
Drug-Induced Lysosomal Impairment Is Associated with the Release of Extracellular Vesicles Carrying Autophagy Markers

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
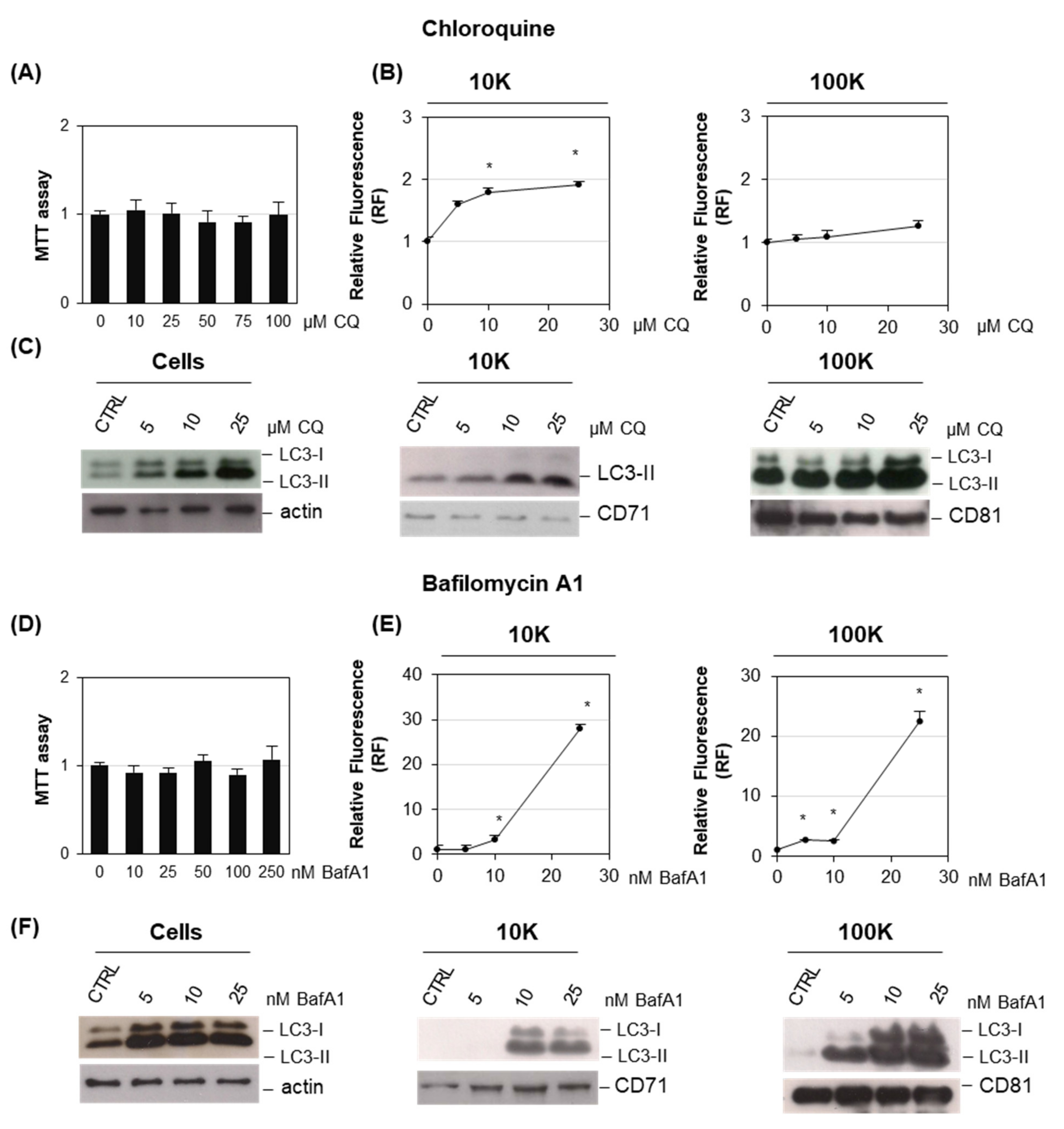
2. Results
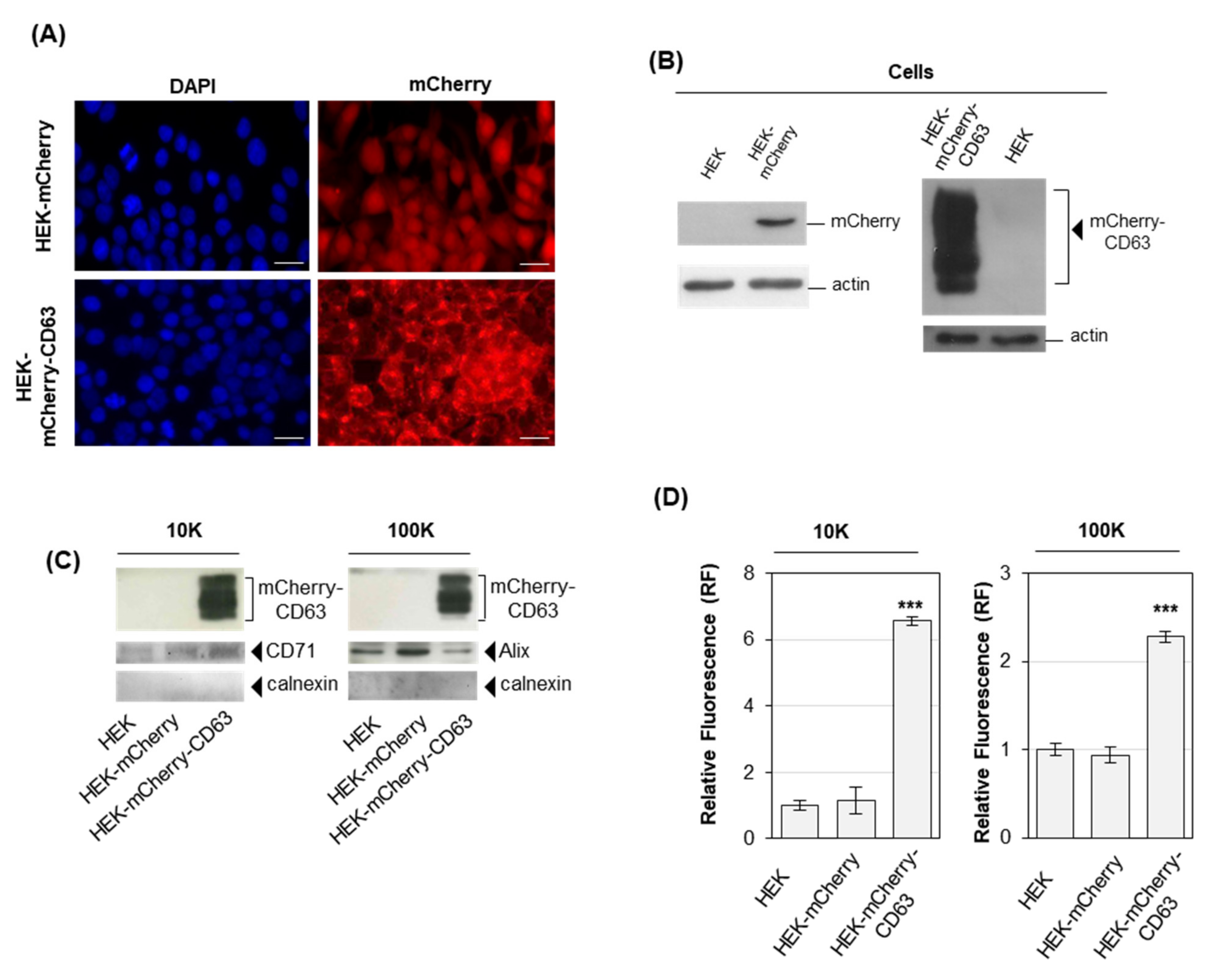
2.1. Cells Overexpressing Fluorescent Proteins Release Fluorescent EVs
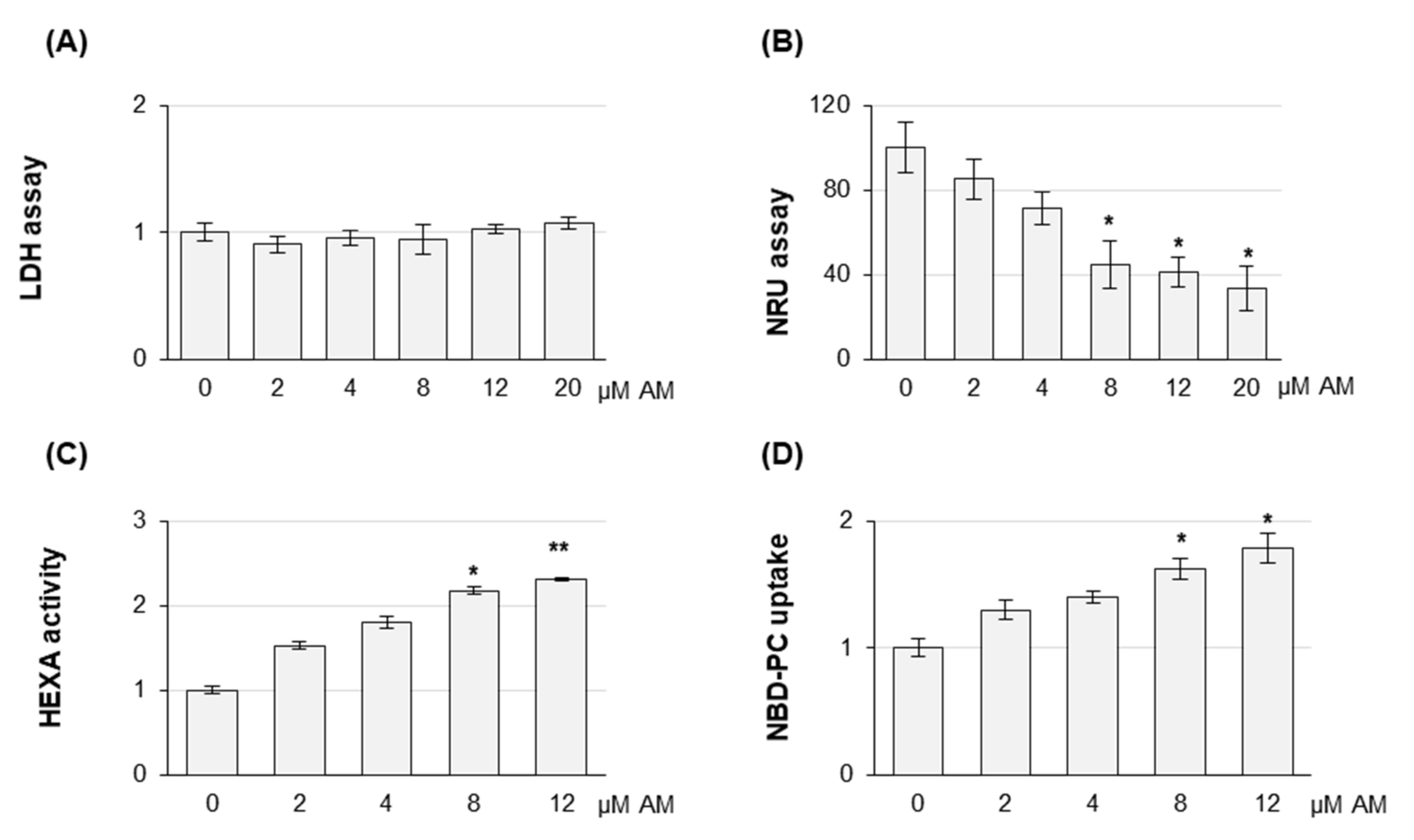
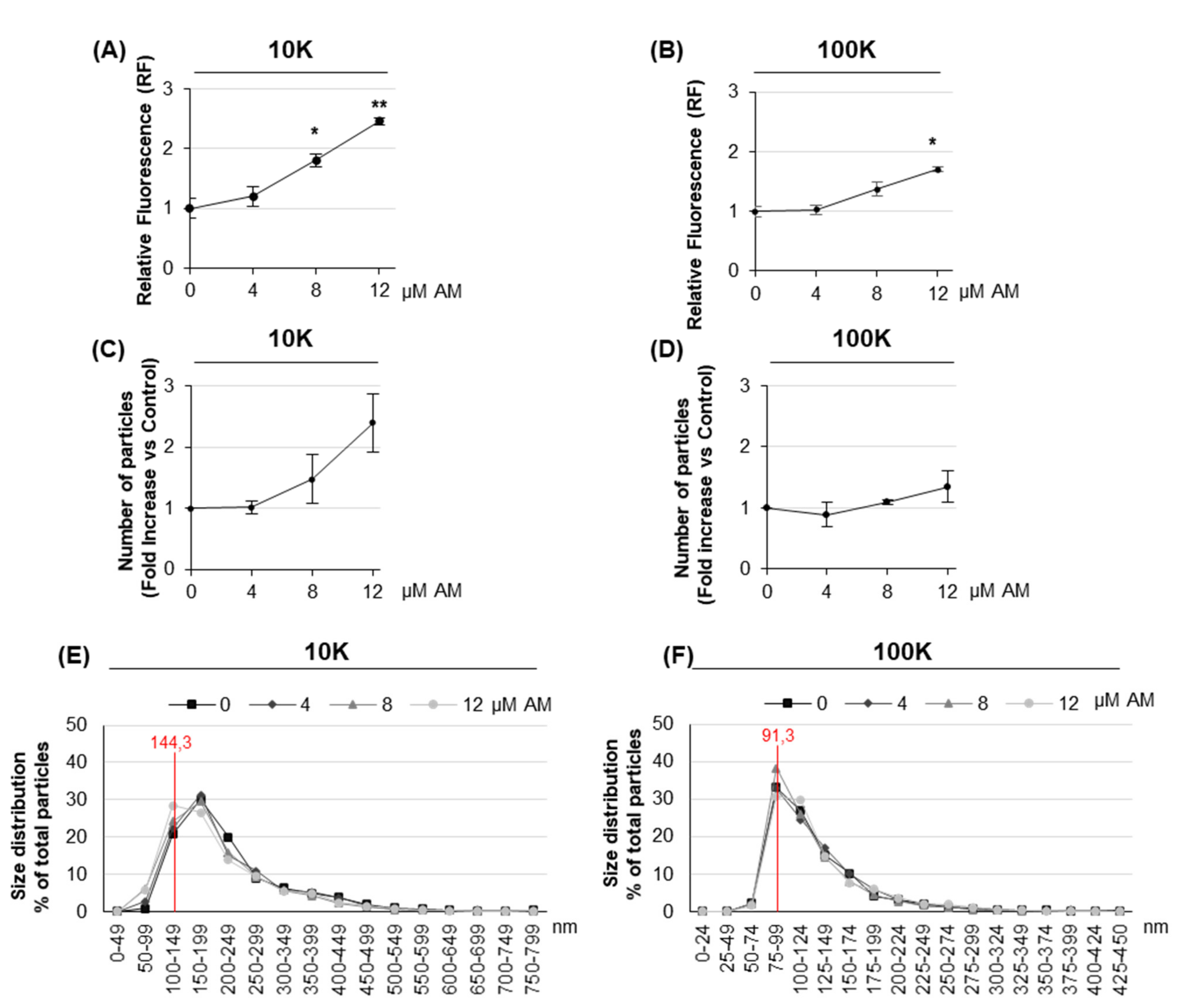
2.2. Amiodarone Affects the Release of Large/Medium and Small EVs in a Dose-Dependent Manner
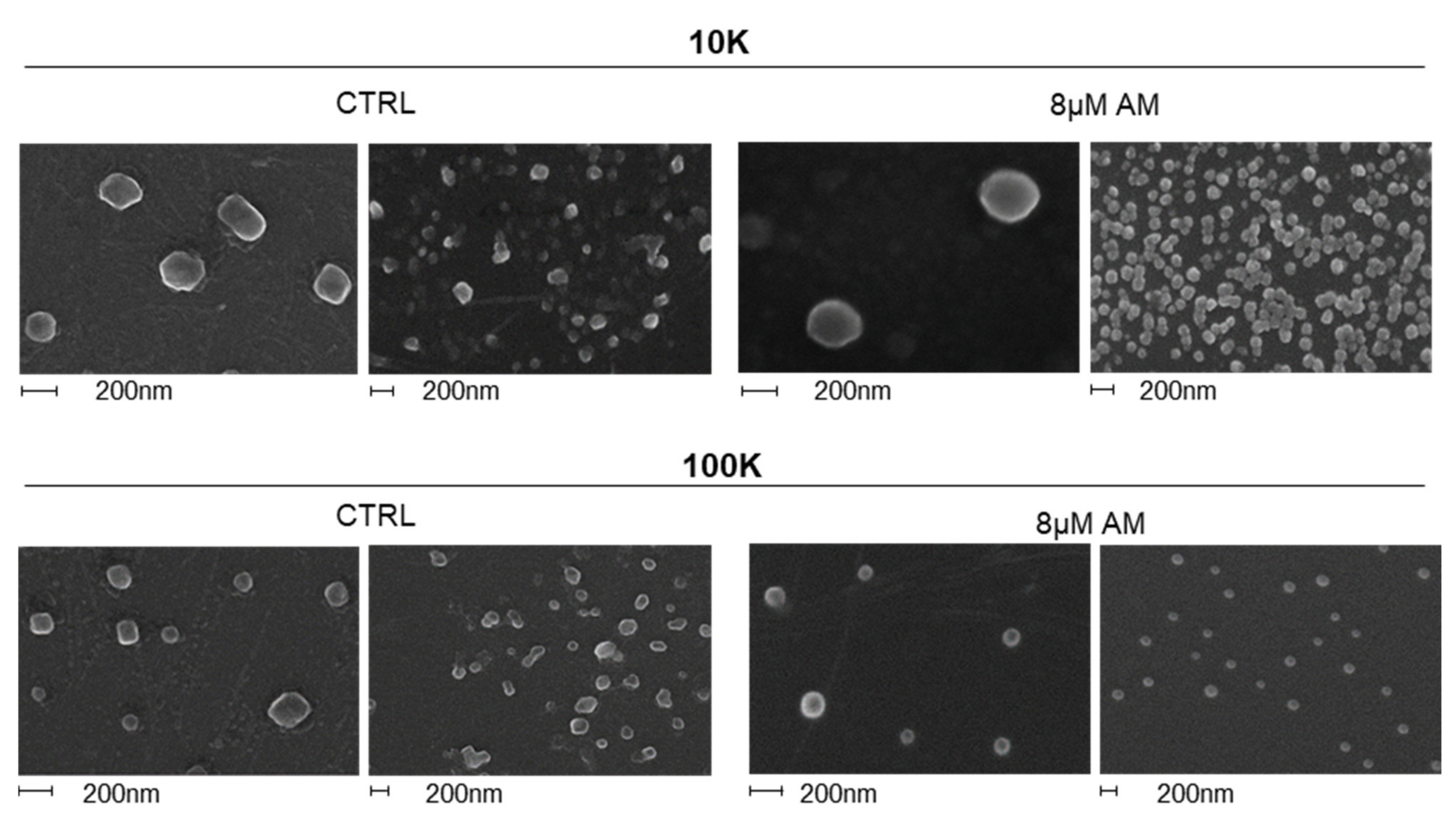
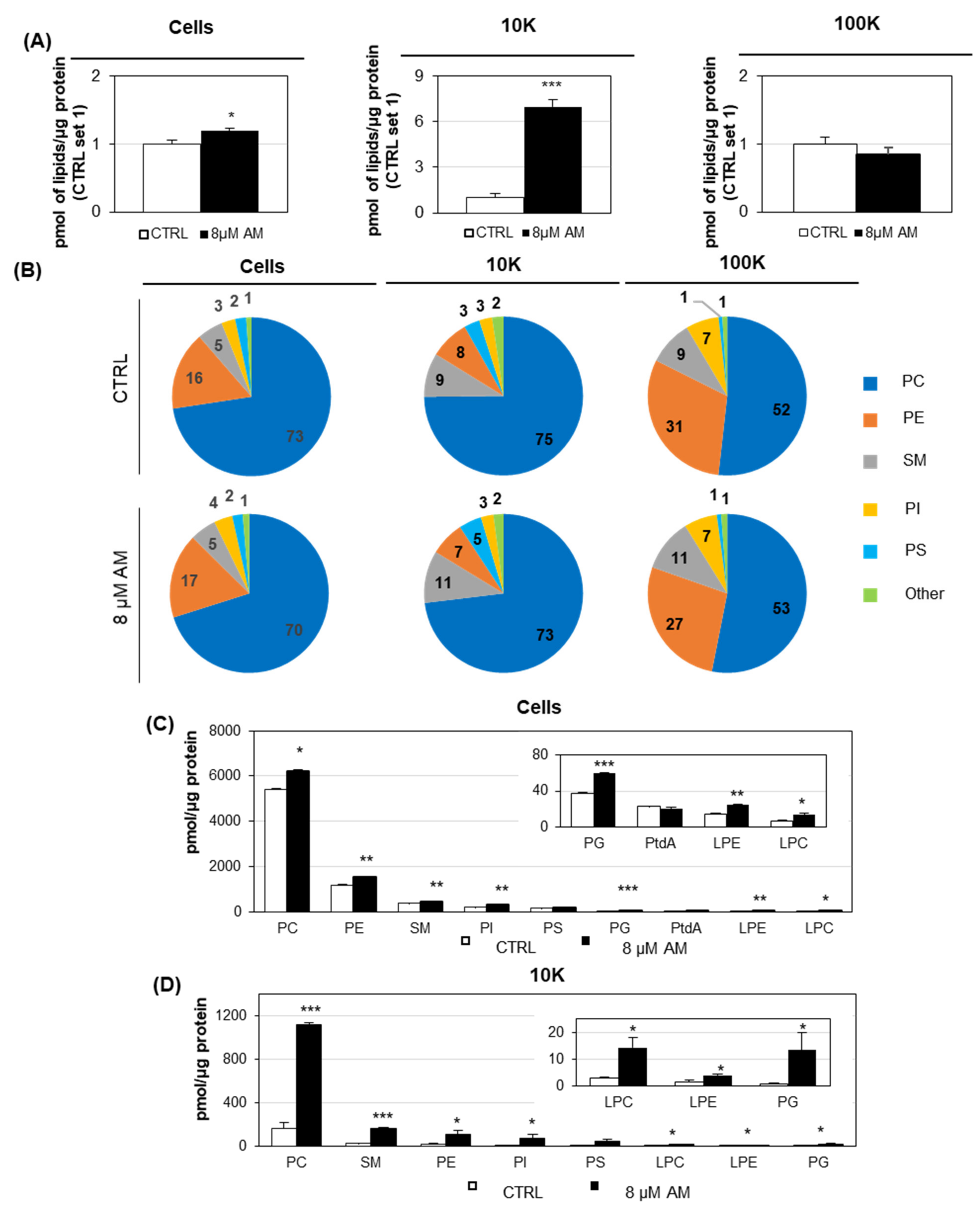
2.3. Amiodarone Modifies the Content of Large/Medium EVs
3. Discussion
4. Materials and Methods
4.1. Constructs
4.2. Cell Culture and Transfection
4.3. Extracellular Vesicle Purification
4.3.1. Extracellular Vesicle Fluorescence Measurement
4.3.2. Extracellular Vesicle Scanning Electron Microscopy
4.3.3. Extracellular Vesicle Nanoparticle Tracking Analysis
4.3.4. Extracellular Vesicles Proteinase K Protection Assay
4.4. LDH Assay
4.5. NRU Assay
4.6. Phospholipidosis Evaluation Using NBD-PC Uptake
4.7. Lysosomal β-Hexosaminidase A Assay
4.8. Fluorescence Microscopy
4.9. Immunoblotting
4.10. Cells Preparation for Phospholipid Profile
4.11. Phospholipid Profile by Liquid Chromatography-Tandem Mass Spectrometry (LC-MS)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, N.; Borlak, J. Drug-induced phospholipidosis. FEBS Lett. 2006, 580, 5533–5540. [Google Scholar] [CrossRef]
- Shayman, J.A.; Abe, A. Drug induced phospholipidosis: An acquired lysosomal storage disorder. Biochim. Biophys. Acta 2013, 1831, 602–611. [Google Scholar] [CrossRef] [Green Version]
- Nonoyama, T.; Fukuda, R. Drug induced phospholipidosis pathological aspects and its prediction. J. Toxicol. Pathol. 2008, 21, 9–24. [Google Scholar] [CrossRef] [Green Version]
- Abe, A.; Hiraoka, M.; Shayman, J.A. A role for lysosomal phospholipase A2 in drug induced phospholipidosis. Drug Metab. Lett. 2007, 1, 49–53. [Google Scholar] [CrossRef]
- Ikeda, K.; Hirayama, M.; Hirota, Y.; Asa, E.; Seki, J.; Tanaka, Y. Drug-induced phospholipidosis is caused by blockade of mannose 6-phosphate receptor-mediated targeting of lysosomal enzymes. Biochem. Biophys. Res. Commun. 2008, 377, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Goracci, L.; Buratta, S.; Urbanelli, L.; Ferrara, G.; Di Guida, R.; Emiliani, C.; Cross, S. Evaluating the risk of phospholipidosis using a new multidisciplinary pipeline approach. Eur. J. Med. Chem. 2015, 92, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Staudt, C.; Puissant, E.; Boonen, M. Subcellular Trafficking of Mammalian Lysosomal Proteins: An Extended View. Int. J. Mol. Sci. 2017, 18, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buratta, S.; Urbanelli, L.; Ferrara, G.; Sagini, K.; Goracci, L.; Emiliani, C. A role for the autophagy regulator Transcription Factor EB in amiodarone-induced phospholipidosis. Biochem. Pharmacol. 2015, 95, 201–209. [Google Scholar] [CrossRef]
- Urbanelli, L.; Magini, A.; Buratta, S.; Brozzi, A.; Sagini, K.; Polchi, A.; Tancini, B.; Emiliani, C. Signaling pathways in exosomes biogenesis, secretion and fate. Genes 2013, 4, 152–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanelli, L.; Buratta, S.; Sagini, K.; Ferrara, G.; Lanni, M.; Emiliani, C. Exosome-based strategies for Diagnosis and Therapy. Recent Pat. CNS Drug Discov. 2015, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, J.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, R.M. Maturation of reticulocytes: Formation of exosomes as a mechanism for shedding membrane proteins. Biochem. Cell Biol. 1992, 70, 179–190. [Google Scholar] [CrossRef]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [Green Version]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012, 12, 421. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Paine, M.S.; Brooks, A.M.; McCubrey, J.A.; Renegar, R.H.; Wang, R.; Terrian, D.M. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008, 68, 7864–7871. [Google Scholar] [CrossRef] [Green Version]
- Buratta, S.; Urbanelli, L.; Sagini, K.; Giovagnoli, S.; Caponi, S.; Fioretto, D.; Mitro, N.; Caruso, D.; Emiliani, C. Extracellular vesicles released by fibroblasts undergoing H-Ras induced senescence show changes in lipid profile. PLoS ONE 2017, 12, e0188840. [Google Scholar] [CrossRef] [PubMed]
- Crow, J.; Atay, S.; Banskota, S.; Artale, B.; Schmitt, S.; Godwin, A.K. Exosomes as mediators of platinum resistance in ovarian cancer. Oncotarget 2017, 8, 11917–11936. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, C.; Rani, S.; O’Brien, K.; O’Neill, A.; Prencipe, M.; Sheikh, R.; Webb, G.; McDermott, R.; Watson, W.; Crown, J.; et al. Docetaxel-resistance in prostate cancer: Evaluating associated phenotypic changes and potential for resistance transfer via exosomes. PLoS ONE 2012, 7, e50999. [Google Scholar]
- Vassallo, P.; Trohman, R.G. Prescribing amiodarone: An evidence-based review of clinical indications. J. Am. Med. Assoc. 2007, 298, 1312–1322. [Google Scholar] [CrossRef]
- Sawada, H.; Takami, K.; Asahi, S. A toxicogenomic approach to drug-induced phospholipidosis: Analysis of its induction mechanism and establishment of a novel in vitro screening system. Toxicol. Sci. 2005, 83, 282–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccoli, E.; Nadai, M.; Caretta, C.M.; Bergonzini, V.; Del Vecchio, C.; Ha, H.R.; Bigler, L.; Dal Zoppo, D.; Faggin, E.; Pettenazzo, A. Amiodarone impairs trafficking through late endosomes inducing a Niemann-Pick C-like phenotype. Biochem. Pharmacol. 2011, 82, 1234–1249. [Google Scholar] [CrossRef] [PubMed]
- Pisonero-Vaquero, S.; Medina, D.L. Lysosomotropic Drugs: Pharmacological Tools to Study Lysosomal Function. Curr. Drug Metab. 2017, 18, 1147–1158. [Google Scholar] [CrossRef]
- Tancini, B.; Buratta, S.; Sagini, K.; Costanzi, E.; Delo, F.; Urbanelli, L.; Emiliani, C. Insight into the Role of Extracellular Vesicles in Lysosomal Storage Disorders. Genes 2019, 10, 510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eitan, E.; Suire, C.; Zhang, S.; Mattson, M.P. Impact of lysosome status on extracellular vesicle content and release. Ageing Res. Rev. 2016, 32, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baixauli, F.; López-Otín, C.; Mittelbrunn, M. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Camfield, R.; Gorski, S.M. The interplay between exosomes and autophagy—Partners in crime. J. Cell Sci. 2018, 131, jcs215210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal exocytosis, exosome release and secretory autophagy: The autophagic- and endo-lysosomal systems go extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef] [Green Version]
- Strauss, K.; Goebel, C.; Runz, H.; Möbius, W.; Weiss, S.; Feussner, I.; Simons, M.; Schneider, A. Exosome secretion ameliorates lysosomal storage of cholesterol in Niemann-Pick type C disease. J. Biol. Chem. 2010, 285, 26279–26288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canfrán-Duque, A.; Pastor, O.; Reina, M.; Lerma, M.; Cruz-Jentoft, A.J.; Lasunción, M.A.; Busto, R. Curcumin Mitigates the Intracellular Lipid Deposit Induced by Antipsychotics In Vitro. PLoS ONE 2015, 10, e0141829. [Google Scholar]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.G.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Pols, M.S.; Klumperman, J. Trafficking and function of the tetraspanin CD63. Exp. Cell Res. 2009, 315, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Cobbs, A.; Chen, X.; Zhang, Y.; George, J.; Huang, M.B.; Bond, V.; Thompson, W.; Zhao, X. Saturated fatty acid stimulates production of extracellular vesicles by renal tubular epithelial cells. Mol. Cell. Biochem. 2019, 458, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Buratta, S.; Shimanaka, Y.; Costanzi, E.; Ni, S.; Urbanelli, L.; Kono, N.; Morena, F.; Sagini, S.; Giovagnoli, S.; Romani, R.; et al. Lipotoxic stress alters the membrane lipid profile of extracellular vesicles released by Huh-7 hepatocarcinoma cells. Sci. Rep. 2021, 11, 4613. [Google Scholar] [CrossRef] [PubMed]
- Bissig, C.; Gruenberg, J. ALIX and the multivesicular endosome: ALIX in Wonderland. Trends Cell Biol. 2014, 24, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Gonda, A.; Kabagwira, J.; Senthil, G.N.; Wall, N.R. Internalization of Exosomes through Receptor-Mediated Endocytosis. Mol. Cancer Res. 2019, 17, 337–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Mauvezin, C.; Nagy, P.; Juhász, G.; Neufeld, T.P. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 2015, 6, 7007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morissette, G.; Ammoury, A.; Rusu, D.; Marguery, M.C.; Lodge, R.; Poubelle, P.E.; Marceau, F. Intracellular sequestration of amiodarone: Role of vacuolar ATPase and macroautophagic transition of the resulting vacuolar cytopathology. Br. J. Pharmacol. 2009, 157, 1531–1540. [Google Scholar] [CrossRef] [Green Version]
- Rabia, M.; Leuzy, V.; Soulage, C.; Durand, A.; Fourmaux, B.; Errazuriz-Cerda, E.; Köffel, R.; Draeger, A.; Colosetti, P.; Jalabert, A.; et al. Bis(monoacylglycero)phosphate, a new lipid signature of endosome-derived extracellular vesicles. Biochimie 2020, 178, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.D.; Fang, Y.T.; Cheng, Y.L.; Lin, C.F.; Hsu, L.J.; Wang, S.Y.; Anderson, R.; Chang, C.P.; Lin, Y.S. Exophagy of annexin A2 via RAB11, RAB8A and RAB27A in IFN-γ-stimulated lung epithelial cells. Sci. Rep. 2017, 7, 5676. [Google Scholar] [CrossRef]
- Marques, A.R.A.; Saftig, P. Lysosomal Storage Disorders—Challenges, Concepts and Avenues for Therapy: Beyond Rare Diseases. J. Cell Sci. 2019, 132, jcs221739. [Google Scholar] [CrossRef]
- Lee, H.H.; Elia, N.; Ghirlando, R.; Lippincott-Schwartz, J.; Hurley, J.H. Midbody targeting of the ESCRT machinery by a noncanonical coiled coil in CEP55. Science 2008, 322, 576–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borenfreund, E.; Puerner, J.A. Toxicity determined in vitro by morphological alterations and neutral red absorption. Toxicol. Lett. 1985, 24, 119–124. [Google Scholar] [CrossRef]
- Kasahara, T.; Tomita, K.; Murano, H.; Harada, T.; Tsubakimoto, K.; Ogihara, T.; Ohnishi, S.; Kakinuma, C. Establishment of an in vitro high-throughput screening assay for detecting phospholipidosis-inducing potential. Toxicol. Sci. 2006, 90, 133–141. [Google Scholar] [CrossRef]
- Urbanelli, L.; Magini, A.; Ercolani, L.; Sagini, K.; Polchi, A.; Tancini, B.; Brozzi, A.; Armeni, T.; Principato, G.; Emiliani, C. Oncogenic H-Ras up-regulates acid β-hexosaminidase by a mechanism dependent on the autophagy regulator TFEB. PLoS ONE 2014, 9, e89485. [Google Scholar] [CrossRef] [PubMed]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sagini, K.; Buratta, S.; Delo, F.; Pellegrino, R.M.; Giovagnoli, S.; Urbanelli, L.; Emiliani, C. Drug-Induced Lysosomal Impairment Is Associated with the Release of Extracellular Vesicles Carrying Autophagy Markers. Int. J. Mol. Sci. 2021, 22, 12922. https://doi.org/10.3390/ijms222312922
Sagini K, Buratta S, Delo F, Pellegrino RM, Giovagnoli S, Urbanelli L, Emiliani C. Drug-Induced Lysosomal Impairment Is Associated with the Release of Extracellular Vesicles Carrying Autophagy Markers. International Journal of Molecular Sciences. 2021; 22(23):12922. https://doi.org/10.3390/ijms222312922
Chicago/Turabian StyleSagini, Krizia, Sandra Buratta, Federica Delo, Roberto Maria Pellegrino, Stefano Giovagnoli, Lorena Urbanelli, and Carla Emiliani. 2021. "Drug-Induced Lysosomal Impairment Is Associated with the Release of Extracellular Vesicles Carrying Autophagy Markers" International Journal of Molecular Sciences 22, no. 23: 12922. https://doi.org/10.3390/ijms222312922