
Genetic Variants of the NF-κB Pathway: Unraveling the Genetic Architecture of Psoriatic Disease



Abstract
:1. Introduction
2. The NF-κB Signaling Pathway
3. Role in the Etiology of Psoriatic Disease
4. NF-κB Pathway Variants and Drug Response
5. NF-κB Genetic Variants and Cardiometabolic Comorbidity
6. Conclusions
Funding
Conflicts of Interest
References
- Queiro, R.; Lorenzo, A.; Pardo, E.; Cañete, J.D. Psoriatic arthritis: A current vision. In Encyclopedia of Biomedical Gerontology; Rattan, S.I.S., Ed.; Elsevier, Academic Press: Cambridge, MA, USA, 2020; Volume 3, pp. 105–119. [Google Scholar]
- Pérez, A.R.; Queiro, R.; Seoane-Mato, D.; Graell, E.; Chamizo, E.; Chaparro, L.C.; Herrera, S.R.; Dolset, J.P.; Ostáriz, M.A.P.; Garrido, S.R.-A.; et al. Higher prevalence of Psoriatic arthritis in the adult population in Spain? A population-based cross-sectional study. PLoS ONE 2020, 15, e0234556. [Google Scholar]
- Perez-Chada, L.M.; Merola, J.F. Comorbidities associated with Psoriatic arthritis: Review and update. Clin. Immunol. 2020, 214, 108397. [Google Scholar] [CrossRef]
- Polachek, A.; Touma, Z.; Anderson, M.; Eder, L. Risk of cardiovascular morbidity in patients with psoriatic arthritis: A meta-analysis of observational studies. Arthritis Care Res. 2017, 69, 67–74. [Google Scholar] [CrossRef]
- Gracey, E.; Vereecke, L.; McGovern, D.; Fröhling, M.; Schett, G.; Danese, S.; De Vos, M.; Bosch, F.V.D.; Elewaut, D. Revisiting the gut-joint axis: Links between gut inflammation and spondyloarthritis. Nat. Rev. Rheumatol. 2020, 16, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Lonnberg, A.S.; Skov, L.; Skytthe, A.; Kyvik, K.O.; Pedersen, O.B.; Thomsen, S.F. Heritability of Psoriasis in a large twin sample. Br. J. Dermatol. 2013, 169, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, S.; Li, Q.; Rahman, P.; Chandran, V. Insights into the pathogenesis of Psoriatic arthritis from genetic studies. Semin. Immunopathol. 2021, 43, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, K.; Okada, Y. The current landscape of Psoriasis genetics in 2020. J. Dermatol. Sci. 2020, 99, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Goldminz, A.M.; Au, S.C.; Kim, N.; Gottlieb, A.B.; Lizzul, P.F. NF-κB: An essential transcription factor in Psoriasis. J. Dermatol. Sci. 2013, 69, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Rebholz, B.; Haase, I.; Eckelt, B.; Paxian, S.; Flaig, M.J.; Ghoreschi, K.; Nedospasov, S.A.; Mailhammer, R.; Debey-Pascher, S.; Schultze, J.; et al. Crosstalk between keratinocytes and adaptive immune cells in an IκBα protein-mediated inflammatory disease of the skin. Immunity 2007, 27, 296–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizzul, P.F.; Aphale, A.; Malaviya, R.; Sun, Y.; Masud, S.; Dombrovskiy, V.; Gottlieb, A.B. Differential expression of phosphorylated NF-κB/RelA in normal and psoriatic epidermis and downregulation of NF-κB in response to treatment with etanercept. J. Invest. Dermatol. 2005, 124, 1275–1283. [Google Scholar] [CrossRef] [Green Version]
- Howes, A.; O’Sullivan, P.A.; Breyer, F.; Ghose, A.; Cao, L.; Krappmann, D.; Bowcock, A.M.; Ley, S.C. Psoriasis mutations disrupt CARD14 autoinhibition promoting BCL10-MALT1-dependent NF-kappaB activation. Biochem. J. 2016, 473, 1759–1768. [Google Scholar] [CrossRef]
- Mellett, M.; Meier-Schiesser, B.; Mohanan, D.; Schairer, R.; Cheng, P.; Satoh, T.; Kiefer, B.; Ospelt, C.; Nobbe, S.; Thome, M.; et al. CARD14 gain-of-function mutation alone is sufficient to drive IL-23/IL-17—mediated psoriasiform skin inflammation in vivo. J. Invest. Dermatol. 2018, 138, 2010–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, C.T.; Cao, L.; Roberson, E.; Pierson, K.C.; Yang, C.-F.; Joyce, C.E.; Ryan, C.; Duan, S.; Helms, C.A.; Liu, Y.; et al. PSORS2 is due to mutations in CARD14. Am. J. Hum. Genet. 2012, 90, 784–795. [Google Scholar] [CrossRef] [Green Version]
- Jordan, C.T.; Cao, L.; Roberson, E.; Duan, S.; Helms, C.A.; Nair, R.P.; Duffin, K.C.; Stuart, P.E.; Goldgar, D.; Hayashi, G.; et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in Psoriasis. Am. J. Hum. Genet. 2012, 90, 796–808. [Google Scholar] [PubMed] [Green Version]
- Tsoi, L.C.; Spain, S.L.; Knight, J.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Ding, J.; Li, Y.; Tejasvi, T.; Gudjonsson, J.E.; et al. Identification of 15 new Psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012, 44, 1341–1348. [Google Scholar] [PubMed] [Green Version]
- Shi, G.; Li, S.J.; Wang, T.T.; Cheng, C.M.; Fan, Y.M.; Zhu, K.J. The common CARD14 gene missense polymorphism rs11652075 (c.C2458T/p.Arg820Trp) is associated with Psoriasis: A meta-analysis. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef]
- González-Lara, L.; Coto-Segura, P.; Penedo, A.; Eiris, N.; Díaz, M.; Santos-Juanes, J.; Queiro, R.; Coto, E. SNP rs11652075 in the CARD14 gene as a risk factor for Psoriasis (PSORS2) in a Spanish cohort. DNA Cell Biol. 2013, 32, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Morita, C.; Horiuchi, T.; Tsukamoto, H.; Hatta, N.; Kikuchi, Y.; Arinobu, Y.; Otsuka, T.; Sawabe, T.; Harashima, S.I.; Nagasawa, K.; et al. Association of tumor necrosis factor receptor type II polymorphism 196R with systemic lupus erythematosus in the Japanese: Molecular and functional analysis. Arthritis Rheum. 2001, 44, 2819–2827. [Google Scholar] [CrossRef]
- Tolusso, B.; Sacco, S.; Gremese, E.; La Torre, G.; Tomietto, P.; Ferraccioli, G.F. Relationship between the tumor necrosis factor receptor II (TNF-RII) gene polymorphism and sTNF-RII plasma levels in healthy controls and in rheumatoid arthritis. Hum. Immunol. 2004, 65, 1420–1426. [Google Scholar] [CrossRef] [PubMed]
- González-Lara, L.; Batalla, A.; Coto, E.; Gómez, J.; Eiris, N.; Santos-Juanes, J.; Queiro, R.; Coto-Segura, P. The TNFRSF1B rs1061622 polymorphism (p.M196R) is associated with biological drug outcome in Psoriasis patients. Arch. Dermatol. Res. 2015, 307, 405–412. [Google Scholar]
- Karban, A.S.; Okazaki, T.; Panhuysen, C.I.; Gallegos, T.; Potter, J.J.; Bailey-Wilson, J.E.; Silverberg, M.S.; Duerr, R.H.; Cho, J.H.; Gregersen, P.K.; et al. Functional annotation of a novel NFKB1 promoter polymorphism that increases risk for ulcerative colitis. Hum. Mol. Genet. 2004, 13, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Jin, X.; Xu, J.; Gao, J.; Du, X.; Duan, D.; Li, B.; Zhao, J.; Zhan, W.; Tang, H.; et al. Sequencing-based approach identified three new susceptibility loci for Psoriasis. Nat. Commun. 2014, 5, 4331. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhu, Z.; Zhu, C.; Zheng, X.; Zuo, X.; Chen, G.; Zhou, F.; Liang, B.; Tang, H.; Wang, Z.; et al. A genetic variant rs1020760at NFKB1 is associated with clinical features of Psoriasis vulgaris in a Han Chinese population. Ann. Hum. Genet. 2016, 80, 197–202. [Google Scholar] [CrossRef]
- González-Lara, L. Genetic Variants within the TNF-Alpha/TNFRSF1A, B/CARD14/NFKB Signaling Network and Psoriasis Risk. Ph.D. Thesis, Oviedo University, Oviedo, Spain, 2018. [Google Scholar]
- Seidi, A.; Mirzaahmadi, S.; Mahmoodi, K.; Soleiman-Soltanpour, M. The association between NFKB1 -94ATTG ins/del and NFKB1A 826C/T genetic variations and coronary artery disease risk. Mol. Biol. Res. Commun. 2018, 7, 17–24. [Google Scholar]
- Hong, M.; Ye, B.D.; Yang, S.-K.; Jung, S.; Lee, H.-S.; Kim, B.M.; Bin Lee, S.; Hong, J.; Baek, J.; Park, S.H.; et al. Immunochip meta-analysis of inflammatory Bowel disease identifies three novel loci and four novel associations in previously reported loci. J. Crohns Colitis 2018, 12, 730–741. [Google Scholar] [PubMed]
- Stuart, P.E.; Nair, R.P.; Ellinghaus, E.; Ding, J.; Tejasvi, T.; Gudjonsson, S.A.; Li, Y.; Weidinger, S.; Eberlein, B.; Gieger, C.; et al. Genome-wide association analysis identifies three Psoriasis susceptibility loci. Nat. Genet. 2010, 42, 1000–1004. [Google Scholar] [CrossRef]
- Capon, F.; Spencer, C.C.; Knight, J.; Weale, M.E.; Allen, M.H.; Barton, A.; Band, G.; Bellenguez, C.; Bergboer, J.G.; Palmer, C.N.; et al. A genome-wide association study identifies new Psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010, 42, 985–990. [Google Scholar]
- Zuo, X.; Sun, L.; Yin, X.; Gao, J.; Sheng, Y.; Xu, J.; Zhang, J.; He, C.; Qiu, Y.; Wen, G.; et al. Whole-exome SNP array identifies 15 new susceptibility loci for Psoriasis. Nat. Commun. 2015, 6, 6793. [Google Scholar] [CrossRef]
- Coto-Segura, P.; Coto, E.; González-Lara, L.; Alonso, B.; Gómez, J.; Cuesta-Llavona, E.; Queiro, R. Gene variant in the NF-κB pathway inhibitor NFKBIA distinguishes patients with Psoriatic arthritis within the spectrum of Psoriatic disease. Biomed. Res. Int. 2019, 2019, 1030256. [Google Scholar] [CrossRef] [Green Version]
- Stuart, P.E.; Nair, R.P.; Tsoi, L.C.; Tejasvi, T.; Das, S.; Kang, H.M.; Ellinghaus, E.; Chandran, V.; Callis-Duffin, K.; Ike, R.; et al. Genome-wide association analysis of Psoriatic arthritis and Cutaneous Psoriasis reveals differences in their genetic architecture. Am. J. Hum. Genet. 2015, 97, 816–836. [Google Scholar] [CrossRef] [Green Version]
- Johansen, C. IκBζ: A key protein in the pathogenesis of Psoriasis. Cytokine 2016, 78, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Spain, S.L.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Knight, J.; Tejasvi, T.; Kang, H.M.; Allen, M.H.; Lambert, S.; et al. Enhanced meta-analysis and replication studies identify five new Psoriasis susceptibility loci. Nat. Commun. 2015, 6, 7001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coto-Segura, P.; Gonzalez-Lara, L.; Gómez, J.; Eiris, N.; Batalla, A.; Gómez, C.; Requena, S.; Queiro, R.; Alonso, B.; Iglesias, S.; et al. NFKBIZ in Psoriasis: Assessing the association with gene polymorphisms and report of a new transcript variant. Hum. Immunol. 2017, 78, 435–440. [Google Scholar] [CrossRef]
- Herrington, F.D.; Carmody, R.J.; Goodyear, C.S. Modulation of NF-κB signaling as a therapeutic target in autoimmunity. J. Biomol. Screen. 2016, 21, 223–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelsen, T.; Ljungberg, C.; Litman, T.; Huppertz, C.; Hennze, R.; Rønholt, K.; Iversen, L.; Johansen, C. IκBζ is a key player in the antipsoriatic effects of secukinumab. J. Allergy Clin. Immunol. 2020, 145, 379–390. [Google Scholar] [CrossRef] [Green Version]
- Coto-Segura, P.; González-Fernández, D.; Batalla, A.; Gómez, J.; González-Lara, L.; Queiro, R.; Alonso, B.; Iglesias, S.; Coto, E. Common and rare CARD14 gene variants affect the antitumour necrosis factor response among patients with Psoriasis. Br. J. Dermatol. 2016, 175, 134–141. [Google Scholar] [CrossRef]
- Bek, S.; Nielsen, J.V.; Bojesen, A.B.; Franke, A.; Bank, S.; Vogel, U.; Andersen, V. Systematic review: Genetic biomarkers associated with anti-TNF treatment response in inflammatory bowel diseases. Aliment. Pharmacol. Ther. 2016, 44, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Coto-Segura, P.; González-Lara, L.; Batalla, A.; Eiris, N.; Queiro, R.; Coto, E. NFKBIZ and CW6 in Adalimumab response among Psoriasis patients: Genetic association and alternative transcript analysis. Mol. Diagn. Ther. 2019, 23, 627–633. [Google Scholar] [CrossRef] [PubMed]
- O’Rielly, D.D.; Rahman, P. Genetic, epigenetic and pharmacogenetic aspects of Psoriasis and Psoriatic arthritis. Rheum. Dis. Clin. North. Am. 2015, 41, 623–642. [Google Scholar] [CrossRef] [PubMed]
- Batalla, A.; Coto, E.; González-Fernández, D.; González-Lara, L.; Gómez, J.; Santos-Juanes, J.; Queiro, R.; Coto-Segura, P. The Cw6 and late-cornified envelope genotype plays a significant role in anti-tumor necrosis factor response among psoriatic patients. Pharmacogenet. Genom. 2015, 25, 313–316. [Google Scholar]
- Novelli, L.; Lubrano, E.; Venerito, V.; Perrotta, F.M.; Marando, F.; Curradi, G.; Iannone, F. Extra-articular manifestations and comorbidities in Psoriatic Disease: A journey into the immunologic crosstalk. Front. Med. (Lausanne) 2021, 8, 737079. [Google Scholar] [CrossRef] [PubMed]
- Sundarrajan, S.; Arumugam, M. Comorbidities of Psoriasis—Exploring the links by network approach. PLoS ONE 2016, 11, e0149175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, M.; Baurecht, H.; Ried, J.S.; Rodriguez, E.; Schlesinger, S.; Volks, N.; Gieger, C.; Rückert, I.-M.; Heinrich, L.; Willenborg, C.; et al. Psoriasis and cardiometabolic traits: Modest association but distinct genetic architectures. J. Invest. Dermatol. 2015, 135, 1283–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coto, E.; Díaz-Corte, C.; Tranche, S.; Gómez, J.; Alonso, B.; Iglesias, S.; Reguero, J.R.; López-Larrea, C.; Coto-Segura, P. Gene variants in the NF-KB pathway (NFKB1, NFKBIA, NFKBIZ) and their association with type 2 diabetes and impaired renal function. Hum. Immunol. 2018, 79, 494–498. [Google Scholar] [CrossRef]
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a key player in the crosstalk between inflammation and cardiovascular diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar]
- Coto, E.; Reguero, J.R.; Avanzas, P.; Pascual, I.; Martín, M.; Hevia, S.; Morís, C.; Díaz-Molina, B.; Lambert, J.L.; Alonso, B.; et al. Gene variants in the NF-KB pathway (NFKB1, NFKBIA, NFKBIZ) and risk for early-onset coronary artery disease. Immunol. Lett. 2019, 208, 39–43. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, B.; Zhang, M.; Miao, H.; Sun, J. Significant association between rs28362491 polymorphism in NF-kappaB1 gene and coronary artery disease: A meta-analysis. BMC Cardiovasc. Disord. 2020, 20, 278. [Google Scholar] [CrossRef]
- Kracht, M.; Müller-Ladner, U.; Schmitz, M.L. Mutual regulation of metabolic processes and proinflammatory NF-κB signaling. J. Allergy Clin. Immunol. 2020, 146, 694–705. [Google Scholar]
- Alonso, S.; Villa, I.; Fernández, S.; Martín, J.L.; Charca, L.; Pino, M.; Riancho, L.; Morante, I.; Santos, M.; Brandy, A.; et al. Multicenter study of secukinumab survival and safety in Spondyloarthritis and Psoriatic arthritis: SEcukinumab in cantabria and ASTURias study. Front. Med. (Lausanne) 2021, 8, 679009. [Google Scholar] [CrossRef] [PubMed]
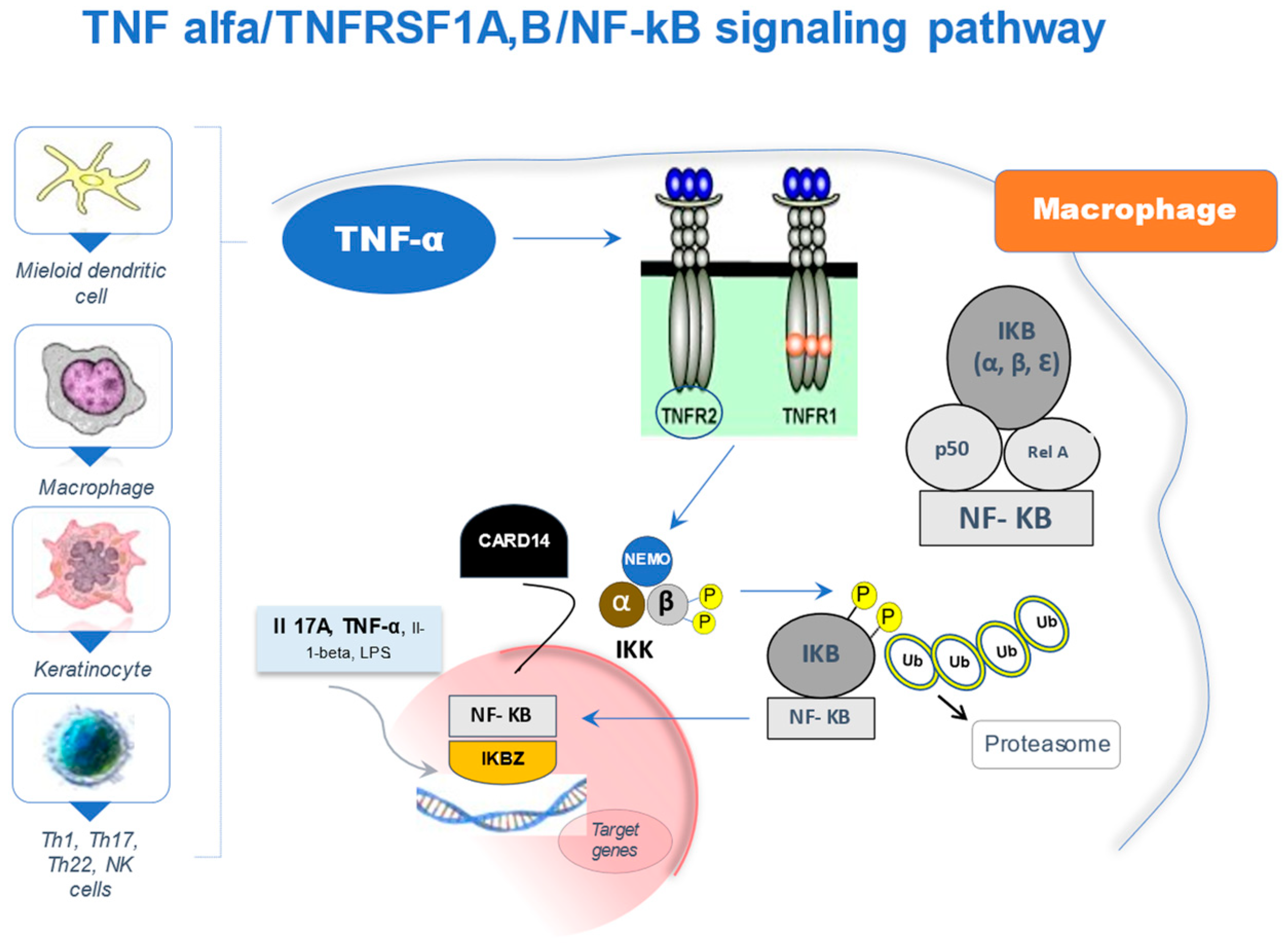

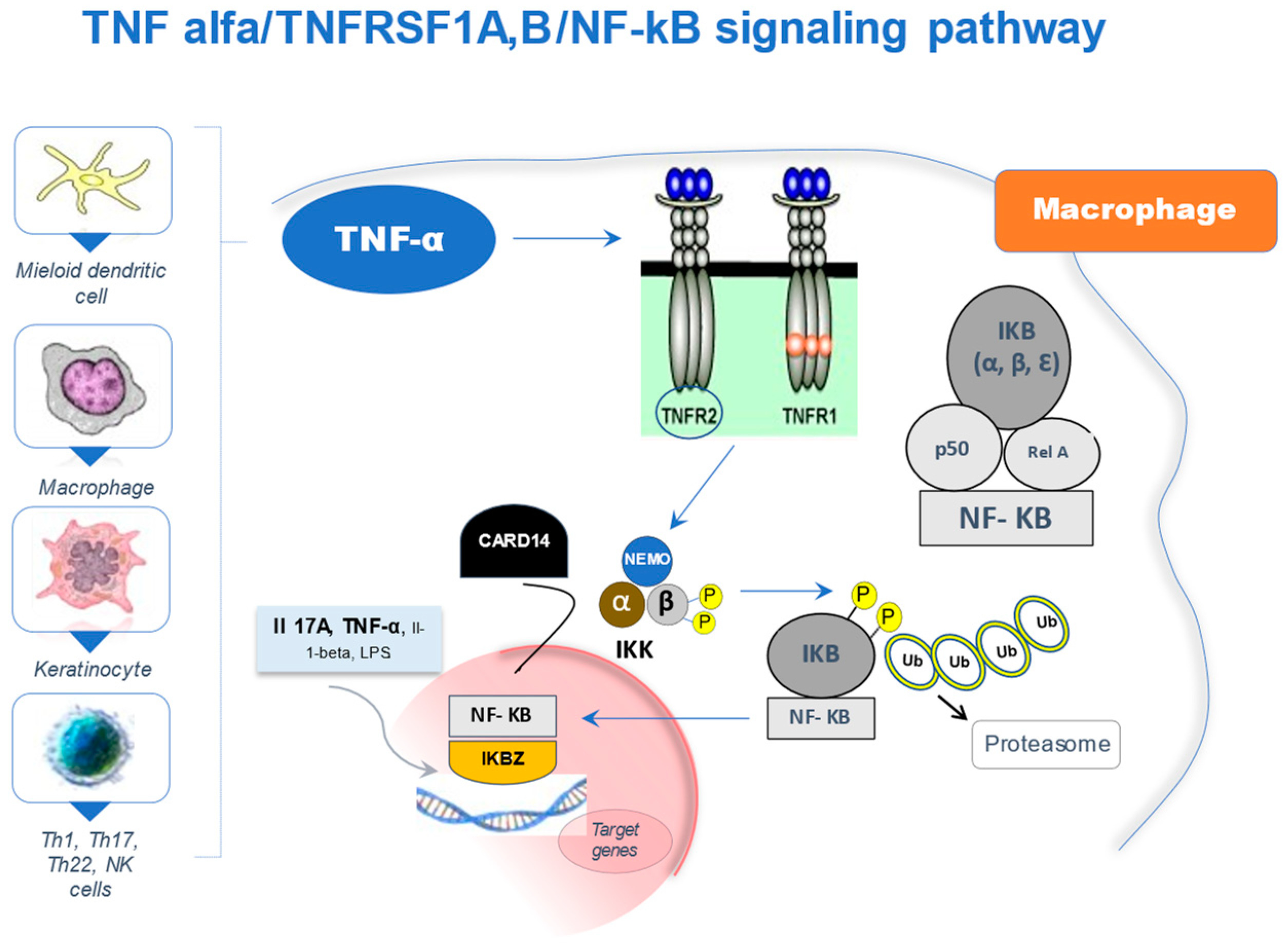{kind=link}
| Genetic Variant | Disease Feature |
|---|---|
| CARD14 rsll652075 (p. Arg820Trp) |
|
| TNFSRF1B rs1061622 (p. Met196Arg) |
|
| NFKB1 * rs28362491 (−94 of ATTG) NFKB1 rs7667496/rs28362491 |
|
| NFKBIA rs7152376 C (complete LD with rs12883343) |
|
| NFKBIZ rs7637230 NFKBIZ rs3217713 ins/ins NFKBIZ rs3217713 del/del |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Queiro, R.; Coto, P.; González-Lara, L.; Coto, E. Genetic Variants of the NF-κB Pathway: Unraveling the Genetic Architecture of Psoriatic Disease. Int. J. Mol. Sci. 2021, 22, 13004. https://doi.org/10.3390/ijms222313004
Queiro R, Coto P, González-Lara L, Coto E. Genetic Variants of the NF-κB Pathway: Unraveling the Genetic Architecture of Psoriatic Disease. International Journal of Molecular Sciences. 2021; 22(23):13004. https://doi.org/10.3390/ijms222313004
Chicago/Turabian StyleQueiro, Rubén, Pablo Coto, Leire González-Lara, and Eliecer Coto. 2021. "Genetic Variants of the NF-κB Pathway: Unraveling the Genetic Architecture of Psoriatic Disease" International Journal of Molecular Sciences 22, no. 23: 13004. https://doi.org/10.3390/ijms222313004
APA StyleQueiro, R., Coto, P., González-Lara, L., & Coto, E. (2021). Genetic Variants of the NF-κB Pathway: Unraveling the Genetic Architecture of Psoriatic Disease. International Journal of Molecular Sciences, 22(23), 13004. https://doi.org/10.3390/ijms222313004