pathway critical for resistance to arginine-deprivation therapy. Theranostics 2021, 11, 3624–3641. [Google Scholar] [CrossRef] [PubMed]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A Novel Antiandrogen for Prostate Cancer Treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in Metastatic Prostate Cancer before Chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Gomes, A.J.P.D.S.; Given, R.; Soto, A.J.; Merseburger, A.S.; Özgüroglu, M.; Uemura, H.; et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2019, 381, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A. PARP inhibitors in ovarian cancer. Ann. Oncol. 2016, 27, i40–i44. [Google Scholar] [CrossRef]
- Das, M. Olaparib provides benefit in metastatic breast cancer. Lancet Oncol. 2017, 18, e376. [Google Scholar] [CrossRef]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komura, K.; Yoshikawa, Y.; Shimamura, T.; Chakraborty, G.; Gerke, T.; Hinohara, K.; Chadalavada, K.; Jeong, S.H.; Armenia, J.; Du, S.-Y.; et al. ATR inhibition controls aggressive prostate tumors deficient in Y-linked histone demethylase KDM5D. J. Clin. Investig. 2018, 128, 2979–2995. [Google Scholar] [CrossRef]
- Neeb, A.; Herranz, N.; Arce-Gallego, S.; Miranda, S.; Buroni, L.; Yuan, W.; Athie, A.; Casals, T.; Carmichael, J.; Rodrigues, D.N.; et al. Advanced Prostate Cancer with ATM Loss: PARP and ATR Inhibitors. Eur. Urol. 2021, 79, 200–211. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gout, J.; Perkhofer, L.; Morawe, M.; Arnold, F.; Ihle, M.; Biber, S.; Lange, S.; Roger, E.; Kraus, J.M.; Stifter, K.; et al. Synergistic targeting and resistance to PARP inhibition in DNA damage repair-deficient pancreatic cancer. Gut 2021, 70, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Zhou, J.; Gorak, E.J.; Quddus, F. Metformin Is Associated with Survival Benefit in Cancer Patients with Concurrent Type 2 Diabetes: A Systematic Review and Meta--Analysis. Oncologist 2013, 18, 1248–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, H.K.; Lee, Y.H.; Koo, K.C. Current Status and Application of Metformin for Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 8540. [Google Scholar] [CrossRef] [PubMed]
- Rothermundt, C.; Hayoz, S.; Templeton, A.J.; Winterhalder, R.; Strebel, R.T.; Bärtschi, D.; Pollak, M.; LuI, L.; Endt, K.; Gillessen, S.; et al. Metformin in chemotherapy-naive castration-resistant prostate cancer: A multicenter phase 2 trial (SAKK 08/09). Eur. Urol. 2014, 66, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, B.K.; Thomson, J.A.; Bomalaski, J.S.; Diaz, M.; Akande, T.; Mahaffey, N.; Li, T.; Dutia, M.P.; Kelly, K.; Gong, I.-Y.; et al. Phase I trial of arginine deprivation therapy with ADI-PEG 20 plus docetaxel in patients with advanced malignant solid tumors. Clin. Cancer Res. 2015, 21, 2480–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szlosarek, P.W.; Steele, J.P.; Nolan, L.; Gilligan, D.; Taylor, P.; Spicer, J.; Lind, M.; Mitra, S.; Shamash, J.; Hackshaw, A.; et al. Arginine Deprivation with Pegylated Arginine Deiminase in Patients with Argininosuccinate Synthetase 1-Deficient Malignant Pleural Mesothelioma: A Randomized Clinical Trial. JAMA Oncol. 2017, 3, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.E.; Lewis, R.; Syed, N.; Shaffer, R.; Evanson, J.; Ellis, S.; Williams, M.; Feng, X.; Johnston, A.; Thomson, J.; et al. A Phase I Study of Pegylated Arginine Deiminase (Pegargiminase), Cisplatin, and Pemetrexed in Argininosuccinate Synthetase 1-Deficient Recurrent High-grade Glioma. Clin. Cancer Res. 2019, 25, 2708–2716. [Google Scholar] [CrossRef] [Green Version]
- Beddowes, E.; Spicer, J.; Chan, P.Y.; Khadeir, R.; Corbacho, J.G.; Repana, D.; Steele, J.P.; Schmid, P.; Szyszko, T.; Cook, G.; et al. Phase 1 Dose-Escalation Study of Pegylated Arginine Deiminase, Cisplatin, and Pemetrexed in Patients with Argininosuccinate Synthetase 1–Deficient Thoracic Cancers. J. Clin. Oncol. 2017, 35, 1778–1785. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Janku, F.; Subbiah, V.; Stewart, J.; Patel, S.P.; Kaseb, A.; Westin, S.N.; Naing, A.; Tsimberidou, A.M.; Hong, D.; et al. Phase 1 trial of ADI-PEG20 plus cisplatin in patients with pretreated metastatic melanoma or other advanced solid malignancies. Br. J. Cancer 2021, 124, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chung, S.-F.; Tam, S.-Y.; Leung, Y.-C.; Guan, X. Arginine deprivation as a strategy for cancer therapy: An insight into drug design and drug combination. Cancer Lett. 2021, 502, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.H.; Pritchard, C.C.; Boyd, T.; Nelson, P.S.; Montgomery, B. Biallelic Inactivation of BRCA2 in Platinum-sensitive Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2016, 69, 992–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.N.; Rescigno, P.; Liu, D.; Yuan, W.; Carreira, S.; Lambros, M.B.; Seed, G.; Mateo, J.; Riisnaes, R.; Mullane, S.; et al. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. J. Clin. Investig. 2018, 128, 4441–4453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreira, S.; Romanel, A.; Goodall, J.; Grist, E.; Ferraldeschi, R.; Miranda, S.; Prandi, D.; Lorente, D.; Frenel, J.-S.; Pezaro, C.; et al. Tumor clone dynamics in lethal prostate cancer. Sci. Transl. Med. 2014, 6, 254ra125. [Google Scholar] [CrossRef] [Green Version]
- Ku, S.-Y.; Gleave, M.; Beltran, H. Towards precision oncology in advanced prostate cancer. Nat. Rev. Urol. 2019, 16, 645–654. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsujino, T.; Komura, K.; Inamoto, T.; Azuma, H. CRISPR Screen Contributes to Novel Target Discovery in Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 12777. https://doi.org/10.3390/ijms222312777
Tsujino T, Komura K, Inamoto T, Azuma H. CRISPR Screen Contributes to Novel Target Discovery in Prostate Cancer. International Journal of Molecular Sciences. 2021; 22(23):12777. https://doi.org/10.3390/ijms222312777
Chicago/Turabian StyleTsujino, Takuya, Kazumasa Komura, Teruo Inamoto, and Haruhito Azuma. 2021. "CRISPR Screen Contributes to Novel Target Discovery in Prostate Cancer" International Journal of Molecular Sciences 22, no. 23: 12777. https://doi.org/10.3390/ijms222312777
APA StyleTsujino, T., Komura, K., Inamoto, T., & Azuma, H. (2021). CRISPR Screen Contributes to Novel Target Discovery in Prostate Cancer. International Journal of Molecular Sciences, 22(23), 12777. https://doi.org/10.3390/ijms222312777