
Epigenome and Epitranscriptome: Potential Resources for Crop Improvement


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Epigenetics and Epitranscriptomics
3. Epigenomic and Epitranscriptomic Changes during Development
4. Epigenomic and Epitranscriptomic Changes in Response to Abiotic and Biotic Stresses
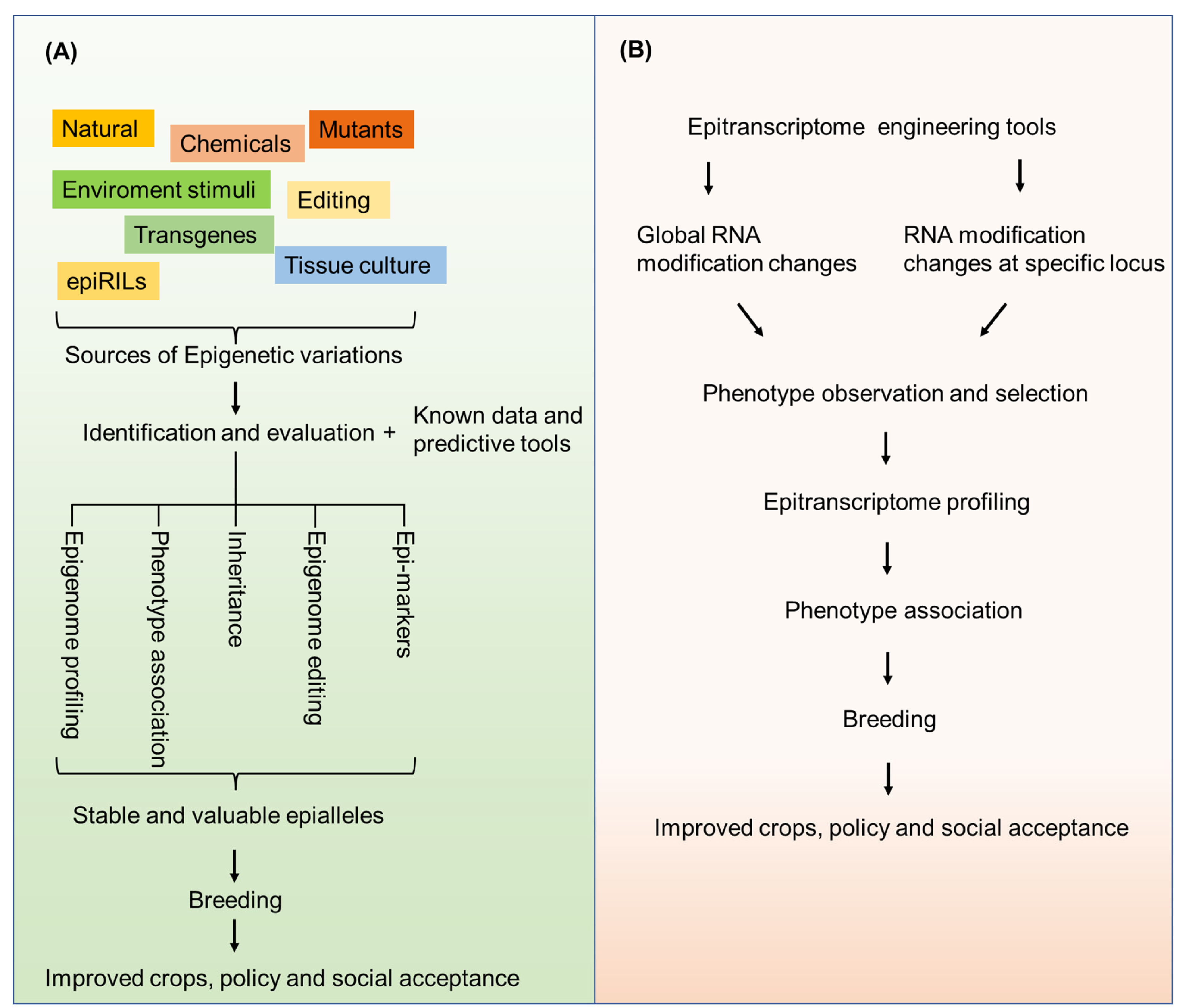
5. Sources for Epiallele Formation
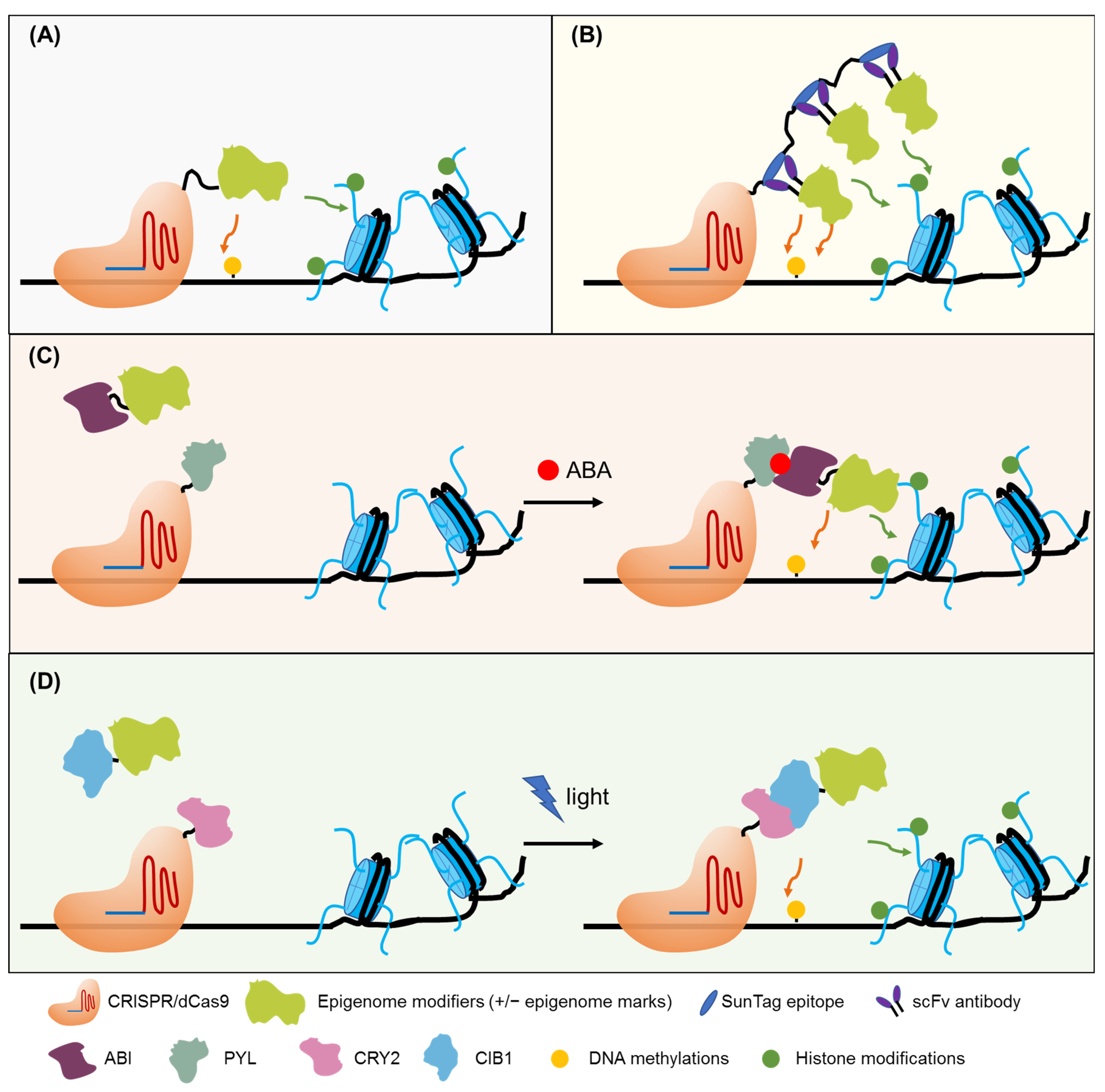
6. Epigenome and Epitranscriptome Engineering for Crop Improvement
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hickey, L.T.; Hafeez, A.N.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.M.; Tester, M.; Gao, C.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B.H. Breeding crops to feed 10 billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, A.; Ghosh, S.; Williams, M.J.; Cuddy, W.S.; Simmonds, J.; Rey, M.-D.; Hatta, M.A.M.; Hinchliffe, A.; Steed, A.; Reynolds, D.; et al. Speed breeding is a powerful tool to accelerate crop research and breeding. Nat. Plants 2018, 4, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Duan, X.; Wang, P.; Li, X.; Yuan, X.; Wang, Z.; Wan, L.; Yang, G.; Hong, D. Comprehensive speed breeding: A high-throughput and rapid generation system for long-day crops. Plant Biotechnol. J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Crouch, J.H. Marker-Assisted Selection in Plant Breeding: From Publications to Practice. Crop. Sci. 2008, 48, 391–407. [Google Scholar] [CrossRef] [Green Version]
- Araus, J.L.; Kefauver, S.C.; Zaman-Allah, M.; Olsen, M.S.; Cairns, J. Translating High-Throughput Phenotyping into Genetic Gain. Trends Plant Sci. 2018, 23, 451–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, S.J.; Furbank, R.T. Explainable machine learning models of major crop traits from satellite-monitored continent-wide field trial data. Nat. Plants 2021, 7, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Q.; Meng, X.-P.; Zhang, Y.; Xia, M.; Wang, X.-P. Over-expression of OsDREB genes lead to enhanced drought tolerance in rice. Biotechnol. Lett. 2008, 30, 2191–2198. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, L.; Shi, H.; Chern, M.; Yu, H.; Yi, H.; He, M.; Yin, J.; Zhu, X.; Li, Y.; et al. A single transcription factor promotes both yield and immunity in rice. Science 2018, 361, 1026–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Tian, Y.; Wu, K.; Ye, Y.; Yu, J.; Zhang, J.; Liu, Q.; Hu, M.; Li, H.; Tong, Y.; et al. Modulating plant growth–metabolism coordination for sustainable agriculture. Nature 2018, 560, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.S.E.A.; Mukhtar, S.; Mansoor, S. Genome Editing: Targeting Susceptibility Genes for Plant Disease Resistance. Trends Biotechnol. 2018, 36, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Galli, M.; Martiny, E.; Imani, J.; Kumar, N.; Koch, A.; Steinbrenner, J.; Kogel, K.-H. CRISPR/SpCas9-mediated double knockout of barley Microrchidia MORC1 and MORC6a reveals their strong involvement in plant immunity, transcriptional gene silencing and plant growth. Plant Biotechnol. J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.Z.; Henry, I.M.; Lynagh, P.G.; Comai, L.; Cahoon, E.B.; Weeks, D.P. Significant enhancement of fatty acid composition in seeds of the allohexaploid, Camelina sativa, using CRISPR /Cas9 gene editing. Plant Biotechnol. J. 2017, 15, 648–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Xian, P.; Cheng, Y.; Ma, Q.; Lian, T.; Nian, H.; Ge, L. CRISPR/Cas9-mediated gene editing of GmJAGGED1 increased yield in the low-latitude soybean variety Huachun 6. Plant Biotechnol. J. 2021, 19, 1898–1900. [Google Scholar] [CrossRef]
- Yu, H.; Lin, T.; Meng, X.; Du, H.; Zhang, J.; Liu, G.; Chen, M.; Jing, Y.; Kou, L.; Li, X.; et al. A route to de novo domestication of wild allotetraploid rice. Cell 2021, 184, 1156–1170. [Google Scholar] [CrossRef]
- Frye, M.; Harada, B.T.; Behm, M.; He, C. RNA modifications modulate gene expression during development. Science 2018, 361, 1346–1349. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Liu, S.; Yu, L.; Xiao, Y.; Zhang, S.; Wang, X.; Xu, Y.; Yu, H.; Li, Y.; Yang, J.; et al. RNA demethylation increases the yield and biomass of rice and potato plants in field trials. Nat. Biotechnol. 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, D.; Sun, F.; Guo, W.; Wang, W.; Li, X.; Lan, Y.; Du, L.; Li, S.; Fan, Y.; et al. ARGONAUTE 2 increases rice susceptibility to rice black-streaked dwarf virus infection by epigenetically regulating HEXOKINASE 1 expression. Mol. Plant Pathol. 2021, 22, 1029–1040. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Zhang, N.; Wang, W.-Q.; Shen, S.-Y.; Bai, C.; Song, X.-J. The ubiquitin-interacting motif-type ubiquitin receptor HDR3 interacts with and stabilizes the histone acetyltransferase GW6a to control the grain size in rice. Plant Cell 2021, 33, 3331–3347. [Google Scholar] [CrossRef] [PubMed]
- Habig, M.; Lorrain, C.; Feurtey, A.; Komluski, J.; Stukenbrock, E.H. Epigenetic modifications affect the rate of spontaneous mutations in a pathogenic fungus. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Meng, X.; Yuan, C.; Harrison, A.P.; Chen, M. The roles of cross-talk epigenetic patterns inArabidopsis thaliana. Brief. Funct. Genom. 2016, 15, 278–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raiber, E.-A.; Hardisty, R.; van Delft, P.; Balasubramanian, S. Mapping and elucidating the function of modified bases in DNA. Nat. Rev. Chem. 2017, 1, 69. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bewick, A.J.; Ji, L.; Niederhuth, C.E.; Willing, E.-M.; Hofmeister, B.T.; Shi, X.; Wang, L.; Lu, Z.; Rohr, N.A.; Hartwig, B.; et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 9111–9116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Wang, C.; Liu, H.; Zhou, Q.; Liu, Q.; Guo, Y.; Peng, T.; Song, J.; Zhang, J.; Chen, L.; et al. Identification and analysis of adenine N6-methylation sites in the rice genome. Nat. Plants 2018, 4, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Shen, L.; Cui, X.; Bao, S.; Geng, Y.; Yu, G.; Liang, F.; Xie, S.; Lu, T.; Gu, X.; et al. DNA N6-Adenine Methylation in Arabidopsis thaliana. Dev. Cell 2018, 45, 406–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Liang, Z.; Cui, X.; Ji, C.; Li, Y.; Zhang, P.; Liu, J.; Riaz, A.; Yao, P.; Liu, M.; et al. N6-Methyladenine DNA Methylation in Japonica and Indica Rice Genomes and Its Association with Gene Expression, Plant Development, and Stress Responses. Mol. Plant 2018, 11, 1492–1508. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Lu, F.; Cui, X.; Cao, X. Histone Methylation in Higher Plants. Annu. Rev. Plant Biol. 2010, 61, 395–420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Sridhar, V.V.; Zhu, J.; Kapoor, A.; Zhu, J.-K. Distinctive Core Histone Post-Translational Modification Patterns in Arabidopsis thaliana. PLoS ONE 2007, 2, e1210. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Grunstein, M. Functions of Site-Specific Histone Acetylation and Deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, R.; Zhou, M.-M. Writers and Readers of Histone Acetylation: Structure, Mechanism, and Inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef] [Green Version]
- Saleh, A.; Alvarez-Venegas, R.; Yilmaz, M.; Le, O.; Hou, G.; Sadder, M.; Al-Abdallat, A.; Xia, Y.; Lu, G.; Ladunga, I.; et al. The Highly Similar Arabidopsis Homologs of Trithorax ATX1 and ATX2 Encode Proteins with Divergent Biochemical Functions. Plant Cell 2008, 20, 568–579. [Google Scholar] [CrossRef] [Green Version]
- Mozgova, I.; Hennig, L. The Polycomb Group Protein Regulatory Network. Annu. Rev. Plant Biol. 2015, 66, 269–296. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Qian, S.; Scheid, R.N.; Lu, L.; Chen, X.; Liu, R.; Du, X.; Lv, X.; Boersma, M.D.; Scalf, M.; et al. EBS is a bivalent histone reader that regulates floral phase transition in Arabidopsis. Nat. Genet. 2018, 50, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, S.; Li, H.; Bodi, Z.; Button, J.; Vespa, L.; Herzog, M.; Fray, R.G. MTA Is an Arabidopsis Messenger RNA Adenosine Methylase and Interacts with a Homolog of a Sex-Specific Splicing Factor. Plant Cell 2008, 20, 1278–1288. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Liang, Z.; Gu, X.; Chen, Y.; Teo, Z.W.N.; Hou, X.; Cai, W.M.; Dedon, P.C.; Liu, L.; Yu, H. N6-Methyladenosine RNA Modification Regulates Shoot Stem Cell Fate in Arabidopsis. Dev. Cell 2016, 38, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Růžička, K.; Zhang, M.; Campilho, A.; Bodi, Z.; Kashif, M.; Saleh, M.; Eeckhout, D.; El-Showk, S.; Li, H.; Zhong, S.; et al. Identification of factors required for m 6 A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. N. Phytol. 2017, 215, 157–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Zhang, Y.-C.; Liao, J.-Y.; Yu, Y.; Zhou, Y.-F.; Feng, Y.-Z.; Yang, Y.-W.; Lei, M.-Q.; Bai, M.; Wu, H.; et al. The subunit of RNA N6-methyladenosine methyltransferase OsFIP regulates early degeneration of microspores in rice. PLoS Genet. 2019, 15, e1008120. [Google Scholar] [CrossRef] [Green Version]
- Duan, H.-C.; Wei, L.-H.; Zhang, C.; Wang, Y.; Chen, L.; Lu, Z.; Chen, P.R.; He, C.; Jia, G. ALKBH10B Is an RNA N6-Methyladenosine Demethylase Affecting Arabidopsis Floral Transition. Plant Cell 2017, 29, 2995–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Pérez, M.; Aparicio, F.; López-Gresa, M.P.; Bellés, J.M.; Sánchez-Navarro, J.A.; Pallás, V. Arabidopsis m6A demethylase activity modulates viral infection of a plant virus and the m6A abundance in its genomic RNAs. Proc. Natl. Acad. Sci. USA 2017, 114, 10755–10760. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.-H.; Song, P.; Wang, Y.; Lu, Z.; Tang, Q.; Yu, Q.; Xiao, Y.; Zhang, X.; Duan, H.-C.; Jia, G. The m6A Reader ECT2 Controls Trichome Morphology by Affecting mRNA Stability in Arabidopsis. Plant Cell 2018, 30, 968–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Liang, Z.; Shen, L.; Zhang, Q.; Bao, S.; Geng, Y.; Zhang, B.; Leo, V.; Vardy, L.; Lu, T.; et al. 5-Methylcytosine RNA Methylation in Arabidopsis Thaliana. Mol. Plant 2017, 10, 1387–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, R.; Burgess, A.; Parker, B.; Li, J.; Pulsford, K.; Sibbritt, T.; Preiss, T.; Searle, I.R. Transcriptome-Wide Mapping of RNA 5-Methylcytosine in Arabidopsis mRNAs and Noncoding RNAs. Plant Cell 2017, 29, 445–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Riaz, A.; Chachar, S.; Ding, Y.; Du, H.; Gu, X. Epigenetic Modifications of mRNA and DNA in Plants. Mol. Plant 2020, 13, 14–30. [Google Scholar] [CrossRef]
- Arango, D.; Sturgill, D.; Alhusaini, N.; Dillman, A.A.; Sweet, T.J.; Hanson, G.; Hosogane, M.; Sinclair, W.R.; Nanan, K.K.; Mandler, M.D.; et al. Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell 2018, 175, 1872–1886. [Google Scholar] [CrossRef] [Green Version]
- Kudrin, P.; Meierhofer, D.; Vågbø, C.B.; Ørom, U.A.V. Nuclear RNA-acetylation can be erased by the deacetylase SIRT7. bioRxiv 2021. [Google Scholar] [CrossRef]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Coruh, C.; Xu, G.; Bourbousse, C.; Lambolez, A.; Law, J.A. The CLASSY family controls tissue-specific DNA methylation patterns in Arabidopsis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gutzat, R.; Rembart, K.; Nussbaumer, T.; Hofmann, F.; Pisupati, R.; Bradamante, G.; Daubel, N.; Gaidora, A.; Lettner, N.; Donà, M.; et al. Arabidopsis shoot stem cells display dynamic transcription and DNA methylation patterns. EMBO J. 2020, 39, 103667. [Google Scholar] [CrossRef] [PubMed]
- Higo, A.; Saihara, N.; Miura, F.; Higashi, Y.; Yamada, M.; Tamaki, S.; Ito, T.; Tarutani, Y.; Sakamoto, T.; Fujiwara, M.; et al. DNA methylation is reconfigured at the onset of reproduction in rice shoot apical meristem. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Stuart, T.; Valdes, M.; Breakfield, N.; Schmitz, R.; Nery, J.R.; Urich, M.A.; Han, X.; Lister, R.; Benfey, P.N.; et al. Unique cell-type-specific patterns of DNA methylation in the root meristem. Nat. Plants 2016, 2, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, J.P.B.; Lister, R. Epigenome plasticity in plants. Nat. Rev. Genet. 2021, 1–14. [Google Scholar] [CrossRef]
- Slotkin, R.K.; Vaughn, M.; Borges, F.; Tanurdžić, M.; Becker, J.D.; Feijó, J.A.; Martienssen, R.A. Epigenetic Reprogramming and Small RNA Silencing of Transposable Elements in Pollen. Cell 2009, 136, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Walker, J.; She, W.; Aldridge, B.; Gao, H.; Deans, S.; Vickers, M.; Feng, X. Nurse cell –derived small RNAs define paternal epigenetic inheritance in Arabidopsis. Science 2021, 373, 556. [Google Scholar] [CrossRef]
- Walker, J.; Gao, H.; Zhang, J.; Aldridge, B.; Vickers, M.; Higgins, J.D.; Feng, X. Sexual-lineage-specific DNA methylation regulates meiosis in Arabidopsis. Nat. Genet. 2018, 50, 130–137. [Google Scholar] [CrossRef]
- Lang, Z.; Wang, Y.; Tang, K.; Tang, D.; Datsenka, T.; Cheng, J.; Zhang, Y.; Handa, A.K.; Zhu, J.-K. Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. USA 2017, 114, E4511–E4519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Q.-X.; Lu, X.; Li, Q.-T.; Chen, H.; Hu, X.-Y.; Ma, B.; Zhang, W.-K.; Chen, S.-Y.; Zhang, J.-S. Genome-Wide Analysis of DNA Methylation in Soybean. Mol. Plant 2013, 6, 1961–1974. [Google Scholar] [CrossRef] [Green Version]
- Makarevitch, I.; Eichten, S.; Briskine, R.; Waters, A.J.; Danilevskaya, O.N.; Meeley, R.B.; Myers, C.L.; Vaughn, M.; Springer, N.M. Genomic Distribution of Maize Facultative Heterochromatin Marked by Trimethylation of H3K27. Plant Cell 2013, 25, 780–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.; Wengier, D.L.; Bergmann, D.C. Cell-type–specific transcriptome and histone modification dynamics during cellular reprogramming in the Arabidopsis stomatal lineage. Proc. Natl. Acad. Sci. USA 2019, 116, 21914–21924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeuchi, M.; Iwase, A.; Rymen, B.; Harashima, H.; Shibata, M.; Ohnuma, M.; Breuer, C.; Morao, A.K.; De Lucas, M.; De Veylder, L.; et al. PRC2 represses dedifferentiation of mature somatic cells in Arabidopsis. Nat. Plants 2015, 1, 15089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deal, R.B.; Henikoff, S. A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev. Cell 2010, 18, 1030–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollmann, H.; Holec, S.; Alden, K.; Clarke, N.D.; Jacques, P.-E.; Berger, F. Dynamic Deposition of Histone Variant H3.3 Accompanies Developmental Remodeling of the Arabidopsis Transcriptome. PLoS Genet. 2012, 8, e1002658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Tang, K.; Zhang, D.; Xie, S.; Zhu, X.; Wang, Z.; Lang, Z. Transcriptome-wide high-throughput deep m6A-seq reveals unique differential m6A methylation patterns between three organs in Arabidopsis thaliana. Genome Biol. 2015, 16, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wang, X.; Li, C.; Hu, S.; Yu, J.; Song, S. Transcriptome-wide N6-methyladenosine profiling of rice callus and leaf reveals the presence of tissue-specific competitors involved in selective mRNA modification. RNA Biol. 2014, 11, 1180–1188. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Tian, S.; Qin, G. RNA methylomes reveal the m6A-mediated regulation of DNA demethylase gene SlDML2 in tomato fruit ripening. Genome Biol. 2019, 20, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Vespa, L.; Vachon, G.; Berger, F.; Perazza, D.; Faure, J.-D.; Herzog, M. The Immunophilin-Interacting Protein AtFIP37 from Arabidopsis Is Essential for Plant Development and Is Involved in Trichome Endoreduplication. Plant Physiol. 2004, 134, 1283–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, D.; Zhang, Z.; Wu, H.; Huang, C.; Shuai, P.; Ye, C.-Y.; Tang, S.; Wang, Y.; Yang, L.; Wang, J.; et al. Single-base-resolution methylomes of populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet. 2014, 15, S9. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-S.; Pan, Y.-J.; Zhao, X.-Q.; Dwivedi, D.; Zhu, L.-H.; Ali, J.; Fu, B.-Y.; Li, Z.-K. Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J. Exp. Bot. 2010, 62, 1951–1960. [Google Scholar] [CrossRef]
- Ganguly, D.R.; Crisp, P.A.; Eichten, S.R.; Pogson, B.J. The Arabidopsis DNA Methylome Is Stable under Transgenerational Drought Stress. Plant Physiol. 2017, 175, 1893–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, D.R.; Crisp, P.A.; Eichten, S.R.; Pogson, B.J. Maintenance of pre-existing DNA methylation states through recurring excess-light stress. Plant Cell Environ. 2018, 41, 1657–1672. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Prado, J.S.; Latrasse, D.; Rodriguez-Granados, N.Y.; Huang, Y.; Manza-Mianza, D.; Brik-Chaouche, R.; Jaouannet, M.; Citerne, S.; Bendahmane, A.; Hirt, H.; et al. The Polycomb protein LHP 1 regulates Arabidopsis thaliana stress responses through the repression of the MYC 2-dependent branch of immunity. Plant J. 2019, 100, 1118–1131. [Google Scholar] [CrossRef] [Green Version]
- Yolcu, S.; Ozdemir, F.; Güler, A.; Bor, M. Histone acetylation influences the transcriptional activation of POX in Beta vulgaris L. and Beta maritima L. under salt stress. Plant Physiol. Biochem. 2016, 100, 37–46. [Google Scholar] [CrossRef]
- Chen, L.-T.; Luo, M.; Wang, Y.-Y.; Wu, K. Involvement of Arabidopsis histone deacetylase HDA6 in ABA and salt stress response. J. Exp. Bot. 2010, 61, 3345–3353. [Google Scholar] [CrossRef] [Green Version]
- Sokol, A.; Kwiatkowska, A.; Jerzmanowski, A.; Prymakowska-Bosak, M. Up-regulation of stress-inducible genes in tobacco and Arabidopsis cells in response to abiotic stresses and ABA treatment correlates with dynamic changes in histone H3 and H4 modifications. Planta 2007, 227, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Rus, A.; Yokoi, S.; Sharkhuu, A.; Reddy, M.; Lee, B.-H.; Matsumoto, T.K.; Koiwa, H.; Zhu, J.-K.; Bressan, R.A.; Hasegawa, P.M. AtHKT1 is a salt tolerance determinant that controls Na+ entry into plant roots. Proc. Natl. Acad. Sci. USA 2001, 98, 14150–14155. [Google Scholar] [CrossRef] [Green Version]
- Sani, E.; Herzyk, P.; Perrella, G.; Colot, V.; Amtmann, A. Hyperosmotic priming of Arabidopsis seedlings establishes a long-term somatic memory accompanied by specific changes of the epigenome. Genome Biol. 2013, 14, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Beena, A.S.; Awana, M.; Singh, A. Salt-Induced Tissue-Specific Cytosine Methylation Downregulates Expression of HKT Genes in Contrasting Wheat (Triticum aestivum L.) Genotypes. DNA Cell Biol. 2017, 36, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Wang, Y.; Zheng, H.; Lu, W.; Wu, C.; Huang, J.; Yan, K.; Yang, G.; Zheng, C. Salt-induced transcription factorMYB74is regulated by the RNA-directed DNA methylation pathway inArabidopsis. J. Exp. Bot. 2015, 66, 5997–6008. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; de Jonge, J.; Trejo-Arellano, M.S.; Santos-González, J.; Köhler, C.; Hennig, L. Role of H1 and DNA methylation in selective regulation of transposable elements during heat stress. N. Phytol. 2021, 229, 2238–2250. [Google Scholar] [CrossRef]
- Sanchez, D.H.; Paszkowski, J. Heat-Induced Release of Epigenetic Silencing Reveals the Concealed Role of an Imprinted Plant Gene. PLoS Genet. 2014, 10, e1004806. [Google Scholar] [CrossRef] [Green Version]
- Kwon, C.S.; Lee, D.; Choi, G.; Chung, W.-I. Histone occupancy-dependent and -independent removal of H3K27 trimethylation at cold-responsive genes in Arabidopsis. Plant J. 2009, 60, 112–121. [Google Scholar] [CrossRef]
- Park, J.; Lim, C.J.; Shen, M.; Park, H.J.; Cha, J.-Y.; Iniesto, E.; Rubio, V.; Mengiste, T.; Zhu, J.-K.; Bressan, R.A.; et al. Epigenetic switch from repressive to permissive chromatin in response to cold stress. Proc. Natl. Acad. Sci. USA 2018, 115, E5400–E5409. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Wang, Q.; Yuan, H.; Huang, X. Chilling-induced DNA Demethylation is associated with the cold tolerance of Hevea brasiliensis. BMC Plant Biol. 2018, 18, 70. [Google Scholar] [CrossRef] [Green Version]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez-Prado, J.S.; Piquerez, S.J.M.; Bendahmane, A.; Hirt, H.; Raynaud, C.; Benhamed, M. Modify the Histone to Win the Battle: Chromatin Dynamics in Plant—Pathogen Interactions. Front. Plant Sci. 2018, 9, 355. [Google Scholar] [CrossRef] [PubMed]
- Annacondia, M.L.; Markovic, D.; Reig-Valiente, J.L.; Scaltsoyiannes, V.; Pieterse, C.M.J.; Ninkovic, V.; Slotkin, R.K.; Martinez, G. Aphid feeding induces the relaxation of epigenetic control and the associated regulation of the defense response in Arabidopsis. N. Phytol. 2021, 230, 1185–1200. [Google Scholar] [CrossRef]
- Liu, J.; Zhi, P.; Wang, X.; Fan, Q.; Chang, C. Wheat WD40-repeat protein TaHOS15 functions in a histone deacetylase complex to fine-tune defense responses to Blumeria graminis f.sp. tritici. J. Exp. Bot. 2019, 70, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Qiu, X.; Kang, J.; Wang, Y.; Chen, H.; Huang, J.; Qiu, M.; Zhao, Y.; Kong, G.; Ma, Z.; et al. A Phytophthora Effector Manipulates Host Histone Acetylation and Reprograms Defense Gene Expression to Promote Infection. Curr. Biol. 2017, 27, 981–991. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, L.; Wang, L.; Liu, L.; Li, L.; Sun, L.; Rao, Q.; Zhang, J.; Huang, S. JMJ704 positively regulates rice defense response against Xanthomonas oryzae pv. oryzae infection via reducing H3K4me2/3 associated with negative disease resistance regulators. BMC Plant Biol. 2015, 15, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Manduzio, S.; Kang, H. Epitranscriptomic RNA Methylation in Plant Development and Abiotic Stress Responses. Front. Plant Sci. 2019, 10, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, S.J.; Kramer, M.C.; Gosai, S.J.; Yu, X.; Vandivier, L.E.; Nelson, A.D.; Anderson, Z.D.; Beilstein, M.A.; Fray, R.G.; Lyons, E.; et al. N6-Methyladenosine Inhibits Local Ribonucleolytic Cleavage to Stabilize mRNAs in Arabidopsis. Cell Rep. 2018, 25, 1146–1157. [Google Scholar] [CrossRef] [Green Version]
- Kramer, M.C.; Janssen, K.A.; Palos, K.; Nelson, A.D.L.; Vandivier, L.E.; Garcia, B.A.; Lyons, E.; Beilstein, M.A.; Gregory, B.D. N 6 -methyladenosine and RNA secondary structure affect transcript stability and protein abundance during systemic salt stress in Arabidopsis. Plant Direct 2020, 4, e00239. [Google Scholar] [CrossRef] [PubMed]
- Ok, S.H.; Jeong, H.J.; Bae, J.M.; Shin, J.-S.; Luan, S.; Kim, K.-N. Novel CIPK1-Associated Proteins in Arabidopsis Contain an Evolutionarily Conserved C-Terminal Region That Mediates Nuclear Localization. Plant Physiol. 2005, 139, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Scutenaire, J.; Deragon, J.-M.; Jean, V.; Benhamed, M.; Raynaud, C.; Favory, J.-J.; Merret, R.; Bousquet-Antonelli, C. The YTH Domain Protein ECT2 Is an m6A Reader Required for Normal Trichome Branching in Arabidopsis. Plant Cell 2018, 30, 986–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taudt, A.; Tatche, M.C.; Johannes, F. Genetic sources of population epigenomic variation. Nat. Rev. Genet. 2016, 17, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, J.; Cao, X.; Song, X. Epigenetic Mutation of RAV6 Affects Leaf Angle and Seed Size in Rice. Plant Physiol. 2015, 169, 2118–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, K.; Tor, M.; Poole, M.; Hong, Y.; Thompson, A.; King, G.; Giovannoni, J.J.; Seymour, G. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 2006, 38, 948–952. [Google Scholar] [CrossRef]
- He, L.; Wu, W.; Zinta, G.; Yang, L.; Wang, D.; Liu, R.; Zhang, H.; Zheng, Z.; Huang, H.; Zhang, Q.; et al. A naturally occurring epiallele associates with leaf senescence and local climate adaptation in Arabidopsis accessions. Nat. Commun. 2018, 9, 460. [Google Scholar] [CrossRef]
- Johannes, F.; Schmitz, R.J. Spontaneous epimutations in plants. N. Phytol. 2018, 221, 1253–1259. [Google Scholar] [CrossRef]
- Cubas, P.; Vincent, C.; Coen, E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature 1999, 401, 157–161. [Google Scholar] [CrossRef]
- Luff, B.; Pawlowski, L.; Bender, J. An Inverted Repeat Triggers Cytosine Methylation of Identical Sequences in Arabidopsis. Mol. Cell 1999, 3, 505–511. [Google Scholar] [CrossRef]
- Stuart, T.; Eichten, S.R.; Cahn, J.; Karpievitch, Y.V.; Borevitz, J.O.; Lister, R. Population scale mapping of transposable element diversity reveals links to gene regulation and epigenomic variation. eLife 2016, 5, e20777. [Google Scholar] [CrossRef] [PubMed]
- Hollick, J.B. Paramutation and related phenomena in diverse species. Nat. Rev. Genet. 2016, 18, 5–23. [Google Scholar] [CrossRef]
- Denkena, J.; Johannes, F.; Colomé-Tatché, M. Region-level epimutation rates in Arabidopsis thaliana. Heredity 2021, 127, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.J.; Schultz, M.D.; Lewsey, M.G.; O’Malley, R.C.; Urich, M.A.; Libiger, O.; Schork, N.J.; Ecker, J.R. Transgenerational Epigenetic Instability Is a Source of Novel Methylation Variants. Science 2011, 334, 369–373. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Purugganan, M.D. Evolutionary Epigenomics of Retrotransposon-Mediated Methylation Spreading in Rice. Mol. Biol. Evol. 2018, 35, 365–382. [Google Scholar] [CrossRef] [Green Version]
- Hofmeister, B.T.; Denkena, J.; Colomé-Tatché, M.; Shahryary, Y.; Hazarika, R.; Grimwood, J.; Mamidi, S.; Jenkins, J.; Grabowski, P.P.; Sreedasyam, A.; et al. A genome assembly and the somatic genetic and epigenetic mutation rate in a wild long-lived perennial Populus trichocarpa. Genome Biol. 2020, 21, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef]
- Fraga, H.P.F.; Vieira, L.N.; Caprestano, C.A.; Steinmacher, D.A.; Micke, G.A.; Spudeit, D.A.; Pescador, R.; Guerra, M.P. 5-Azacytidine combined with 2,4-D improves somatic embryogenesis of Acca sellowiana (O. Berg) Burret by means of changes in global DNA methylation levels. Plant Cell Rep. 2012, 31, 2165–2176. [Google Scholar] [CrossRef]
- Cheng, Y.-H.; Peng, X.-Y.; Yu, Y.-C.; Sun, Z.-Y.; Han, L. The Effects of DNA Methylation Inhibition on Flower Development in the Dioecious Plant Salix Viminalis. Forests 2019, 10, 173. [Google Scholar] [CrossRef] [Green Version]
- Konečná, K.; Sováková, P.P.; Anteková, K.; Fajkus, J.; Fojtová, M. Distinct Responses of Arabidopsis Telomeres and Transposable Elements to Zebularine Exposure. Int. J. Mol. Sci. 2021, 22, 468. [Google Scholar] [CrossRef]
- Yamagishi, K.; Kikuta, Y. Nucleoside derivatives of 5-methylcytosine suppress 5-azacytidine-induced reactivation of a silent transgene in suspension-cultured tobacco cells. Plant Biotechnol. 2021, 38, 173–178. [Google Scholar] [CrossRef]
- González, A.P.R.; Preite, V.; Verhoeven, K.J.F.; Latzel, V. Transgenerational Effects and Epigenetic Memory in the Clonal Plant Trifolium repens. Front. Plant Sci. 2018, 9, 1677. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowska, B.; Botor, M.; Morończyk, J.; Wójcik, A.; Nodzynski, T.; Karcz, J.; Gaj, M.D. Trichostatin A Triggers an Embryogenic Transition in Arabidopsis Explants via an Auxin-Related Pathway. Front. Plant Sci. 2018, 9, 1353. [Google Scholar] [CrossRef]
- Reinders, J.; Wulff, B.B.; Mirouze, M.; Marí-Ordóñez, A.; Dapp, M.; Rozhon, W.; Bucher, E.; Theiler, G.; Paszkowski, J. Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev. 2009, 23, 939–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Li, N.; Xu, C.; Zhong, S.; Lin, X.; Yang, J.; Zhou, T.; Yuliang, A.; Wu, Y.; Chen, Y.-R.; et al. Mutation of a major CG methylase in rice causes genome-wide hypomethylation, dysregulated genome expression, and seedling lethality. Proc. Natl. Acad. Sci. USA 2014, 111, 10642–10647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Eichten, S.R.; Hermanson, P.J.; Zaunbrecher, V.M.; Song, J.; Wendt, J.; Rosenbaum, H.; Madzima, T.F.; Sloan, A.E.; Huang, J.; et al. Genetic Perturbation of the Maize Methylome. Plant Cell 2014, 26, 4602–4616. [Google Scholar] [CrossRef] [Green Version]
- Springer, N.M.; Schmitz, R. Exploiting induced and natural epigenetic variation for crop improvement. Nat. Rev. Genet. 2017, 18, 563–575. [Google Scholar] [CrossRef]
- Stroud, H.; Ding, B.A.; Simon, S.; Feng, S.; Bellizzi, M.; Pellegrini, M.; Wang, G.-L.; Meyers, B.E.; Jacobsen, S. Plants regenerated from tissue culture contain stable epigenome changes in rice. eLife 2013, 2, e00354. [Google Scholar] [CrossRef]
- Han, Z.; Crisp, P.; Stelpflug, S.; Kaeppler, S.M.; Li, Q.; Springer, N.M. Heritable Epigenomic Changes to the Maize Methylome Resulting from Tissue Culture. Genetics 2018, 209, 983–995. [Google Scholar] [CrossRef]
- Ong-Abdullah, M.; Ordway, J.M.; Jiang, N.; Ooi, S.-E.; Kok, S.-Y.; Sarpan, N.; Azimi, N.; Hashim, A.T.; Ishak, Z.; Rosli, S.K.; et al. Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature 2015, 525, 533–537. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Gao, D.; Zhang, R.; Zeng, G.; Yan, H.; Lim, E.; Liang, F.-S. Chemically Controlled Epigenome Editing through an Inducible dCas9 System. J. Am. Chem. Soc. 2017, 139, 11337–11340. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Wang, Y.; Liang, F.-S. Chemical and Light Inducible Epigenome Editing. Int. J. Mol. Sci. 2020, 21, 998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soppe, W.J.J.; Jacobsen, S.E.; Alonso-Blanco, C.; Jackson, J.P.; Kakutani, T.; Koornneef, M.; Peeters, A.J.M. The Late Flowering Phenotype of fwa Mutants Is Caused by Gain-of-Function Epigenetic Alleles of a Homeodomain Gene. Mol. Cell 2000, 6, 791–802. [Google Scholar] [CrossRef]
- Johnson, L.M.; Du, J.; Hale, C.J.; Bischof, S.; Feng, S.; Chodavarapu, R.K.; Zhong, X.; Marson, G.; Pellegrini, M.; Segal, D.J.; et al. SRA- and SET-domain-containing proteins link RNA polymerase V occupancy to DNA methylation. Nature 2014, 507, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallego-Bartolome, J.; Liu, W.; Kuo, P.H.; Feng, S.; Ghoshal, B.; Gardiner, J.; Zhao, J.M.-C.; Park, S.Y.; Chory, J.; Jacobsen, S.E. Co-targeting RNA Polymerases IV and V Promotes Efficient De Novo DNA Methylation in Arabidopsis. Cell 2019, 176, 1068–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallego-Bartolomé, J.; Gardiner, J.; Liu, W.; Papikian, A.; Ghoshal, B.; Kuo, H.Y.; Zhao, J.M.-C.; Segal, D.J.; Jacobsen, S.E. Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc. Natl. Acad. Sci. USA 2018, 115, E2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papikian, A.; Liu, W.; Gallego-Bartolomé, J.; Jacobsen, S.E. Site-specific manipulation of Arabidopsis loci using CRISPR-Cas9 SunTag systems. Nat. Commun. 2019, 10, 729. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Wong, C.E.; Shen, L.; Yu, H. N6-methyladenosine modification underlies messenger RNA metabolism and plant development. Curr. Opin. Plant Biol. 2021, 63, 102047. [Google Scholar] [CrossRef]
- Zhou, L.; Tang, R.; Li, X.; Tian, S.; Li, B.; Qin, G. N6-methyladenosine RNA modification regulates strawberry fruit ripening in an ABA-dependent manner. Genome Biol. 2021, 22, 168. [Google Scholar] [CrossRef]
- Tang, Y.; Gao, C.-C.; Gao, Y.; Yang, Y.; Shi, B.; Yu, J.-L.; Lyu, C.; Sun, B.-F.; Wang, H.-L.; Xu, Y.; et al. OsNSUN2-Mediated 5-Methylcytosine mRNA Modification Enhances Rice Adaptation to High Temperature. Dev. Cell 2020, 53, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cai, J.; Park, S.J.; Lee, K.; Li, Y.; Chen, Y.; Yun, J.; Xu, T.; Kang, H. N 6 -Methyladenosine mRNA methylation is important for salt stress tolerance in Arabidopsis. Plant J. 2021, 106, 1759–1775. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, Y.; He, Q.; Qi, Z.; Zhang, G.; Xu, W.; Yi, T.; Wu, G.; Li, R. MTA, an RNA m6A Methyltransferase, Enhances Drought Tolerance by Regulating the Development of Trichomes and Roots in Poplar. Int. J. Mol. Sci. 2020, 21, 2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Yu, H. Epitranscriptome engineering in crop improvement. Mol. Plant 2021, 14, 1418–1420. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Chen, P.J.; Miao, Z.; Liu, D.R. Programmable m6A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat. Biotechnol. 2020, 38, 1431–1440. [Google Scholar] [CrossRef]
- Kim, J.-S. Precision genome engineering through adenine and cytosine base editing. Nat. Plants 2018, 4, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Mulqueen, R.M.; Pokholok, D.; Norberg, S.J.; Torkenczy, K.A.; Fields, A.J.; Sun, D.; Sinnamon, J.R.; Shendure, J.; Trapnell, C.; O’Roak, B.J.; et al. Highly scalable generation of DNA methylation profiles in single cells. Nat. Biotechnol. 2018, 36, 428–431. [Google Scholar] [CrossRef]
- Ku, W.L.; Nakamura, K.; Gao, W.; Cui, K.; Hu, G.; Tang, Q.; Ni, B.; Zhao, K. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat. Methods 2019, 16, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, P.; Guo, W.; Liu, H.; Li, X.; Zhang, Q.; Du, Z.; Hu, G.; Han, X.; Pu, L.; et al. A deep learning approach to automate whole-genome prediction of diverse epigenomic modifications in plants. N. Phytol. 2021, 232, 880–897. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, Q.; Wan, X. Epigenome and Epitranscriptome: Potential Resources for Crop Improvement. Int. J. Mol. Sci. 2021, 22, 12912. https://doi.org/10.3390/ijms222312912
Hou Q, Wan X. Epigenome and Epitranscriptome: Potential Resources for Crop Improvement. International Journal of Molecular Sciences. 2021; 22(23):12912. https://doi.org/10.3390/ijms222312912
Chicago/Turabian StyleHou, Quancan, and Xiangyuan Wan. 2021. "Epigenome and Epitranscriptome: Potential Resources for Crop Improvement" International Journal of Molecular Sciences 22, no. 23: 12912. https://doi.org/10.3390/ijms222312912
APA StyleHou, Q., & Wan, X. (2021). Epigenome and Epitranscriptome: Potential Resources for Crop Improvement. International Journal of Molecular Sciences, 22(23), 12912. https://doi.org/10.3390/ijms222312912