
Regulation of Neural Stem Cell Competency and Commitment during Indirect Neurogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
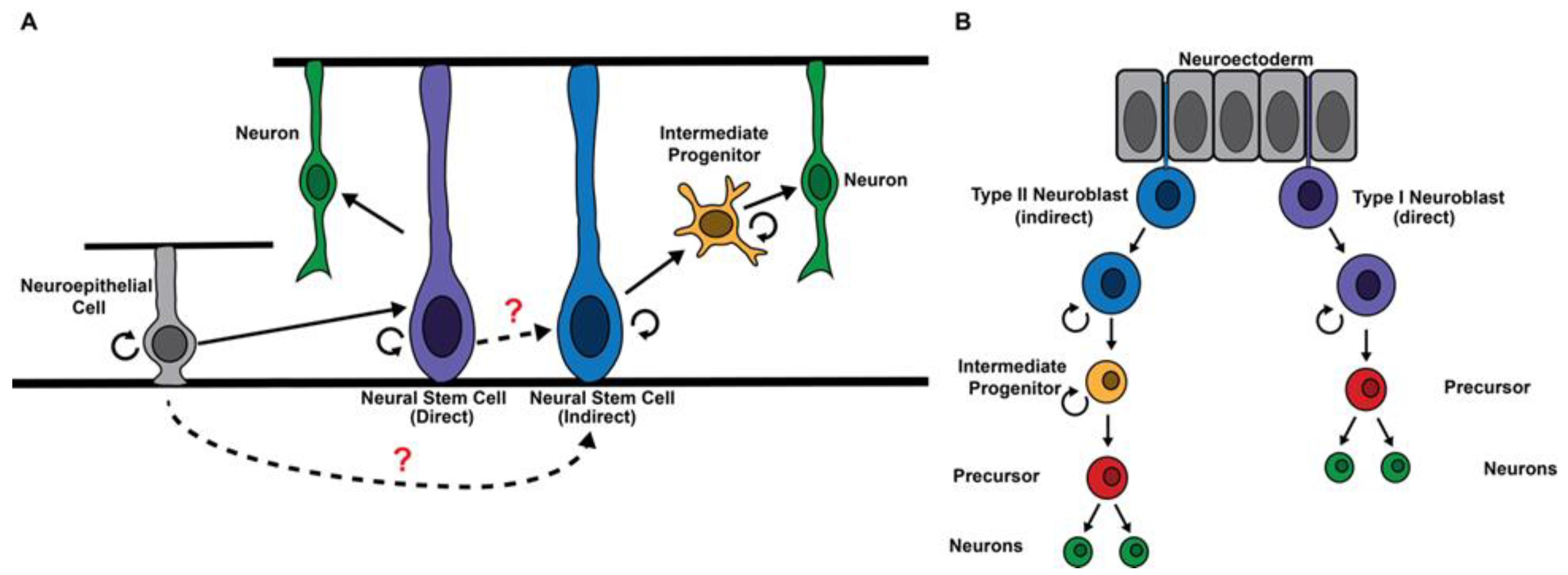
:1. Introduction
2. Regulation of the Competency to Generate Intermediate Progenitors
3. Regulation of the Commitment to Intermediate Progenitor Identity
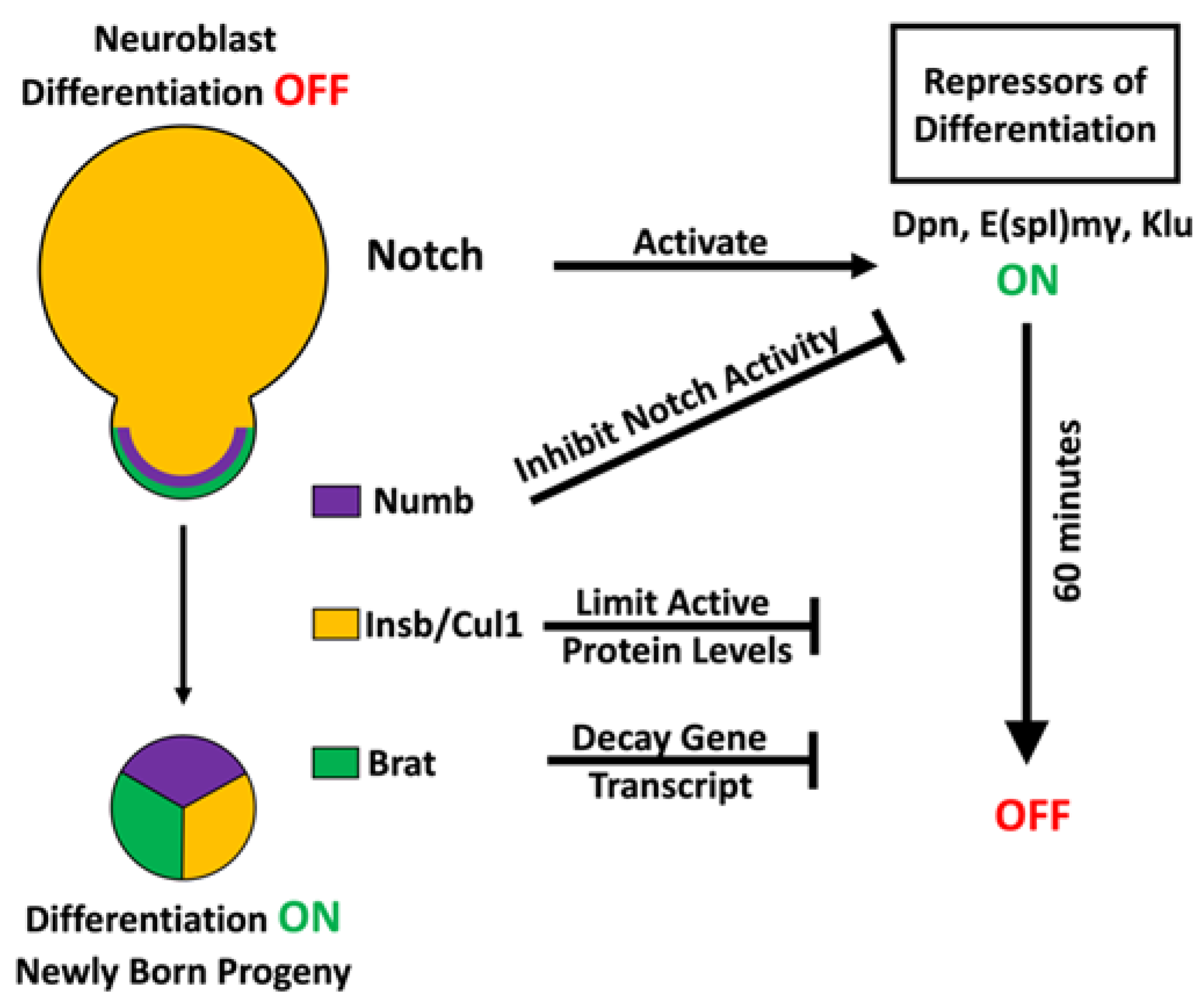
3.1. Multilayered Control of Timely Termination of Stem Cell Genes
3.2. Sequential Repression to Drive Robust Commitment to an Intermediate Progenitor Identity
4. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schnack, H.G.; van Haren, N.E.; Brouwer, R.M.; Evans, A.; Durston, S.; Boomsma, D.I.; Kahn, R.S.; Hulshoff Pol, H.E. Changes in thickness and surface area of the human cortex and their relationship with intelligence. Cereb. Cortex 2015, 25, 1608–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briscoe, S.D.; Ragsdale, C.W. Homology, neocortex, and the evolution of developmental mechanisms. Science 2018, 362, 190–193. [Google Scholar] [CrossRef] [Green Version]
- Cárdenas, A.; Borrell, V. Molecular and cellular evolution of corticogenesis in amniotes. Cell Mol. Life Sci. 2020, 77, 1435–1460. [Google Scholar] [CrossRef] [PubMed]
- Penisson, M.; Ladewig, J.; Belvindrah, R.; Francis, F. Genes and Mechanisms Involved in the Generation and Amplification of Basal Radial Glial Cells. Front. Cell Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef]
- Kageyama, R.; Ochi, S.; Sueda, R.; Shimojo, H. The significance of gene expression dynamics in neural stem cell regulation. Proc. Jpn Acad. Ser. B Phys. Biol. Sci. 2020, 96, 351–363. [Google Scholar] [CrossRef]
- Imayoshi, I.; Sakamoto, M.; Yamaguchi, M.; Mori, K.; Kageyama, R. Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J. Neurosci. 2010, 30, 3489–3498. [Google Scholar] [CrossRef] [Green Version]
- Baumgardt, M.; Karlsson, D.; Salmani, B.Y.; Bivik, C.; MacDonald, R.B.; Gunnar, E.; Thor, S. Global programmed switch in neural daughter cell proliferation mode triggered by a temporal gene cascade. Dev. Cell 2014, 30, 192–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnsworth, D.R.; Doe, C.Q. Opportunities lost and gained: Changes in progenitor competence during nervous system development. Neurogenesis 2017, 4, e1324260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homem, C.C.; Reichardt, I.; Berger, C.; Lendl, T.; Knoblich, J.A. Long-term live cell imaging and automated 4D analysis of drosophila neuroblast lineages. PLoS ONE 2013, 8, e79588. [Google Scholar] [CrossRef] [Green Version]
- Janssens, D.H.; Lee, C.Y. It takes two to tango, a dance between the cells of origin and cancer stem cells in the Drosophila larval brain. Semin. Cell Dev. Biol. 2014, 28, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Janssens, D.H.; Hamm, D.C.; Anhezini, L.; Xiao, Q.; Siller, K.H.; Siegrist, S.E.; Harrison, M.M.; Lee, C.Y. An Hdac1/Rpd3-Poised Circuit Balances Continual Self-Renewal and Rapid Restriction of Developmental Potential during Asymmetric Stem Cell Division. Dev. Cell 2017, 40, 367–380 e367. [Google Scholar] [CrossRef] [Green Version]
- Komori, H.; Xiao, Q.; McCartney, B.M.; Lee, C.Y. Brain tumor specifies intermediate progenitor cell identity by attenuating β-catenin/Armadillo activity. Development 2014, 141, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Li, X.; Zhang, X.; Mei, S.; Li, H.; Urso, A.; Zhu, S. The Drosophila Sp8 transcription factor Buttonhead prevents premature differentiation of intermediate neural progenitors. Elife 2014, 3, e03596. [Google Scholar] [CrossRef]
- Zhu, S.; Barshow, S.; Wildonger, J.; Jan, L.Y.; Jan, Y.N. Ets transcription factor Pointed promotes the generation of intermediate neural progenitors in Drosophila larval brains. Proc. Natl. Acad. Sci. USA 2011, 108, 20615–20620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez, J.A.; Díaz-Benjumea, F.J. Origin and specification of type II neuroblasts in the Drosophila embryo. Development 2018, 145, dev158394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, K.T.; Doe, C.Q. Drosophila embryonic type II neuroblasts: Origin, temporal patterning, and contribution to the adult central complex. Development 2017, 144, 4552–4562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakes, A.E.; Brand, A.H. Tailless/TLX reverts intermediate neural progenitors to stem cells driving tumourigenesis via repression of asense/ASCL1. Elife 2020, 9, e53377. [Google Scholar] [CrossRef] [PubMed]
- Komori, H.; Xiao, Q.; Janssens, D.H.; Dou, Y.; Lee, C.Y. Trithorax maintains the functional heterogeneity of neural stem cells through the transcription factor buttonhead. Elife 2014, 3, e03502. [Google Scholar] [CrossRef]
- Rives-Quinto, N.; Komori, H.; Ostgaard, C.M.; Janssens, D.H.; Kondo, S.; Dai, Q.; Moore, A.W.; Lee, C.Y. Sequential activation of transcriptional repressors promotes progenitor commitment by silencing stem cell identity genes. Elife 2020, 9. [Google Scholar] [CrossRef]
- Xie, Y.; Li, X.; Deng, X.; Hou, Y.; O’Hara, K.; Urso, A.; Peng, Y.; Chen, L.; Zhu, S. The Ets protein Pointed prevents both premature differentiation and dedifferentiation of Drosophila intermediate neural progenitors. Development 2016, 143, 3109–3118. [Google Scholar] [CrossRef] [Green Version]
- Krogan, N.J.; Dover, J.; Khorrami, S.; Greenblatt, J.F.; Schneider, J.; Johnston, M.; Shilatifard, A. COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J. Biol. Chem. 2002, 277, 10753–10755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.; Krogan, N.J.; Dover, J.; Erdjument-Bromage, H.; Tempst, P.; Johnston, M.; Greenblatt, J.F.; Shilatifard, A. COMPASS: A complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci. USA 2001, 98, 12902–12907. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Hou, Y.; Connell, M.; Zhu, S. Homeodomain protein Six4 prevents the generation of supernumerary Drosophila type II neuroblasts and premature differentiation of intermediate neural progenitors. PLoS Genet. 2021, 17, e1009371. [Google Scholar] [CrossRef] [PubMed]
- Sobhan, P.K.; Funa, K. TLX-Its Emerging Role for Neurogenesis in Health and Disease. Mol. Neurobiol. 2017, 54, 272–280. [Google Scholar] [CrossRef] [Green Version]
- Lim, D.A.; Huang, Y.C.; Swigut, T.; Mirick, A.L.; Garcia-Verdugo, J.M.; Wysocka, J.; Ernst, P.; Alvarez-Buylla, A. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 2009, 458, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albagli, O.; Klaes, A.; Ferreira, E.; Leprince, D.; Klambt, C. Function of ets genes is conserved between vertebrates and Drosophila. Mech. Dev. 1996, 59, 29–40. [Google Scholar] [CrossRef]
- Borello, U.; Berarducci, B.; Delahaye, E.; Price, D.J.; Dehay, C. SP8 Transcriptional Regulation of Cyclin D1 During Mouse Early Corticogenesis. Front. Neurosci. 2018, 12, 119. [Google Scholar] [CrossRef]
- Zhang, X.M.; Cai, Y.; Wang, F.; Wu, J.; Mo, L.; Zhang, F.; Patrylo, P.R.; Pan, A.; Ma, C.; Fu, J.; et al. Sp8 expression in putative neural progenitor cells in guinea pig and human cerebrum. Dev. Neurobiol. 2016, 76, 939–955. [Google Scholar] [CrossRef] [Green Version]
- Kiyota, T.; Kato, A.; Kato, Y. Ets-1 regulates radial glia formation during vertebrate embryogenesis. Organogenesis 2007, 3, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Chichung Lie, D.; Taupin, P.; Nakashima, K.; Ray, J.; Yu, R.T.; Gage, F.H.; Evans, R.M. Expression and function of orphan nuclear receptor TLX in adult neural stem cells. Nature 2004, 427, 78–83. [Google Scholar] [CrossRef]
- Liu, H.K.; Belz, T.; Bock, D.; Takacs, A.; Wu, H.; Lichter, P.; Chai, M.; Schütz, G. The nuclear receptor tailless is required for neurogenesis in the adult subventricular zone. Genes Dev. 2008, 22, 2473–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Sun, G.; Yang, S.; Qu, Q.; Nakashima, K.; Shi, Y. Nuclear receptor TLX regulates cell cycle progression in neural stem cells of the developing brain. Mol. Endocrinol. 2008, 22, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Sun, G.; Murai, K.; Ye, P.; Shi, Y. Characterization of TLX expression in neural stem cells and progenitor cells in adult brains. PLoS ONE 2012, 7, e43324. [Google Scholar] [CrossRef] [Green Version]
- Obernier, K.; Simeonova, I.; Fila, T.; Mandl, C.; Hölzl-Wenig, G.; Monaghan-Nichols, P.; Ciccolini, F. Expression of Tlx in both stem cells and transit amplifying progenitors regulates stem cell activation and differentiation in the neonatal lateral subependymal zone. Stem. Cells 2011, 29, 1415–1426. [Google Scholar] [CrossRef]
- Niu, W.; Zou, Y.; Shen, C.; Zhang, C.L. Activation of postnatal neural stem cells requires nuclear receptor TLX. J. Neurosci. 2011, 31, 13816–13828. [Google Scholar] [CrossRef] [PubMed]
- Murai, K.; Qu, Q.; Sun, G.; Ye, P.; Li, W.; Asuelime, G.; Sun, E.; Tsai, G.E.; Shi, Y. Nuclear receptor TLX stimulates hippocampal neurogenesis and enhances learning and memory in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2014, 111, 9115–9120. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Park, S.S.; Giaimo, B.D.; Hall, D.; Ferrante, F.; Ho, D.M.; Hori, K.; Anhezini, L.; Ertl, I.; Bartkuhn, M.; et al. RBPJ/CBF1 interacts with L3MBTL3/MBT1 to promote repression of Notch signaling via histone demethylase KDM1A/LSD1. EMBO J. 2017, 36, 3232–3249. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Namihira, M. LSD1 Mediates Neuronal Differentiation of Human Fetal Neural Stem Cells by Controlling the Expression of a Novel Target Gene, HEYL. Stem. Cells 2016, 34, 1872–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Wildonger, J.; Barshow, S.; Younger, S.; Huang, Y.; Lee, T. The bHLH repressor Deadpan regulates the self-renewal and specification of Drosophila larval neural stem cells independently of Notch. PLoS ONE 2012, 7, e46724. [Google Scholar] [CrossRef]
- Zacharioudaki, E.; Magadi, S.S.; Delidakis, C. bHLH-O proteins are crucial for Drosophila neuroblast self-renewal and mediate Notch-induced overproliferation. Development 2012, 139, 1258–1269. [Google Scholar] [CrossRef] [Green Version]
- Janssens, D.H.; Komori, H.; Grbac, D.; Chen, K.; Koe, C.T.; Wang, H.; Lee, C.Y. Earmuff restricts progenitor cell potential by attenuating the competence to respond to self-renewal factors. Development 2014, 141, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Berger, C.; Harzer, H.; Burkard, T.R.; Steinmann, J.; van der Horst, S.; Laurenson, A.S.; Novatchkova, M.; Reichert, H.; Knoblich, J.A. FACS purification and transcriptome analysis of drosophila neural stem cells reveals a role for Klumpfuss in self-renewal. Cell Rep. 2012, 2, 407–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Komori, H.; Lee, C.Y. klumpfuss distinguishes stem cells from progenitor cells during asymmetric neuroblast division. Development 2012, 139, 2670–2680. [Google Scholar] [CrossRef] [Green Version]
- Zacharioudaki, E.; Housden, B.E.; Garinis, G.; Stojnic, R.; Delidakis, C.; Bray, S. Genes implicated in stem-cell identity and temporal-program are directly targeted by Notch in neuroblast tumours. Development 2016, 143, 219–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San-Juan, B.P.; Baonza, A. The bHLH factor deadpan is a direct target of Notch signaling and regulates neuroblast self-renewal in Drosophila. Dev. Biol. 2011, 352, 70–82. [Google Scholar] [CrossRef]
- Lee, C.Y.; Robinson, K.J.; Doe, C.Q. Lgl, Pins and aPKC regulate neuroblast self-renewal versus differentiation. Nature 2006, 439, 594–598. [Google Scholar] [CrossRef]
- Rolls, M.M.; Albertson, R.; Shih, H.P.; Lee, C.Y.; Doe, C.Q. Drosophila aPKC regulates cell polarity and cell proliferation in neuroblasts and epithelia. J. Cell. Biol. 2003, 163, 1089–1098. [Google Scholar] [CrossRef] [Green Version]
- Atwood, S.X.; Prehoda, K.E. aPKC phosphorylates Miranda to polarize fate determinants during neuroblast asymmetric cell division. Curr. Biol. 2009, 19, 723–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, M.J.; Prehoda, K.E. Establishment of Par-Polarized Cortical Domains via Phosphoregulated Membrane Motifs. Dev. Cell 2015, 35, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Bello, B.; Reichert, H.; Hirth, F. The brain tumor gene negatively regulates neural progenitor cell proliferation in the larval central brain of Drosophila. Development 2006, 133, 2639–2648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betschinger, J.; Mechtler, K.; Knoblich, J.A. Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell 2006, 124, 1241–881253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.Y.; Wilkinson, B.D.; Siegrist, S.E.; Wharton, R.P.; Doe, C.Q. Brat is a Miranda cargo protein that promotes neuronal differentiation and inhibits neuroblast self-renewal. Dev. Cell 2006, 10, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, H.; Golden, K.L.; Kobayashi, T.; Kageyama, R.; Lee, C.Y. Multilayered gene control drives timely exit from the stem cell state in uncommitted progenitors during Drosophila asymmetric neural stem cell division. Genes Dev. 2018, 32, 1550–1561. [Google Scholar] [CrossRef] [Green Version]
- Laver, J.D.; Li, X.; Ray, D.; Cook, K.B.; Hahn, N.A.; Nabeel-Shah, S.; Kekis, M.; Luo, H.; Marsolais, A.J.; Fung, K.Y.Y.; et al. Brain tumor is a sequence-specific RNA-binding protein that directs maternal mRNA clearance during the Drosophila maternal-to-zygotic transition. Genome Biol. 2015, 16, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laver, J.D.; Marsolais, A.J.; Smibert, C.A.; Lipshitz, H.D. Regulation and Function of Maternal Gene Products During the Maternal-to-Zygotic Transition in Drosophila. Curr. Top Dev. Biol. 2015, 113, 43–84. [Google Scholar]
- Loedige, I.; Jakob, L.; Treiber, T.; Ray, D.; Stotz, M.; Treiber, N.; Hennig, J.; Cook, K.B.; Morris, Q.; Hughes, T.R.; et al. The Crystal Structure of the NHL Domain in Complex with RNA Reveals the Molecular Basis of Drosophila Brain-Tumor-Mediated Gene Regulation. Cell Rep. 2015, 13, 1206–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loedige, I.; Stotz, M.; Qamar, S.; Kramer, K.; Hennig, J.; Schubert, T.; Löffler, P.; Längst, G.; Merkl, R.; Urlaub, H.; et al. The NHL domain of BRAT is an RNA-binding domain that directly contacts the hunchback mRNA for regulation. Genes Dev. 2014, 28, 749–764. [Google Scholar] [CrossRef] [Green Version]
- Reichardt, I.; Bonnay, F.; Steinmann, V.; Loedige, I.; Burkard, T.R.; Meister, G.; Knoblich, J.A. The tumor suppressor Brat controls neuronal stem cell lineages by inhibiting Deadpan and Zelda. EMBO Rep. 2018, 19, 102–117. [Google Scholar] [CrossRef]
- Haenfler, J.M.; Kuang, C.; Lee, C.Y. Cortical aPKC kinase activity distinguishes neural stem cells from progenitor cells by ensuring asymmetric segregation of Numb. Dev. Biol. 2012, 365, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Somers, G.W.; Bashirullah, A.; Heberlein, U.; Yu, F.; Chia, W. Aurora-A acts as a tumor suppressor and regulates self-renewal of Drosophila neuroblasts. Genes Dev. 2006, 20, 3453–3463. [Google Scholar] [CrossRef] [Green Version]
- Wirtz-Peitz, F.; Nishimura, T.; Knoblich, J.A. Linking cell cycle to asymmetric division: Aurora-A phosphorylates the Par complex to regulate Numb localization. Cell 2008, 135, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.Y.; Andersen, R.O.; Cabernard, C.; Manning, L.; Tran, K.D.; Lanskey, M.J.; Bashirullah, A.; Doe, C.Q. Drosophila Aurora-A kinase inhibits neuroblast self-renewal by regulating aPKC/Numb cortical polarity and spindle orientation. Genes Dev. 2006, 20, 3464–3474. [Google Scholar] [CrossRef] [Green Version]
- Weng, M.; Golden, K.L.; Lee, C.Y. dFezf/Earmuff maintains the restricted developmental potential of intermediate neural progenitors in Drosophila. Dev. Cell 2010, 18, 126–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magadi, S.S.; Voutyraki, C.; Anagnostopoulos, G.; Zacharioudaki, E.; Poutakidou, I.K.; Efraimoglou, C.; Stapountzi, M.; Theodorou, V.; Nikolaou, C.; Koumbanakis, K.A.; et al. Dissecting Hes-centred transcriptional networks in neural stem cell maintenance and tumorigenesis in Drosophila. Development 2020, 147, dev191544. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, C.; Sandanaraj, E.; Aw, S.S.; Koe, C.T.; Wong, J.J.; Yu, F.; Ang, B.T.; Tang, C.; Wang, H. The SCFSlimb E3 ligase complex regulates asymmetric division to inhibit neuroblast overgrowth. EMBO Rep. 2014, 15, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kageyama, R.; Ohtsuka, T.; Kobayashi, T. The Hes gene family: Repressors and oscillators that orchestrate embryogenesis. Development 2007, 134, 1243–1251. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.A.; Hannenhalli, S.; Tobias, J.W.; Cooch, N.; Shiekhattar, R.; Kadesch, T. Functional analysis of Hes-1 in preadipocytes. Mol. Endocrinol. 2006, 20, 698–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isomura, A.; Kageyama, R. Ultradian oscillations and pulses: Coordinating cellular responses and cell fate decisions. Development 2014, 141, 3627–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.; Bessho, Y.; Hojo, M.; Kageyama, R. The bHLH gene Hes6, an inhibitor of Hes1, promotes neuronal differentiation. Development 2000, 127, 2933–2943. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, C.; Wu, H.; Ma, Y.; Luo, X.; Gong, X.; Jiang, F.; Gui, Y.; Zhang, H.; Lu, F. The E3 ubiquitin ligase SCF(FBXL14) complex stimulates neuronal differentiation by targeting the Notch signaling factor HES1 for proteolysis. J. Biol. Chem. 2017, 292, 20100–20112. [Google Scholar] [CrossRef] [Green Version]
- Gratton, M.O.; Torban, E.; Jasmin, S.B.; Theriault, F.M.; German, M.S.; Stifani, S. Hes6 promotes cortical neurogenesis and inhibits Hes1 transcription repression activity by multiple mechanisms. Mol. Cell Biol. 2003, 23, 6922–6935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, H.; Yoshiura, S.; Ohtsuka, T.; Bessho, Y.; Harada, T.; Yoshikawa, K.; Kageyama, R. Oscillatory expression of the bHLH factor Hes1 regulated by a negative feedback loop. Science 2002, 298, 840–843. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Iwamoto, Y.; Takashima, K.; Isomura, A.; Kosodo, Y.; Kawakami, K.; Nishioka, T.; Kaibuchi, K.; Kageyama, R. Deubiquitinating enzymes regulate Hes1 stability and neuronal differentiation. FEBS J. 2015, 282, 2411–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roese-Koerner, B.; Stappert, L.; Brüstle, O. Notch/Hes signaling and miR-9 engage in complex feedback interactions controlling neural progenitor cell proliferation and differentiation. Neurogenesis 2017, 4, e1313647. [Google Scholar] [CrossRef] [Green Version]
- Homem, C.C.F.; Steinmann, V.; Burkard, T.R.; Jais, A.; Esterbauer, H.; Knoblich, J.A. Ecdysone and mediator change energy metabolism to terminate proliferation in Drosophila neural stem cells. Cell 2014, 158, 874–888. [Google Scholar] [CrossRef] [Green Version]
- Farnsworth, D.R.; Bayraktar, O.A.; Doe, C.Q. Aging Neural Progenitors Lose Competence to Respond to Mitogenic Notch Signaling. Curr. Biol. 2015, 25, 3058–3068. [Google Scholar] [CrossRef] [Green Version]
- Larson, E.D.; Komori, H.; Gibson, T.J.; Ostgaard, C.M.; Hamm, D.C.; Schnell, J.M.; Lee, C.-Y.; Harrison, M.M. Cell-type-specific chromatin occupancy by the pioneer factor Zelda drives key developmental transitions in Drosophila. bioRxiv 2021. [Google Scholar] [CrossRef]
- Cenik, B.K.; Shilatifard, A. COMPASS and SWI/SNF complexes in development and disease. Nat. Rev. Genet. 2021, 22, 38–58. [Google Scholar] [CrossRef]
- Jancewicz, I.; Siedlecki, J.A.; Sarnowski, T.J.; Sarnowska, E. BRM: The core ATPase subunit of SWI/SNF chromatin-remodelling complex-a tumour suppressor or tumour-promoting factor? Epigenetics Chromatin 2019, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Allis, C.D. SWI/SNF complex in cancer. Nat. Genet. 2017, 49, 178–179. [Google Scholar] [CrossRef] [PubMed]
- Runge, J.S.; Raab, J.R.; Magnuson, T. Epigenetic Regulation by ATP-Dependent Chromatin-Remodeling Enzymes: SNF-ing Out Crosstalk. Curr. Top Dev. Biol. 2016, 117, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef]
- Song, Y.; Lu, B. Regulation of cell growth by Notch signaling and its differential requirement in normal vs. tumor-forming stem cells in Drosophila. Genes Dev. 2011, 25, 2644–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, S.K.; Rolland, V.; Betschinger, J.; Kinsey, K.A.; Emery, G.; Knoblich, J.A. The tumor suppressors Brat and Numb regulate transit-amplifying neuroblast lineages in Drosophila. Dev. Cell 2008, 14, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Brunet Avalos, C.; Maier, G.L.; Bruggmann, R.; Sprecher, S.G. Single cell transcriptome atlas of the Drosophila larval brain. Elife 2019, 8, e50354. [Google Scholar] [CrossRef] [PubMed]
- Genovese, S.; Clement, R.; Gaultier, C.; Besse, F.; Narbonne-Reveau, K.; Daian, F.; Foppolo, S.; Luis, N.M.; Maurange, C. Coopted temporal patterning governs cellular hierarchy, heterogeneity and metabolism in Drosophila neuroblast tumors. Elife 2019, 8, e50375. [Google Scholar] [CrossRef] [PubMed]
- Michki, N.S.; Li, Y.; Sanjasaz, K.; Zhao, Y.; Shen, F.Y.; Walker, L.A.; Cao, W.; Lee, C.Y.; Cai, D. The molecular landscape of neural differentiation in the developing Drosophila brain revealed by targeted scRNA-seq and multi-informatic analysis. Cell Rep. 2021, 35, 109039. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajan, A.; Ostgaard, C.M.; Lee, C.-Y. Regulation of Neural Stem Cell Competency and Commitment during Indirect Neurogenesis. Int. J. Mol. Sci. 2021, 22, 12871. https://doi.org/10.3390/ijms222312871
Rajan A, Ostgaard CM, Lee C-Y. Regulation of Neural Stem Cell Competency and Commitment during Indirect Neurogenesis. International Journal of Molecular Sciences. 2021; 22(23):12871. https://doi.org/10.3390/ijms222312871
Chicago/Turabian StyleRajan, Arjun, Cyrina M. Ostgaard, and Cheng-Yu Lee. 2021. "Regulation of Neural Stem Cell Competency and Commitment during Indirect Neurogenesis" International Journal of Molecular Sciences 22, no. 23: 12871. https://doi.org/10.3390/ijms222312871